

Abstract
Mammalian cells can utilize hydrogen sulfide (H2S) to support mitochondrial respiration. The aim of our study was to explore the potential role of S-sulfhydration (a H2S-induced posttranslational modification, also known as S-persulfidation) of the mitochondrial inner membrane protein ATP synthase (F1F0 ATP synthase/Complex V) in the regulation of mitochondrial bioenergetics. Using a biotin switch assay, we have detected S-sulfhydration of the α subunit (ATP5A1) of ATP synthase in response to exposure to H2S in vitro. The H2S generator compound NaHS induced S-sulfhydration of ATP5A1 in HepG2 and HEK293 cell lysates in a concentration-dependent manner (50–300 μM). The activity of immunocaptured mitochondrial ATP synthase enzyme isolated from HepG2 and HEK293 cells was stimulated by NaHS at low concentrations (10–100 nM). Site-directed mutagenesis of ATP5A1 in HEK293 cells demonstrated that cysteine residues at positions 244 and 294 are subject to S-sulfhydration. The double mutant ATP synthase protein (C244S/C294S) showed a significantly reduced enzyme activity compared to control and the single-cysteine-mutated recombinant proteins (C244S or C294S). To determine whether endogenous H2S plays a role in the basal S-sulfhydration of ATP synthase in vivo, we compared liver tissues harvested from wild-type mice and mice deficient in cystathionine-gamma-lyase (CSE, one of the three principal mammalian H2S-producing enzymes). Significantly reduced S-sulfhydration of ATP5A1 was observed in liver homogenates of CSE−/− mice, compared to wild-type mice, suggesting a physiological role for CSE-derived endogenous H2S production in the S-sulfhydration of ATP synthase. Various forms of critical illness (including burn injury) upregulate H2S-producing enzymes and stimulate H2S biosynthesis. In liver tissues collected from mice subjected to burn injury, we detected an increased S-sulfhydration of ATP5A1 at the early time points post-burn. At later time points (when systemic H2S levels decrease) S-sulfhydration of ATP5A1 decreased as well. In conclusion, H2S induces S-sulfhydration of ATP5A1 at C244 and C294. This post-translational modification may be a physiological mechanism to maintain ATP synthase in a physiologically activated state, thereby supporting mitochondrial bioenergetics. The sulfhydration of ATP synthase may be a dynamic process, which may be regulated by endogenous H2S levels under various pathophysiological conditions.
Keywords: H2S, bioenergetics, S-sulfhydration, burn, ATP synthase, cysteine, mitochondria, hydrogen sulfide, burn injury
Graphical abstract
1. Introduction
The biology and pathobiology of the gasotransmitter hydrogen sulfide (H2S) have received significant attention over the last decades. H2S exerts broad range of physiological effects as an antioxidant, cytoprotective agent [1,2], vasorelaxant [3–5], neurotransmitter [6,7], and endogenous promoter of angiogenesis [8,9]. H2S also plays important roles in the regulation of various inflammatory processes via a variety of mechanisms including the regulation of pro-inflammatory signaling [10–13]. Furthermore, H2S plays a role in the control of metabolism, via the regulation of glucose and lipid metabolism, insulin sensitivity and energy balance [14–16].
During the last ten years the role of H2S in the regulation of cellular bioenergetics has been completely re-evaluated. It has been known for several decades high concentrations of H2S reversibly inhibit cytochrome c oxidase (complex IV) of the mitochondrial electron transport chain, resulting in the inhibition of ATP production and metabolic suppression [17]. However, an additional body of more recent data, which has emerged over the last decade, demonstrates that H2S, at lower concentrations, exerts a directionally different effect on mitochondria: it acts as a stimulator of mitochondrial bioenergetics and a mitochondrial protectant through a combination of several different mechanisms [18–35]. First, H2S donates electrons to the mitochondrial electron transport chain through sulfide:quinone oxidoreductase (SQR) and mitochondrial Complex II, thereby stimulating oxidative phosphorylation (OXPHOS) and increasing mitochondrial ATP production [18–27]. Second, H2S as a free radical scavenger can neutralize mitochondrial reactive oxygen and reactive nitrogen species, thereby maintaining mitochondrial integrity [28]. Third, H2S inhibits the intramitochondrial phosphodiesterase 2A isoform, which, in turn, elevates intramitochondrial cAMP level; thus process stimulates mitochondrial electron transport, most likely via cAMP-dependent protein kinases, which phosphorylate key mitochondrial electron transport subunits [31]. Fourth, H2S can stimulate mitochondrial DNA repair [32]. Fifth, H2S can be metabolized to sulfite, sulfate and thiosulfate in the mitochondria; these species can act as endogenous storage ‘pools’ of H2S (physiological “prodrugs”) from which H2S can be liberated [33–35].
ATP synthase (EC 3.6.3.14), also called mitochondrial Complex V, is localized in the mitochondrial inner membrane, and it is responsible for generating adenosine triphosphate (ATP). In order to produce ATP from adenosine diphosphate (ADP) and inorganic phosphate (Pi), ATP synthase requires energy. Carbon-based substrates, such as pyruvate and succinate provide NADH/H+ and FADH2 via the Krebs cycle in the mitochondrial matrix. These molecules donate electrons to mitochondrial Complex I and Complex II, fueling the mitochondrial electron transport chain; electrons flow through Complex III and finally complex IV, where they react with O2 to form water. According to the theory of chemiosmosis, the purpose of this process is the pumping of protons from the mitochondrial matrix into the intermembrane space. This proton gradient is, in turn, “harvested” by ATP synthase via a rotatory mechanism to produce ATP [36–38].
Recent studies revealed a novel H2S-induced posttranslational modification, termed protein S-sulfhydration (also known as S-persulfidation) [39–41]. During this process, the –SH group of cysteine residues become covalently converted to a –SSH group, which can result in changes in the activity of the protein. This process importantly contributes to physiological and pathophysiological H2S- signaling. In 2010, Snyder’s group, using liquid chromatography followed by tandem mass spectrometry (LC-MS/MS), identified 39 S-sulfhydrated proteins in liver lysates. These proteins include GAPDH, beta-tubulin, actin as well as mitochondrial ATP synthase [39]. We have decided to follow up on the last observation, in order to investigate the mechanisms and functional consequences of the sulfhydration of mitochondrial ATP synthase. The data presented in the current report confirm the sulfhydration of ATP synthase; identify the two regulatory cysteines involved and indicate that sulfhydration of ATP synthase serves as an activating post-translational modification and a stimulatory process of cellular bioenergetics.
2. Materials and Methods
2.1. Materials
Sodium hydrosulfide hydrate (NaHS × H2O), neocuproine, deferoxamine mesylate salt (DFO), S-Methyl methanethiosulfonate (MMTS), polyvinylpyrrolidone were obtained from Sigma-Aldrich (St. Louis, MO). StartingBlock™ T20 (TBS) blocking buffer, Lipofectamine 2000, EZ-Link™ HPDP-biotin and Pierce™ streptavidin agarose were purchased from Thermo Fisher Scientific Inc. (Waltham, MA).
2.2. Cell culture
The human hepatocellular carcinoma-derived cell line (HepG2) and human embryonic kidney cells (HEK293) were obtained from the American Type Culture Collection (ATCC, Manassas, VA). Cell cultures were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 1 g/l glucose and 10% fetal bovine serum (Invitrogen, Carlsbad, CA), 4 mM glutamine, 100 IU/ml penicillin and 100 μg/ml streptomycin.
2.3. Animal handling
CSE−/− mice were generated as previously described [42]. For the mouse model of burn injury, male mice (10–12 weeks old) were used. All animal experiments were conducted in compliance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health and approved by the local Animal Care Committee. Animals were maintained on standard rodent chow and had free access to food and water.
2.4. Biotin switch assay of S-sulfhydration
The biotin switch assay was carried out as described previously with some modifications [16]. Cells or liver tissue was homogenized in HEN buffer (250 mM Hepes-NaOH pH 7.7 supplemented with 1 mM EDTA, 0.1 mM neocuproine) containing 150 μM deferoxamine, 1% Nonidet-P40 (NP-40), and protease/phosphatase inhibitors. Homogenized samples were sonicated and centrifuged at 16,000 g for 15 min at 4°C. The supernatant samples were placed into 10K molecular weight concentrator and the retentates were used for protein determination. Lysates were diluted to reach 5.5 mg/ml final protein concentration and were added to HEN buffer supplemented with 2.5% SDS and 20 mM methyl methanethiosulfonate (MMTS). The samples were frequently shaked at 50°C for 25 min. Then, the MMTS was removed by adding acetone and in the meanwhile, the proteins were also precipitated at −20°C for 45 min. Pellets were resuspended in HEN buffer containing 1 % SDS and 4 mM biotin-N-[6-(biotinamido) hexyl]-3′-(2′-pyridyldithio) propinamide (HPDP). After 3-hour-long incubation at 25°C, biotinylated proteins were purified by streptavidin-agarose beads. The biotinylated proteins were eluted in 2× Laemmeli sample buffer and subjected to Western blotting with anti-ATP5A1 antibody (Abcam, Cambridge, MA, USA), anti-GAPDH antibody (Proteintech Group, Inc., Rosemont, IL) or anti-tetra His antibody (Qiagen, Hilden, Germany).
2.5. Western blotting
Mouse liver tissue or cells were lysed (25 μg protein/10 μl) and diluted in Biorad 2× Laemmeli Sample Buffer. Samples were resolved on 4–12% NuPage Bis-Tris acrylamide gels (Thermo Fisher Scientific, Waltham, MA) and transferred to PVDF membranes. Membranes were blocked with 1% polyvinylpyrrolidone and then probed overnight with primary antibodies; such as (1) anti-ATP synthase 5A1 antibody (anti-ATP5A1, 1:1000, Abcam, Cambridge, MA), (2) anti-GAPDH antibody (anti-GAPDH, 1:1000, Proteintech Group, Inc., Rosemont, IL) or (3) anti-tetraHIS antibody (anti-His, 1:500, Qiagen, Hilden, Germany). On the following day, anti-goat (HRP, 1:3000, Abcam, Cambridge, MA) or anti-mouse-horseradish peroxidase conjugate secondary antibody (Sigma, St. Louis, MO) was applied. An enhanced chemiluminescent substrate (ECL, Pierce) was used to detect the signal using a CCD-camera-based chemiluminescence detection system or high sensitivity films (Amersham Hyperfilm ECL). The intensity of Western blot signals was quantified by densitometry using the ImageJ 1.45s software (NIH). The S-sulfhydrated samples were compared to the aliquots of lysate (“loading controls”) that had not been subjected to the S-sulfhydration assay.
2.6. Site-directed mutagenesis
Human ATP5A1 cDNA construct was purchased from Sino Biological Inc. (Beijing, China). ATP5A1 cDNA was cloned into pGEM-T Vector/pCMV2-His tag. A single mutation at cyteine-244 or cysteine-294 in ATP5A1 was conducted using the Quick Change Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA). The oligonucleotides using for mutagenesis were 5′-GAAAAGAAGAAGCTGTACTccATTTATGTTGCTATTGGTC-3′ (forward) and 5′-GACCAATAGCAACATAAATggAGTACAGCTTCTTCTTTTC-3′ (reverse) for cysteine-244, and 5′-CCTTACTCTGGCTcTTCCATGGGAGAG-3′ (forward) and 5′-CTCTCCCATGGAAgAGCCAGAGTAAGG-3′ (reverse) for cysteine-294. The site-directed mutants were confirmed by DNA sequencing at the MOBIX Laboratory of McMaster University, ON, Canada.
2.7. ATP synthase activity measurement
A commercially available ATP synthase enzyme ELISA assay (Abcam, Cambridge, MA) was used with modifications to determined ATP synthase activity from cell lysates or from purified recombinant proteins. The kit is designed to immunocapture ATP synthase proteins using the same anti-ATP5A1 antibody that we previously applied for the biotin-switch S-sulfhydration assay. The immunocaptured ATP synthase enzyme operates in the “reverse” direction as the physiological role of ATP synthase: it hydrolyzes ATP to ADP and phosphate. This production of ADP is, in turn, coupled to the oxidation of NADH to NAD+, which is monitored as a decrease in absorbance at 340 nm. ATP hydrolysis is inhibited by oligomycin (a specific inhibitor of ATP synthase that inhibits through binding to the Fo domain of the protein). During the cell lysate preparation, the entire ATP synthase enzyme is immunocaptured; we considered the oligomycin-inhibitable component of the ATP consumption as specific ATP synthase activity.
2.8. Mouse model of burn injury
Male C57/BL6 mice (10–12 weeks old) were housed at 24–26°C on a 12:12 light: dark cycle. Burn injury was induced as described [43]. Sham mice and mice subjected to burn injury underwent identical experimental procedures except of injury. Following an intraperitoneal (i.p.) injection of 0.1 mg/kg buprenorphine, mice were anesthetized by inhalation of 3–5% isoflurane. Next, ~40% of the dorsum was shaved with electrical clippers and ~ 1 cc of lactated ringers (LR) solution was injected under the skin along the spinal column. The dorsa of burn treated animals were then exposed to ~95°C water for 10 sec to produce a full thickness scald wound covering ~30% of the total body surface area (TBSA). Mice were then resuscitated with 2 cc of LR. Burn and sham-treated mice were individually housed throughout the experimental period. To minimize animal suffering, pain or distress, animals were scored twice daily throughout the post-burn period using an IACUC-approved Rodent Intervention Score Sheet to assess their well-being and clinical status by a certified veterinarian. To minimize the suffering of the animals the analgesic buprenorphine was administered when indicated to reduce pain and distress. Cohorts of mice were sacrificed at 1, 4, 10, 20, and 30 days post-injury. Liver tissues were used for further analysis.
2.9. Statistical analysis
Data are shown as means ± SEM. Student’s t-tests and one-way ANOVA with Dunnett post-hoc test were used to detect differences between groups; *p < 0.05 and **p < 0.01 considered statistically significant. All statistical calculations were performed using the Graphpad Prism 5 analysis software.
3. Results
3.1. H2S induces the S-sulfhydration of the ATP synthase alpha subunit
The S-sulfhydration of ATP synthase was confirmed both in HepG2 and HEK293 cell lysates utilizing a biotin switch S-sulfhydration assay. The α subunit of the F1 domain (ATP5A1), which – together with the ß subunit – is indispensable for the catalytic activity of the enzyme [36–38] – was S-sulfhydrated. ATP5A1 sulfhydration increased in response to treatment of the cell lysates with NaHS (50 – 300 μM) (Fig. 1).
Figure 1.
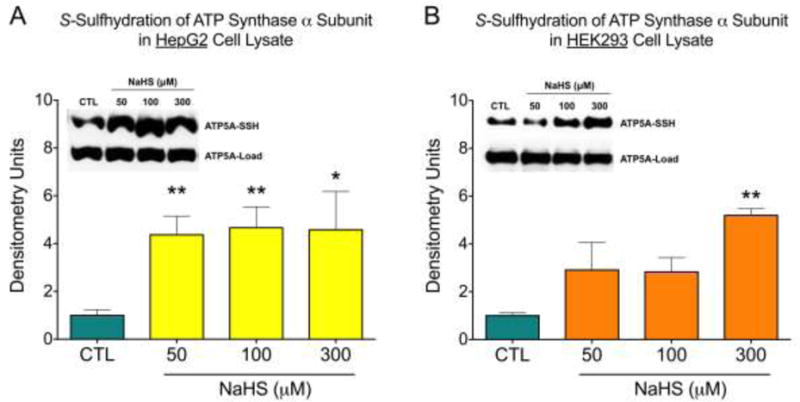
H2S induced S-sulfhydration of ATP5A1. HepG2 (A) and HEK 293 (B) cell lysates were incubated with NaHS at various concentrations (50–300 μM) for 30 min at 37°C. S-sulfhydration of ATP5A1 was detected by the biotin switch assay using anti-ATP5A1 antibody. Densitometry data show mean±SEM of n=5 independent experiments, *p<0.05 and **p<0.01 indicate significantly increased S-sulfhydration after NaHS, compared to the vehicle control (CTL) group.
3.2. H2S increases ATP5A1 activity
Next, we measured the specific activity of ATP synthase with and without NaHS treatment. ATP synthase isolated from both HEK and HepG2 cell lysates showed a concentration-dependent increase of its enzymatic activity in response to NaHS (10 – 100 nM). The effect of NaHS – similar to many other pharmacological effects of H2S – followed a bell-shaped concentration-response curve: at higher concentrations (1–10 μM) the activation was no longer apparent, and a tendency for an inhibition of enzymatic activity was noted (Fig. 2).
Figure 2.
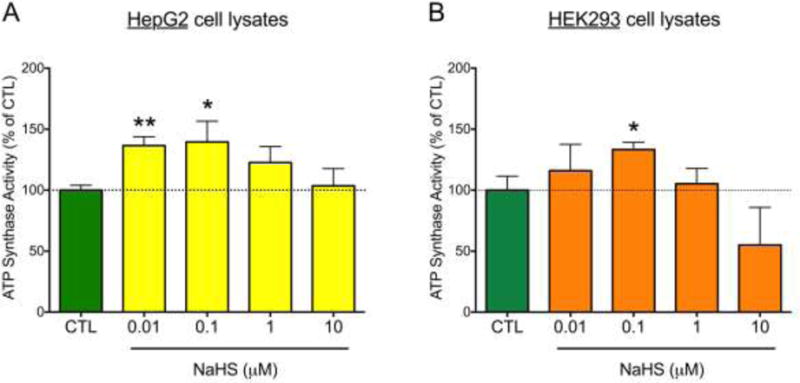
NaHS stimulates ATP5A1 enzyme activity in HepG2 (A) and HEK293 (B) cell lysates. ATP synthase protein, immunocaptured from the cell homogenates, was incubated with NaHS for 30 min at 37°C, and enzyme activity was determined by a kinetic, spectrophotometric assay which measures the consumption of ATP and the consequent production of ADP (which is coupled to the oxidation of NADH to NAD+). The oligomycin-inhibitable component of the ATP consumption was considered specific ATP synthase activity. Data were expressed as percent values of control (CTL) the ATP synthase activity (immunocaptured ATP synthase treated with NaHS vehicle). Data show mean±SEM values from n=3 independent experiments. *p<0.05 and **p<0.01 indicate the stimulatory effect of NaHS on ATP synthase.
3.3. H2S induced S-sulfhydration of ATP5A1 occurs at cysteines 244 and 294
There are two highly conserved cysteine (Cys) residues, Cys 244 or Cys 294, in ATP5A1 (Fig. 3). To determine the cysteines of ATP5A1 that are subject to S-sulfhydration, we mutated Cys 244 and Cys 294 to serine via site-directed mutagenesis. HEK293 cells were then transfected with plasmids encoding three different recombinant mutants (C244S; C294S; C244S and C294S double-mutant). NaHS-induced S-sulfhydration was abolished in HEK293 cells expressing the recombinant double mutant ATP5A1 (C244S/C294S) (Fig. 4).
Figure 3.
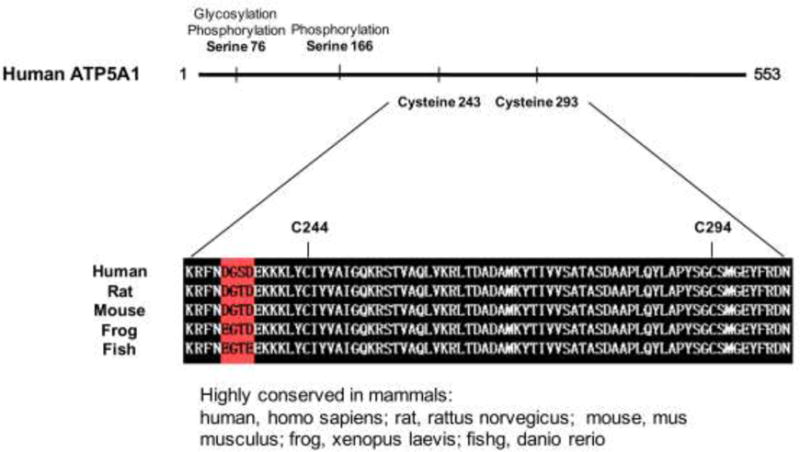
Domains of ATP5A1, indicating the sites of highly conserved cysteine residues across different species.
Figure 4.
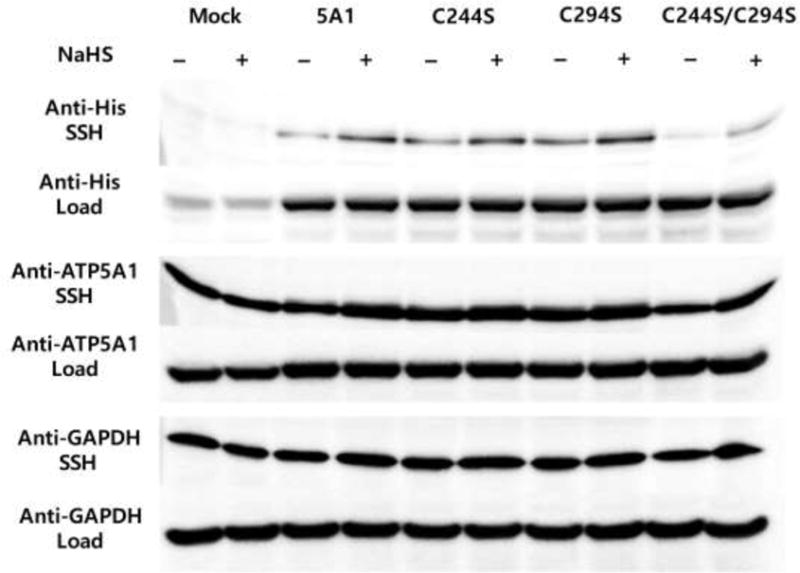
Western blot analysis of HEK-293 cells transfected either with empty vector, or native human ATP5A1 (5A1), or cysteine 244 mutant (C244S) human ATP5A1, or cysteine 294 mutant (C294S) human ATP5A1, or the double mutant C244S/C294S human ATP5A1 plasmids for 48 hrs. Cell lysates were incubated with NaHS (100 μM) for an additional 30 min at 37°C. The biotin switch assay using anti-tetraHis antibody was used to differentiate the recombinant ATP5A1 from the endogenous ATP5A1.
3.4. Double mutation of Cys 244 and 294 suppresses ATP synthase enzyme activity
Comparison of the activity of the HIS-tagged labeled recombinant proteins (C244S; C294S; C244S/C294S) from HEK293 cell lysates demonstrated that double mutant recombinant protein (C244S/C294S) exhibits significantly reduced enzyme activity (over 50% suppression of activity), compared to the single cysteine mutated versions, which exhibit a slight reduction in activity (Fig. 5).
Figure 5.
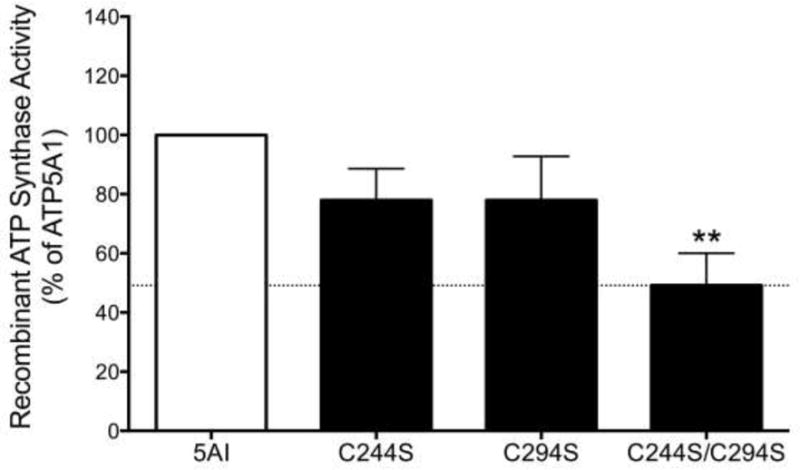
The double mutant recombinant protein (C244S/C294S) exhibits a significantly reduced enzyme activity compared to the intact human ATP5A1 protein. Mean±SEM values are shown from n=3 independent experiments. **p<0.01 indicates significant suppression of the catalytic activity of the double mutant protein, compared to wild-type protein.
3.5. Basal ATP5A1 S-sulfhydration is reduced CSE-KO mice
Wild-type mice exhibited a detectable basal level of S-sulfhydration of ATP5A1; ATP5A1 sulfhydration was significantly lower in liver homogenates harvested from CSE−/− mice than ATP5A1 sulfhydration in wild-type mice (Fig. 6).
Figure 6.
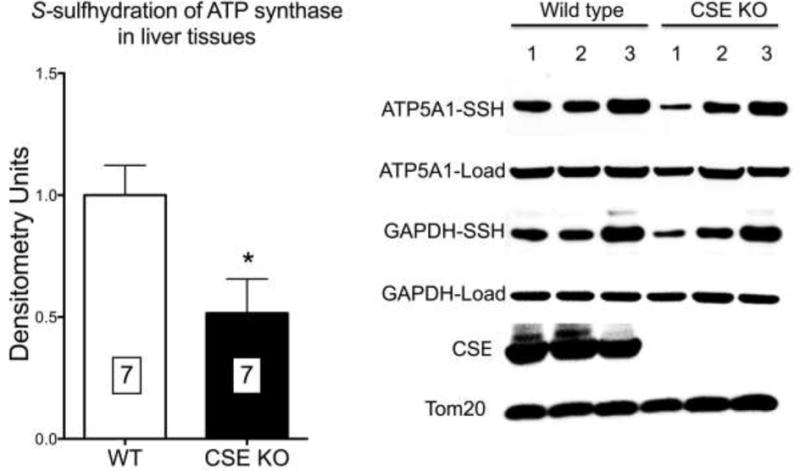
ATP5A1 prepared from livers of CSE−/− mice exhibits reduced S-sulfhydration compared to ATP synthase prepared from livers of wild-type mice. Densitometry analysis represents mean±SEM values of n=7 wild-type or n=7 CSE−/− livers. *p<0.05 shows significant difference between the CSE−/− and the wild-type group.
3.6. S-sulfhydration of ATP5A1 is increased after burn injury
Recent studies demonstrate that CSE is upregulated and H2S plasma levels are increased in mice subjected to burn injury [44,45]. In liver homogenates obtained from mice subjected to burn injury, increased S-sulfhydration of ATP5A1 was detected at the early time points, followed by a return to baseline over time (Fig. 7). S-sulfhydration of GAPDH [39] – a positive control in our S-sulfhydration assay – exhibited a similar time-course (Fig. 7).
Figure 7.
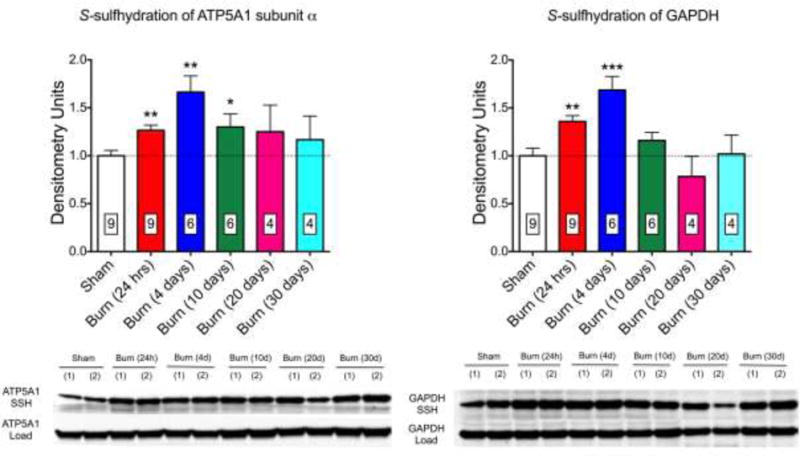
S-sulfhydration of ATP5A1 (A) and GAPDH (B) is increased at early time points after burn injury in livers of mice. A representative western blot and densitometry analysis (mean±SEM) of n=4–9 liver tissues is shown. *p<0.05 and **p<0.01 represent significant increases in the sulfhydration of the target enzyme after burn injury compared to the sham group.
4. Discussion
The main conclusions of the current study are the following: (a) confirming and extending on a the prior observation [39] that ATP synthase is a subject of sulfhydration by H2S, we have demonstrated that the alpha subunit of ATP synthase (ATP5A1) becomes sulfhydrated in response to the H2S donor NaHS; (b) ATP5A1 sulfhydration occurs at Cys 244 and Cys294; (c) H2S, at low concentrations, stimulates the activity of ATP synthase; (d) mutation of either cysteine 244 or 294 slightly decreases the catalytic activity of ATP synthase, while mutation of both cysteines results in a marked inhibition; (e) in vivo, the alpha subunit of ATP synthase is basally sulfhydrated; (f) the basal sulfhydration of ATP synthase is, at least in part, due to CSE-derived H2S, as it is suppressed in CSE−/− mice; and, finally, (g) burn injury, which upregulates CSE and increases H2S production, results in an increase in ATP synthase sulfhydration.
Recent work has identified several distinct H2S-related signaling mechanisms. First, H2S can react with metal centers by binding and transferring electrons. For example, cytochrome c oxidase (Complex IV) contains two heme molecules, a cytochrome ‘a’ and cytochrome ‘a3’, and two copper centers, the CuA, and CuB centers. Reaction of these centers with H2S results in the inhibition of the activity of Complex IV, leading in mitochondrial “poisoning”. This mechanism is generally accepted as the principal mechanism of toxicity of high concentrations of H2S [18,19]. Second, H2S can neutralize reactive oxygen and nitrogen species via direct as well as indirect mechanisms. H2S can directly act as a free radical and oxidant scavenger, although with relatively slow reaction rates; and it can also upregulate endogenous cellular antioxidant responses, for instance via the regulation of the NRF2 response [2,28,40]. Via these mechanisms, H2S can exert cytoprotective and mitochondrial protective effects. A third pathway of H2S signaling occurs via post-transcriptional modulation of reactive cysteine residues via protein S-sulfhydration (also termed S-persulfidation) [39–41,46]. During the S-sulfhydration, H2S covalently modifies the SH groups of cysteines, creating persulfides (-SSH groups). Cysteines can be subject to several different oxidative posttranslational modifications (oxPTMs) (similar to S-nitrosylation, S-sulfonation, S-sulfenylation, and S-glutathionylation) in addition to S-sulfhydration. Persulfide formation does not occur through direct interaction between H2S and the –SH group of cysteines, because sulfur is in its lowest oxidation state, −2, in both H2S and –SH group. Therefore, polysulfides have been identified as the chemical species that are directly responsible for protein sulfhydration [41,46]. Nevertheless, in in vitro experimental systems, S-sulfhydration reactions can be produced by chemical H2S donors [39–41,46–52], presumably because these donors lead to the formation of polysulfide species, which, in turn, result in the formation of the S-sulfhydrated cysteine groups on various protein targets. Several proteins have been identified as targets of S-sulfhydration by H2S. The effect of S-sulfhydration on protein function depends on the pH and the position of the target cysteine amino acids in the protein (e.g. its proximity to the active centrum of the enzyme), the surrounding amino acids, local electric charge and redox status [41,46]. The current report confirms that the ATP5A1 is a S-sulfhydration target and indicate that the functional consequence of this reaction is the stimulation of ATP synthase activity. It remains to be further elucidated whether the S-sulfhydration of ATP synthase is transient or permanent; to our knowledge the reversibility of S-sulfhydration reactions under cellular or physiological conditions has not yet been explored in the literature.
Several groups, including our own, have previously characterized the effect of H2S on mitochondrial activity and cellular bioenergetics [18,19,22–27]. These studies demonstrated that H2S induces a bell-shaped bioenergetic concentration-response in various cell types in vitro, with activation at lower concentrations and inhibition at higher concentrations. In followup studies different parts of the mitochondrial electron transport chain were separately tested. When Complexes I–IV or Complexes II-IV were functional, the bell-shaped effect of H2S persisted; when Complex IV alone was functional, the activating effect of H2S was not seen at lower concentrations, but the inhibitory effect at higher concentrations persisted [27]. In these experiments, however, ATP synthase activity was not measured; the assay used (Extracellular Flux Analysis) measures oxygen consumption, and not ATP production. In other experiments, however, we have also measured ATP generation [24] and showed that NaHS increases it. It would be important to measure the effect of H2S on ATP synthase activity directly in a cellular or mitochondrial preparation; however, unfortunately such assays are currently beyond our technical capabilities. The assay shown in the current paper (which is based on ATP synthase activity measurement using an ELISA system), nevertheless, indicate that H2S has a direct effect on the catalytic activity of ATP synthase.
We have noted in the current study that that there is a significant difference in the concentration-responses to H2S when using whole cell-homogenate-based assays vs. isolated/recombinant enzymes or similar reductionist systems. Based on prior studies in the literature, we believe that the difference between the cell-homogenate-based potency of H2S and the enzyme-based potency of H2S is due to the chemical and pharmacological nature of H2S. When cell-homogenates or whole-cell-based systems are used, first of all, the indicated H2S concentrations only represent nominal and initial concentrations. For instance, in cell-based studies, H2S concentrations will diminish by the time H2S reaches the cells at the bottom of the culture dish, and they will further diminish as they reach the cytosolic – and especially the mitochondrial – compartment. This is believed to happen due to passive degradation/oxidation as well as active metabolism of H2S though various metabolic pathways (e.g. rhodanese). Similarly, in whole cell homogenates, the H2S applied to the homogenate is subject to degradation and reactions by multiple enzymes and non-enzymatic processes, which diminishes its actual concentration that is “seen” by the target enzyme of the experiment. So far, to our knowledge, no investigators have been able to reliably quantify, in absolute terms, the intracellular (e.g. mitochondrial) H2S levels that one can reach when applying exogenous H2S donors (or even when applying mitochondrially targeted ones). We can, nevertheless, assume that the concentrations that ATP synthase “sees” in a whole cell homogenate are markedly lower than the initial (applied) concentrations of H2S. In contrast, in systems where H2S donors are directly mixed with recombinant or isolated enzymes dissolved in simple solutions, the actual H2S concentrations that the enzyme “sees” are likely to be much closer to the initially applied (nominal) concentrations. In the current paper we find that the concentrations of H2S needed to S-sulfhydrate ATP synthase, when cell homogenates were treated with H2S, was approximately 100 μM, while the concentrations of H2S needed to activate immunocaptured H2S in a reductionist in vitro system was approximately 0.1 μM (a 1000-fold concentration difference). Similarly, we have previously demonstrated that the concentration of H2S needed to inhibit Complex IV activity in whole mitochondrial preparation was 100 μM, while the concentration of H2S needed to inhibit purified mitochondrial Complex IV were 0.1 μM [27] (again, a 1000-fold concentration difference). Taken together, the above considerations, together with the finding that mutation of the S-sulfhydrated cysteines diminishes the catalytic activity of ATP synthase are consistent with the suggestion that there is a causative link between the sulfhydration of ATP synthase and its catalytic activation by H2S.
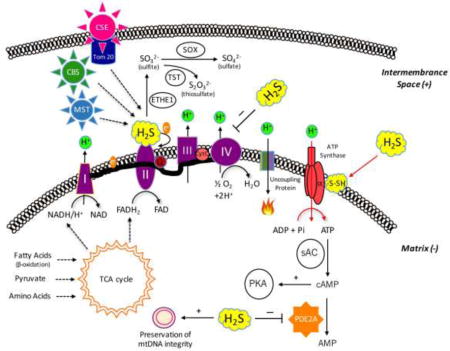
Previous studies have demonstrated that low concentrations of H2S can stimulate mitochondrial function via a variety of mechanisms (Fig. 8). H2S (a diffusible molecule) can enter the mitochondrial compartment either via diffusion after it is produced by cytosolic enzymes – CSE, or cystathionine beta synthase (CBS), or 3-mercaptopyruvate sulfurtransferase (3-MST) – or by constitutive mitochondrial H2S-producing enzymes (3-MST and CBS). Moreover, CSE and CBS can also translocate into the mitochondria under various pathophysiological conditions [20,22]. Once in the mitochondrial compartment, H2S can stimulate mitochondrial function via a number of mechanisms outlined in the Introduction section [18–35]. Based on the results of the current study, H2S, via S-sulfhydration of ATP synthase (Complex V), can also contribute to the activation of mitochondrial function. The latter effect may be necessary to optimize mitochondrial efficiency, because if H2S, through electron donation [18–25] and/or through the stimulation of intramitochondrial cAMP levels [31], increases electron transport, this is expected to produce an increase in the proton gradient across the mitochondrial inner membrane. It makes, therefore, biological sense that an increase in the specific activity of ATP synthase on the “receiving end”, balances these processes, in order to efficiently “harvest” the increased proton gradient. All of these processes are shown in Fig. 8. For reasons of completeness, Fig. 8 also indicates the inhibitory effect of H2S on cytochrome c oxidase (complex IV) [17,27]; this process occurs at higher (pathophysiological or toxicological) levels of H2S.
Figure 8.
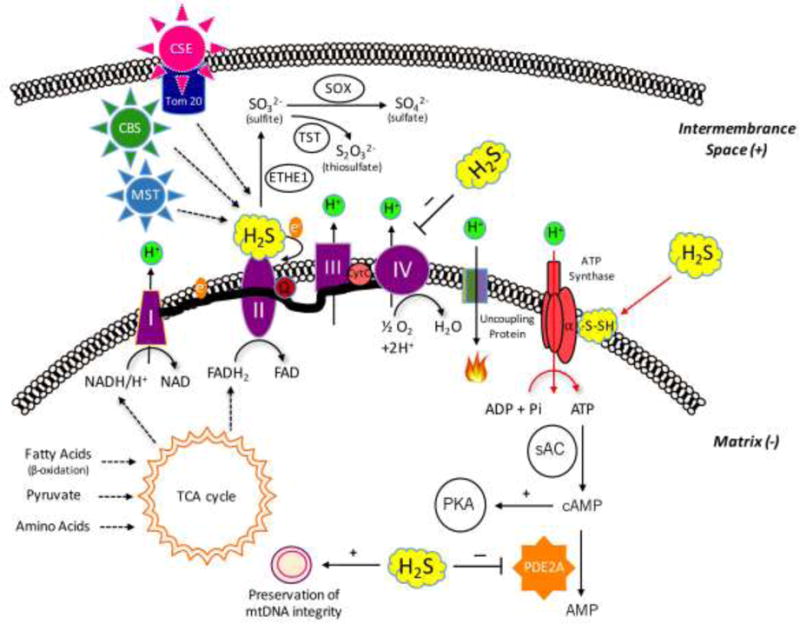
Summary of the various effects of H2S on mitochondrial bioenergetics. (a) The novel findings contained in the current report (S-sulfhydration of ATP synthase) are indicated with red arrows. H2S can be produced in the mitochondrion constitutively by two distinct H2S-producing enzymes, 3-MST, and CBS. Moreover, CSE is capable of translocating to the mitochondrial outer membrane under certain stress condition (e.g.: increased intracellular calcium signal), contributing the growth of intramitochondrial H2S level. Lower concentrations of H2S exert stimulatory effects on mitochondrial function via several different mechanisms: (b) H2S can donate electrons to mitochondrial electron transport chain. (c) H2S can act as an antioxidant neutralizing mitochondrial-derived reactive oxygen and nitrogen species, stabilizing the electron transport chain proteins, as well as preventing mitochondrial DNA damage. (d) H2S oxidization can results in sulfite, sulfate and thiosulfate; some of these species can also act as “pools” or sources of biologically active H2S. (e) H2S can inhibit mitochondrial PDE2A enzyme, which increases intramitochondrial cAMP levels, and stimulates mitochondrial function via activation of the cAMP-dependent protein kinase (PKA). Higher concentrations of H2S can also exert marked inhibitory effects on mitochondrial function: (f) H2S can inhibits cytochrome c oxidase (complex IV) resulting in a reversible inhibition of mitochondrial electron transport and ATP production.
Our data showing that S-sulfhydration of ATP synthase is lower in the CSE−/− mice presented indicate that basal S-sulfhydration of ATP synthase may be a physiological mechanism in response to endogenously produced H2S. In addition to CSE-derived H2S, mitochondrial H2S levels are also affected by 3-MST (a partially mitochondrially localized enzyme), as well as CBS (which is physiologically present in liver mitochondria) [22,23,27]. It is conceivable, therefore, that the production of H2S by 3-MST and/or CBS may also contribute to the sulfhydration of ATP synthase under physiological (or pathophysiological) conditions; this remains to be tested in future studies. Mitochondrially targeted H2S donors are also available now for in vitro and in vivo studies, for instance the compound AP39; this compound is known to increase cellular bioenergetics in vitro [30]. Further work needs to be conducted to test whether mitochondrial delivery of H2S increases the sulfhydration and catalytic activity of ATP synthase.
Previous work has already examined the functional consequences of various modifications of the cysteines in ATP synthase. Cys 294, which is located on the surface of ATP synthase protein, has been shown to form an acid–base motif and is easily targeted by various external redox stimuli. Moreover, Cys 244 and Cys 294 have been shown to form a disulfide bond under oxidative stress conditions [54–56]. In addition – similar to the results of the current study – Wang and colleagues have previously demonstrated that the catalytic activity of the α subunit of ATP synthase is decreased either after the mutation of Cys 244 or in response to the mutation of Cys 294 [54]. Cys 244 is now being viewed as a general ‘redox sensor’ of ATP synthase, which plays a key role in the regulation of the activity of ATP synthase under various physiological and pathophysiological conditions [55]. The exact (patho)physiological consequence(s) of the sulfhydration of ATP synthase – in view of the fact that the same cysteine can also be a target for a variety of other post-translational modifications – remains to be further explored in future studies.
We are aware of the fact that the current study has a number of limitations. First, the exact mechanism of the sulfhydration process (via H2S itself, or, more likely, via intermediary persulfide reactions) remains to be characterized. Second, a difference was noted between the concentrations of H2S needed to S-sulfhydrate ATP synthase and the concentrations needed to activate the enzyme. Although this concentration difference may be explained by the difference in the experimental conditions used, further work may be necessary to further address this issue. Third, we have not investigated the reversibility of the S-sulfhydration reaction. Fourth, we have not distinguished between the activating effect of H2S on ATP synthase activity (occurring at lower concentrations) and the inhibitory effects seen at higher concentrations. Fifth, the mechanism(s) and source(s) of H2S responsible for the endogenous sulfhydration of the enzyme remain to be identified; the experiments using CSE−/− mice indicate that CSE plays an important role; the roles of the other two H2S-generating enzymes (CBS and 3-MST) remain to be explored. Sixth, the role of the increased sulfhydration of ATP synthase – in light of the complex pathomechanisms of burn injury, and the multiple potential roles of H2S in burns [57] – remain to be characterized. Seventh, the current ex vivo studies only investigated one selected organ (the liver); it remains to be examined whether S-sulfhydration of ATP synthase occurs in other organs as well, and if it does, what functional role does it play. Eight, we have only studied one, selected pathophysiological condition (burn injury). Given the important changes in H2S homeostasis in various disease states with either increased or decreased H2S production, the potential changes in the sulfhydration of ATP synthase in various pathophysiological conditions remain to be explored in future studies.
Acknowledgments
K.M. was supported by a postdoctoral fellowship award and A.A.U. was supported by a doctoral research award from the Canadian Institutes of Health Research. This work was supported by a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada to R.W. This work was also supported by grants from the Shriners of North America (#85800) and the National Institutes of Health (R01 GM107846) to C.S. M.K. would like to thank Ms. Rosanne Licskai for her assistance in the preparation of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
The authors declare no conflicts of interest in relationship to this study.
Authors’ contributions
Katalin Módis: conduction of experiments, analysis of data, preparation of manuscript; YoungJun Ju, Akbar Ahmad, Ashley A. Untereiner, Zaid Altaany, Lingyun Wu: conduction of experiments; Csaba Szabo, Rui Wang: experimental design, interpretation of the data, preparation of manuscript.
References
- 1.Szabo C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6:917–35. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- 2.Calvert JW, Coetzee WA, Lefer DJ. Novel insights into hydrogen sulfide–mediated cytoprotection. Antioxid Redox Signal. 2010;12:1203–17. doi: 10.1089/ars.2009.2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J. 2001;20:6008–16. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li L, Rose P, Moore PK. Hydrogen sulfide and cell signaling. Annu Rev Pharmacol Toxicol. 2011;51:169–87. doi: 10.1146/annurev-pharmtox-010510-100505. [DOI] [PubMed] [Google Scholar]
- 5.Mustafa AK, Sikka G, Gazi SK, Steppan J, Jung SM, Bhunia AK, Barodka VM, Gazi FK, Barrow RK, Wang R, Amzel LM, Berkowitz DE, Snyder SH. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ Res. 2011;109:1259–68. doi: 10.1161/CIRCRESAHA.111.240242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abe K, Kimura H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci. 1996;16:1066–71. doi: 10.1523/JNEUROSCI.16-03-01066.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev. 2012;92:791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- 8.Papapetropoulos A, Pyriochou A, Altaany Z, Yang G, Marazioti A, Zhou Z, Jeschke MG, Branski LK, Herndon DN, Wang R, Szabo C. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc Natl Acad Sci USA. 2009;106:21972–7. doi: 10.1073/pnas.0908047106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coletta C, Papapetropoulos A, Erdelyi K, Olah G, Módis K, Panopoulos P, Asimakopoulou A, Gerö D, Sharina I, Martin E, Szabo C. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc Natl Acad Sci USA. 2012;109:9161–6. doi: 10.1073/pnas.1202916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whiteman M, Winyard PG. Hydrogen sulfide and inflammation: the good, the bad, the ugly and the promising. Expert Rev Clin Pharmacol. 2011;4:13–32. doi: 10.1586/ecp.10.134. [DOI] [PubMed] [Google Scholar]
- 11.Li L, Bhatia M, Zhu YZ, Zhu YC, Ramnath RD, Wang ZJ, Anuar FB, Whiteman M, Salto-Tellez M, Moore PK. Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. FASEB J. 2005;19:1196–8. doi: 10.1096/fj.04-3583fje. [DOI] [PubMed] [Google Scholar]
- 12.Badiei A, Chambers ST, Gaddam RR, Bhatia M. Cystathionine-γ-lyase gene silencing with siRNA in monocytes/macrophages attenuates inflammation in cecal ligation and puncture-induced sepsis in the mouse. J Biosci. 41(2016):87–95. doi: 10.1007/s12038-016-9598-9. [DOI] [PubMed] [Google Scholar]
- 13.Ahmad A, Druzhyna N, Szabo C. Delayed treatment with sodium hydrosulfide improves regional blood flow and alleviates cecal ligation and puncture (CLP) – induced septic shock. Shock. 2016 doi: 10.1097/SHK.0000000000000589. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ju Y, Untereiner A, Wu L, Yang G. H2S-induced S-sulfhydration of pyruvate carboxylase contributes to gluconeogenesis in liver cells. Biochim Biophys Acta. 2015;1850:2293–303. doi: 10.1016/j.bbagen.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 15.Untereiner AA, Wang R, Ju Y, Wu L. Decreased gluconeogenesis in the absence of cystathionine gamma-lyase and the underlying mechanisms. Antioxid Redox Signal. 2016;24:129–40. doi: 10.1089/ars.2015.6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Módis K, Karimi Z, Wang R. Mitochondria in Liver Disease. CRC Press; 2015. Regulation of mitochondrial function by hydrogen sulfide; pp. 105–140. [Google Scholar]
- 17.Cooper CE, Brown CG. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: chemical mechanism and physiological significance. J Bioenerg Biomembr. 2008;40:533–9. doi: 10.1007/s10863-008-9166-6. [DOI] [PubMed] [Google Scholar]
- 18.Goubern M, Andriamihaja M, Nübel T, Blachier F, Bouillaud F. Sulfide, the first inorganic substrate for human cells. FASEB J. 2007;8:1699–706. doi: 10.1096/fj.06-7407com. [DOI] [PubMed] [Google Scholar]
- 19.Lagoutte E, Mimoun S, Andriamihaja M, Chaumontet C, Blachier F, Bouillaud F. Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim Biophys Acta. 2010;1797:1500–11. doi: 10.1016/j.bbabio.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 20.Fu M, Zhang W, Wu L, Yang G, Li H, Wang R. Hydrogen sulfide (H2S) metabolism in mitochondria and its regulatory role in energy production. Proc Natl Acad Sci USA. 2012;109:2943–2948. doi: 10.1073/pnas.1115634109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Groeger M, Matallo J, McCook O, Wagner F, Wachter U, Bastian O, Gierer S, Reich V, Stahl B, Huber-Lang M, Szabo C, Georgieff M, Radermacher P, Calzia E, Wagner K. Temperature and cell-type dependency of sulfide effects on mitochondrial respiration. Shock. 2012;38:367–74. doi: 10.1097/SHK.0b013e3182651fe6. [DOI] [PubMed] [Google Scholar]
- 22.Teng H, Wu B, Zhao K, Yang G, Wu L, Wang R. Oxygen-sensitive mitochondrial accumulation of cystathionine beta-synthase mediated by Lon protease. Proc Natl Acad Sci USA. 2013;110:12679–84. 20–32. doi: 10.1073/pnas.1308487110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Módis K, Coletta C, Erdélyi K, Papapetropoulos A, Szabo C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 2013;27:601–11. doi: 10.1096/fj.12-216507. [DOI] [PubMed] [Google Scholar]
- 24.Módis K, Asimakopoulou A, Coletta C, Papapetropoulos A, Szabo C. Oxidative stress suppresses the cellular bioenergetic effect of the 3-mercaptopyruvate sulfurtransferase/hydrogen sulfide pathway. Biochem Biophys Res Commun. 2013;433:401–7. doi: 10.1016/j.bbrc.2013.02.131. [DOI] [PubMed] [Google Scholar]
- 25.Szabo C, Coletta C, Chao C, Módis K, Szczesny B, Papapetropoulos A, Hellmich MR. Tumor-derived hydrogen sulfide, produced by cystathionine-β-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc Natl Acad Sci USA. 2013;110:12474–9. doi: 10.1073/pnas.1306241110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beaumont M, Andriamihaja M, Lan A, Khodorova N, Audebert M, Blouin JM, Grauso M, Lancha L, Benetti PH, Benamouzig R, Tomé D, Bouillaud F, Davila AM, Blachier F. Detrimental effects for colonocytes of an increased exposure to luminal hydrogen sulfide: The adaptive response. Free Radic Biol Med. 2016;93:155–64. doi: 10.1016/j.freeradbiomed.2016.01.028. [DOI] [PubMed] [Google Scholar]
- 27.Szabo C, Ransy C, Módis K, Andriamihaja M, Murghes B, Coletta C, Olah G, Yanagi K, Bouillaud F. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br J Pharmacol. 2014;171:2099–122. doi: 10.1111/bph.12369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suzuki K, Olah G, Modis K, Coletta C, Kulp G, Gerö D, Szoleczky P, Chang T, Zhou Z, Wu L, Wang R, Papapetropoulos A, Szabo C. Hydrogen sulfide replacement therapy protects the vascular endothelium in hyperglycemia by preserving mitochondrial function. Proc Natl Acad Sci USA. 2011;108:13829–34. doi: 10.1073/pnas.1105121108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Módis K, Asimakopoulou A, Coletta C, Papapetropoulos A, Szabo C. Oxidative stress suppresses the cellular bioenergetic effect of the 3-mercaptopyruvate sulfurtransferase/hydrogen sulfide pathway. Biochem Biophys Res Commun. 2013;433:401–7. doi: 10.1016/j.bbrc.2013.02.131. [DOI] [PubMed] [Google Scholar]
- 30.Szczesny B, Módis K, Yanagi K, Coletta C, Le Trionnaire S, Perry A, Wood ME, Whiteman M, Szabo C. AP39, a novel mitochondria-targeted hydrogen sulfide donor, stimulates cellular bioenergetics, exerts cytoprotective effects and protects against the loss of mitochondrial DNA integrity in oxidatively stressed endothelial cells in vitro. Nitric Oxide. 2014;41:120–30. doi: 10.1016/j.niox.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Módis K, Panopoulos P, Coletta C, Papapetropoulos A, Szabo C. Hydrogen sulfide-mediated stimulation of mitochondrial electron transport involves inhibition of the mitochondrial phosphodiesterase 2A, elevation of cAMP and activation of protein kinase A. Biochem Pharmacol. 2013;86:1311–9. doi: 10.1016/j.bcp.2013.08.064. [DOI] [PubMed] [Google Scholar]
- 32.Sakaguchi M, Marutani E, Shin HS, Chen W, Hanaoka K, Xian M, Ichinose F. Sodium thiosulfate attenuates acute lung injury in mice. Anesthesiology. 2014;121:1248–57. doi: 10.1097/ALN.0000000000000456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Snijder PM, Frenay AR, Koning AM, Bachtler M, Pasch A, Kwakernaak AJ, van den Berg E, Bos EM, Hillebrands JL, Navis G, Leuvenink HG, van Goor H. Sodium thiosulfate attenuates angiotensin II-induced hypertension, proteinuria and renal damage. Nitric Oxide. 2014;42:87–98. doi: 10.1016/j.niox.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 34.Marutani E, Yamada M, Ida T, Tokuda K, Ikeda K, Kai S, Shirozu K, Hayashida K, Kosugi S, Hanaoka K, Kaneki M, Akaike T, Ichinose F. Thiosulfate mediates cytoprotective effects of hydrogen sulfide against neuronal ischemia. J Am Heart Assoc. 2015;4 doi: 10.1161/JAHA.115.002125. pii: e002125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Snijder PM, Baratashvili M, Grzeschik NA, Leuvenink HGD, Kuijpers L, Huitema S, Schaap O, Giepmans BNG, Kuipers J, Miljkovic JL, Mitrovic A, Bos EM, Szabo C, Kampinga HH, Dijkers PF, den Dunnen WFA, Filipovic MR, van Goor H, Sibon OCM. Overexpression of cystathionine γ-lyase suppresses detrimental effects of spinocerebellar ataxia type 3. Mol Med. 2016 doi: 10.2119/molmed.2015.00221. in press. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walker JE. The ATP synthase: the understood, the uncertain and the unknown Biochem. Soc Trans. 2013;41:1–16. doi: 10.1042/BST20110773. [DOI] [PubMed] [Google Scholar]
- 37.Rühle T, Leister D. Assembly of F1F0-ATP synthases. Biochim Biophys Acta. 2015;1847:849–86. doi: 10.1016/j.bbabio.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 38.Junge W, Nelson N. ATP synthase. Annu Rev Biochem. 2015;84:631–57. doi: 10.1146/annurev-biochem-060614-034124. [DOI] [PubMed] [Google Scholar]
- 39.Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, Barrow RK, Yang G, Wang R, Snyder SH. H2S signals through protein S-sulfhydration. Sci Signal. 2009;2:ra72. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paul BD, Snyderm SH. H2S: A Novel gasotransmitter that signals by sulfhydration. Trends Biochem Sci. 2015;40:687–700. doi: 10.1016/j.tibs.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kimura H. Signaling molecules: hydrogen sulfide and polysulfide. Antioxid Redox Signal. 2015;22:362–76. doi: 10.1089/ars.2014.5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH, Wang R. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–90. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Szczesny B, Brunyánszki A, Ahmad A, Oláh G, Porter C, Toliver-Kinsky T, Sidossis L, Herndon DH, Szabo C. Time-dependent and organ-specific changes in mitochondrial function, mitochondrial DNA integrity, oxidative stress and mononuclear cell infiltration in a mouse model of burn injury. PLoS One. 2015;10:e0143730. doi: 10.1371/journal.pone.0143730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang J, Sio SE, Moochhala S, Bhatia M. Role of hydrogen sulfide in severe burn injury-induced inflammation in mice. Mol Med. 2010;16:417–24. doi: 10.2119/molmed.2010.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brunyanszki A, Erdelyi K, Szczesny B, Olah G, Salomao R, Herndon DN, Szabo C. Upregulation and mitochondrial sequestration of hemoglobins occurs in circulating leukocytes during critical illness, conferring a cytoprotective phenotype. Mol Med. 2016 doi: 10.2119/molmed.2015.00187. in press. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Filipovi MR. Persulfidation (S-sulfhydration) and H2S. Handb Exp Pharmacol. 2015;230:29–59. doi: 10.1007/978-3-319-18144-8_2. [DOI] [PubMed] [Google Scholar]
- 47.Krishnan N, Fu C, Pappin DJ, Tonks NK. H2S-Induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci Signal. 2011;4:ra86. doi: 10.1126/scisignal.2002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang G, Zhao K, Ju Y, Mani S, Cao Q, Puukila S, Khaper N, Wu L, Wang R. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid Redox Signal. 2013;18:1906–19. doi: 10.1089/ars.2012.4645. [DOI] [PubMed] [Google Scholar]
- 49.Vandiver MS, Paul BD, Xu R, Karuppagounder S, Rao F, Snowman AM, Ko HS, Lee YI, Dawson VL, Dawson TM, Sen N, Snyder SH. Sulfhydration mediates neuroprotective actions of parkin. Nat Commun. 2013;4:1626. doi: 10.1038/ncomms2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao K, Ju Y, Li S, Altaany Z, Wang R, Yang G. S-sulfhydration of MEK1 leads to PARP-1 activation and DNA damage repair. EMBO Rep. 2014;15:792–800. doi: 10.1002/embr.201338213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Altaany Z, Ju Y, Yang G, Wang R. The coordination of S-sulfhydration, S-nitrosylation, and phosphorylation of endothelial nitric oxide synthase by hydrogen sulfide. Sci Signal. 2014;7:ra87. doi: 10.1126/scisignal.2005478. [DOI] [PubMed] [Google Scholar]
- 52.Ju Y, Untereiner A, Wu L, Yang G. H2S-induced S-sulfhydration of pyruvate carboxylase contributes to gluconeogenesis in liver cells. Biochim Biophys Acta. 2015;1850:2293–303. doi: 10.1016/j.bbagen.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 53.Módis K, Bos EM, Calzia E, VanGoor H, Coletta C, Papapetropoulos A, Hellmich MR, Radermacher P, Bouillaud F, Szabo C. Regulation of mitochondrial function by hydrogen sulfide. Part II. Pathophysiological and therapeutic aspects. Br J Pharmacol. 2014;171:2123–46. doi: 10.1111/bph.12368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang SB, Foster DB, Rucker J, O’Rourke B, Kass DA, Van Eyk JE. Redox regulation of mitochondrial ATP synthase: implications for cardiac resynchronization therapy. Circ Res. 2011;109:750–7. doi: 10.1161/CIRCRESAHA.111.246124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang SB, Murray C, Chung HS, VanEyk JE. Redox regulation of mitochondrial ATP synthase. Trends Cardiovasc Med. 2013;23:14–18. doi: 10.1016/j.tcm.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mailloux RJ, Treberg JR. Protein S-glutathionlyation links energy metabolism to redox signaling in mitochondria. Redox Biol. 2015;8:110–118. doi: 10.1016/j.redox.2015.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Akter F. The role of hydrogen sulfide in burns. Burns. 2016;S0305–4179:00204–1. doi: 10.1016/j.burns.2015.07.005. [DOI] [PubMed] [Google Scholar]