
Figure 7.
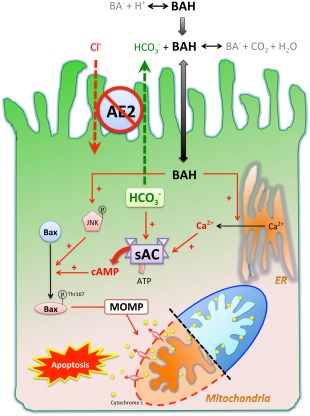
Working hypothesis of how sAC is involved in the pathogenesis of PBC in the context of AE2 down‐regulation. Protonated bile salts are nonpolar and enter cells at a higher rate than their deprotonated counterparts. AE2 on the apical membrane of cholangiocytes generates a bicarbonate umbrella that deprotonates the otherwise proapoptotic bile salts. Reduced AE2 expression in PBC cholangiocytes causes more bile acids to enter. Once inside the cells, bile salts cause JNK phosphorylation and activating phosphorylation of Bax at Thr167, which promote mitochondrial translocation of Bax, leading to MOMP, cytochrome c release, and apoptosis. Inhibition of sAC prevents bile salt‐induced Bax phosphorylation at Thr167 but not JNK phosphorylation. By acting as a calcium ionophore, bile salts cause Ca2+ release from endoplasmic reticulum, which is necessary but not sufficient for BSIA. On the other hand, the intracellular bicarbonate accumulation secondary to impaired secretion increases sAC activity synergistically with bile salt‐triggered Ca2+ release and enhances MOMP and apoptosis. Abbreviations: ATP, adenosine triphosphate; BA‐, deprotonated bile salt; BAH, protonated bile salt; ER, endoplasmic reticulum.