

Abstract
Brassica juncea (AjAjBjBj), is an allotetraploid that arose from two diploid species, B. rapa (ArAr) and B. nigra (BnBn). It is an old oilseed crop with unique favorable traits, but the genetic improvement on this species is limited. We developed an approach to broaden its genetic base within several generations by intensive selection. The Ar subgenome from the Asian oil crop B. rapa (ArAr) and the Bc subgenome from the African oil crop B. carinata (BcBcCcCc) were combined in a synthesized allohexaploid (ArArBcBcCcCc), which was crossed with traditional B. juncea to generate pentaploid F1 hybrids (ArAjBcBjCc), with subsequent self-pollination to obtain newly synthesized B. juncea (Ar/jAr/jBc/jBc/j). After intensive cytological screening and phenotypic selection of fertility and agronomic traits, a population of new-type B. juncea was obtained and was found to be genetically stable at the F6 generation. The new-type B. juncea possesses good fertility and rich genetic diversity and is distinctly divergent but not isolated from traditional B. juncea, as revealed by population genetic analysis with molecular markers. More than half of its genome was modified, showing exotic introgression and novel variation. In addition to the improvement in some traits of the new-type B. juncea lines, a considerable potential for heterosis was observed in inter-subgenomic hybrids between new-type B. juncea lines and traditional B. juncea accessions. The new-type B. juncea exhibited a stable chromosome number and a novel genome composition through multiple generations, providing insight into how to significantly broaden the genetic base of crops with subgenome introgression from their related species and the potential of exploring inter-subgenomic heterosis for hybrid breeding.
Keywords: interspecific hybridization, Brassica juncea, subgenomic introgression, heterosis, genetic diversity, novel population
Keymessage: Development of new-type Brassica juncea with introgression of subgenomes from related oilseed species.
Introduction
Brassica juncea (AjAjBjBj, 2n = 36) is a tetraploid that originated from its diploid progenitors, B. rapa (ArAr, 2n = 20) and B. nigra (BnBn, 2n = 16). It is deduced that B. juncea formed ~0.039−0.055 million years ago, and, based on a recently completed genome sequence, has significantly diverged from its parental species such as frequent gene loss (Yang et al., 2016). Humans have cultivated this species for approximately 6000 years as an oil and vegetable crop (Chen et al., 2013). In some Southern Asian countries such as in India, B. juncea is currently a major oilseed crop (Chauhan et al., 2011; Dr. Jayantilal Patel personal communication). Currently, increasing attention is being paid to B. juncea worldwide, given its unique and favorable traits, such as resistance to biotic and abiotic stress and a low rate of pod shattering. These traits are desirable in the context of global climate change given its capacity to adapt to heat and other stresses that are commonly encountered under field conditions in Canada, Australia, and many other parts of the world (Woods et al., 1991; Cheung et al., 1997; Burton et al., 2004; Huangfu et al., 2009; Amanullah et al., 2011; Chen et al., 2013). However, genetic improvement on this species is still relatively limited even though previous efforts have significantly increased the yield, particularly increases of seed yield in India (Dr. Zhizheng Chen, personal communication). The low seed yield of B. juncea in most of the rapeseed growing area substantially restricts extensive planting of this species worldwide. This is largely due to deleterious changes in its genome from genetic improvement programs and the absence of a novel approach to exploit the benefits associated with heterosis, and an international effort is needed to genetically improve this crop (Pradhan and Pental, 2011). Increasing the seed yield, exploring novel germplasms, and promoting new approaches for hybrid breeding are therefore essential for improving the agricultural production of B. juncea.
Alien genomic introgression via interspecific crossing is a common and powerful approach in crop breeding programs: (1) limited introgression of loci of high agronomic value for introducing advantageous genes to produce desired traits, and (2) large-scale random introgressions for broadening the genetic basis of species (Allard, 1960; Zamir, 2001; Bennett et al., 2012; Yu et al., 2012). Brassica includes three diploid species (B. rapa, B. nigra, and B. oleracea) with three basic genomes (A, B, and C) and three tetraploids (B. juncea, B. napus, and B. carinata) derived from natural crosses between the diploid species, as represented in the “U's triangle” (UN, 1935). Scientists recognized the genomic differentiation of these A, B, and, C genomes in different Brassica species and introduced the concept of a “subgenome,” which can be distinguished for each species by using a superscript (or subscript e.g., Chalhoub et al., 2014) of the first letter of the species' name, such as AjAjBjBj for B. juncea, AnAnCnCn for B. napus, BcBcCcCc for B. carinata, ArAr for B. rapa (Li et al., 2004; Zou et al., 2010, Figure 1). The cytological relationships and genomic homology among the species provide a major opportunity to generate novel Brassica germplasms, or even novel species, such as Brassica hexaploids, via interspecific hybridization in Brassica (Chen et al., 2011; Mason and Batley, 2015). To improve the major rapeseed species, B. napus, extensive and successful efforts have been made to broaden the genetic basis, and introduce several specific traits from related species (Prakash and Raut, 1983; Meng et al., 1998; Ren et al., 2000; Xiao et al., 2010; Girke et al., 2012a,b; Li et al., 2014). Utilizing the subgenomic differentiation within and between Brassica species would also be helpful to broaden the genetic base of B. juncea and explore inter-subgenomic heterosis.
Figure 1.
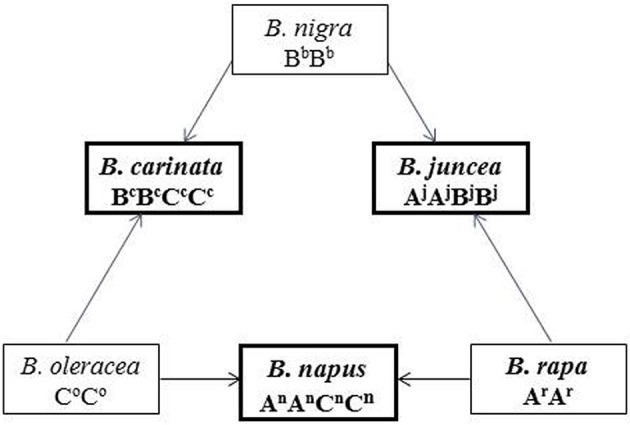
The U's triangle of Brassica presenting the cytological relationship of the relation species and their subgenome constitution. This picture is cited from Zou et al. (2010), which was modified according to UN (1935). The diploid species with the three basic subgenomes (A,B,C) are presented in the three corner of the triangle, and the derived three tetraploid species are presented in the middle of the three lines. Different subgenomes of different species are noted with the superscript of the first letter of each species (Zou et al., 2010).
In a previous study, we obtained allohexaploid Brassica (ArArBcBcCcCc) that was synthesized from the oilseed crops B. rapa and B. carinata (Tian et al., 2010). The Ar subgenome-containing species, B. rapa, is a primary oilseed crop in China, with a long cultivation history prior to the import of B. napus in the 1950s. B. rapa possesses rich genetic diversity and many desirable traits, such as fast growth and tolerance to nutrient-deficient soil and low temperatures (Franks, 2011; Waalen et al., 2011; Ahmed et al., 2012). B. carinata, which contains the B genome, is grown in Ethiopia and the area around Northeastern Africa and has unique traits that are desirable in other Brassica crops, such as disease resistance, shattering resistance, and drought tolerance (Malik, 1990; Warwick, 2011). Considering the divergent evolution of the Ar subgenome and the Bc subgenome in different species and in different continents, in the present study, we aimed to broaden the genetic base of the old oilseed crop B. juncea by developing lines of new-type B. juncea with massive introgression of the Ar and Bc subgenomes.
Materials and methods
Plant materials
Nine allohexaploid lines obtained from crosses between eight accessions of B. rapa from China and nine accessions of B. carinata from Ethiopia, as shown in Supplementary Table 1 (Tian et al., 2010), were used in this study. A total of 45 accessions of traditional B. juncea from various areas of the world were used to perform crosses with the nine hexaploid lines to obtain a new-type of B. juncea and to obtain intersubgenomic hybrids. Furthermore, analyses of genetic diversity were performed, in which an additional nine accessions of B. juncea were also used (Supplementary Table 2).
DNA extraction and molecular marker assays
All of the DNA samples (included 82 plants from nine F2:3 families, five hexaploid lines and eight accessions of traditional B. juncea used as parents of the new-type B. juncea lines, six accessions of traditional B. napus and two accessions of B. carinata of other related species) were extracted from young leaves by a modified version of the DNA extraction method (Doyle, 1990). A total of 208 pairs of fluorescence-labeled primers of simple sequence repeats (SSR, 104 from the A genome, 73 from the C genome and 31 from B genome of Brassica) with clear, reproducible and polymorphic amplification products (Supplementary Table 3) were used to evaluate the genetic diversity and population structure of the novel B. juncea lines and their parental lines (traditional B. juncea) used in this study. The synthesis of the SSR primers, conditions for PCR, and processing of the data generated by capillary sequencing were all described previously (Chen et al., 2010) using an AB3730xl capillary sequencer (Applied Biosystems, USA). An additional set of SSR markers anchored on 31 loci from the B genome in B. carinata (Zou et al., 2014) was used to evaluate the introgression of B genomic components (Supplementary Table 3). The markers were scored according to the polymorphism appeared in the parents and population. Those alleles newly appeared or missing in the population compared with the parents were noted as novel allelic variation.
Cytogenetic observations and GISH and BAC-FISH analyses
The chromosome number in the plants was counted in cells from the wall of the ovary, as described by Tian et al. (2010). For meiotic analysis, young flower buds were fixed in fresh Carnoy's solution for 24 h and then stored in 70% ethanol. Slides of chromosomes from the pollen of mother cells were prepared for GISH (genomic in situ hybridization) and BAC-FISH (bacterial artificial chromosome fluorescence in situ hybridization) based on the procedures of Ge and Li (2007). Genomic DNA from B. nigra cv. Giebra (labeled with digoxigenin-11-dUTP; Roche, Basel, Switzerland) and plasmid DNA from BAC BoB014O06 (labeled with biotin-11-dUTP; provided by Susan J. Armstrong, University of Birmingham, Birmingham, UK, the probe distribute on all C chromosomes) were used to identify B- and C-genome chromosomes using GISH and BAC-FISH analysis. Probe preparation and in situ hybridization were performed using the methods described by Cui et al. (2012).
Field observation of agronomic traits and evaluation of heterosis potential
Plants were mainly grown in Wuhan (located in central China), but some plants in the F4 generation were also grown in Hezheng (northwest China). The proportion of seed sets for each of the new-type B. juncea line and cultivars of traditional B. juncea was accounted for according to the average seed number of 10-30 siliques evaluated with 4-6 plants each line. A total of 445 F4 plants from 72 F3:4 families of new-type B. juncea, and a total of 81 F6 plants from 20 F5:6 families of new-type B. juncea were measured. The kilo-seed weight of each line was evaluated with the average weight of 1000 seeds from each of the investigated plants.
Inter-subgenomic hybrids between the lines of new-type B. juncea in early generations (F3, F4, and F5) and traditional B. juncea were generated by hand crossing and planted along with their parents. The hybrids derived from the F3 lines were grown in Hezheng as a spring-type crop in 2012 and in Wuhan as a semi-winter-type crop from 2012 to 2013 to evaluate heterosis potential for seed yield. All plants were grown in two rows, with each row 1.8 m in length and 0.25 m between rows. The hybrids derived from the F4 lines and F5 lines were grown in Wuhan from 2013 to 2014, and 2014 to 2015, respectively, with two replicates (24 plants with 0.3 m space among plants for each replication) and three replicates (21 plants with 0.3 m among plants for each replication), respectively. The middle parent heterosis and superior parent heterosis of the hybrids were calculated. Individual significant differences and overall significant differences among hybrids and their parents across multiple combinations in each year was evaluated by the F test at the significance level of P < 0.05, using the multiple comparison model under a single factor with repeats and the multiple comparison model under double factors with repeats in SAS 8.1 (SAS Institute, 1999), respectively.
Population genetic analysis
The genetic diversity and genetic distance between different plants was evaluated with molecular markers analyzed using Primer 6 software (Clarke and Gorley, 2006). The Shannon index was calculated using POPGENE 1.31 (Yeh et al., 1999). The phylogenetic tree was analyzed based on Nei's genetic distance (Nei et al., 1983) using the clustering method, employing a neighbor-joining tree with 1000 permutations, using POPTREE 2 (Takezaki et al., 2010). After eliminating improper markers with allelic frequencies less than 10 or >10% missing data, the population structure was analyzed by Bayesian clustering and the “admixture model” with STRUCTURE 2.2 (Pritchard et al., 2000; Falush et al., 2003). The length of burn-in time and replication number were both set to 100,000 in each run. To identify and determine the most probable population number (K), we calculated the ΔK values of K from 1 to 10 replicate runs for each K, and selected the K value corresponding to the peak of the ΔK graph after plotting (Evanno et al., 2005).
Results
The strategy and process of creating the new-type B. juncea
A set of Brassica allohexaploids in which the Ar and Bc subgenomes of B. carinata (BcBcCcCc) and B. rapa (ArAr) (Tian et al., 2010) were combined was used as donors for the introgression of exotic subgenomes into the traditional B. juncea genome. Subsequently, morphological/molecular/cytological identification and intensive selection for generations were performed (Figure 2).
Figure 2.

Strategy used to generate a population of new-type B. juncea. The Brassica allohexaploid in which the Ar and Bc subgenomes from B. carinata (BcBcCcCc) and B. rapa (ArAr) (Tian et al., 2010) were combined was used as the donor of exotic subgenomes. The first step in the current study involved interspecific crosses between various hexaploid and traditional B. juncea to transfer the Ar and Bc subgenomes to B. juncea. Successive phenotypic and cytological selection was then conducted up to the F6 generation, whereas genomic surveying was mainly conducted at the F3 generation.
Initially, nine hexaploid lines were successfully crossed with 45 accessions of traditional B. juncea, which generated thousands of hybrid seeds (approximately 15,000 seeds) derived from 108 combinations. The cross success rate for different pentaploid combinations varied from 0.1 to 1, with an average cross success rate value of 0.8 and 5.7 seeds per silique. The seeds per silique in the hexaploid seeds and traditional B. juncea parents varied from 0.9 to 3.7 and 9.6 to 16.7, respectively. The authenticity of the hybrids was verified using DNA markers after initial identification by morphological observation (Supplementary Figure 1), and the chromosome number of each hybrid was counted. After obtaining pentaploid progeny, self-pollination was performed across generations.
The seeds from 93 identified true hybrids gave rise to the F2 generation. The F2 generation was screened for plants with the correct number (or close to the correct number) of chromosomes (36) as well as good fertility (defined as an abundance of pollen in the flowers and more than eight seeds per silique). The 36 chromosomes were expected to consist of the total 2 A and 2 B genomes, without any of the C genome chromosome. When the genome of A, B and C was integrated in hexaploids, the C genome was more easily lost because the genome stability was B>A>C (Zhou et al., 2016). The pedigrees of the introgressed lines were established according to the original interspecific crosses for the pentaploids. Then, selfing of each pentaploid resulted in different F2 families. Subsequently, the other round of phenotypic selection was performed at the F3 generation with a focus on families descended from plants with 36 chromosomes. A genomic analysis was performed to evaluate whether the F3 plants diverged from the traditional B. juncea lines due to the massive exotic introgression. In the F4 generation, GISH and FISH analyses were used to confirm the correct genomic constitution of the lines of new-type B. juncea (AABB). Chromosomes were further assessed using the lines at the F6 generation.
The consequence of cytogenetic selection on plants with the target number of chromosomes
Because meiosis in the artificially synthesized hexaploids cannot be completely normal (Tian et al., 2010), the F1 hybrids derived from the 108 interspecific crosses between the hexaploid and tetraploid B. juncea might have a segregated chromosome composition deviate from the expected 45 chromosomes. After removing the abnormal seedlings and maternal plants, more than 700 F1 plants bearing self-pollinated seeds were obtained and subjected to chromosome counting. Only 6.73% of the plants had the expected number of chromosomes (45) for pentaploids (Figure 3A). To avoid incorrectly discarding plants with the appropriate number of chromosomes, the plants scored with 44 chromosomes were also regarded as potential pentaploid. Consequently, self-pollinated seeds harvested from 93 F1 plants assumed to be pentaploid, were selected to produce the F2 generation.
Figure 3.

The distribution of plants with different chromosome numbers at four successive segregating generations, from pentaploid F1 to F4. (A) The segregation of F1 plants with different chromosome numbers and seed set. In the F1 generation, the number of seeds produced from each category of plants with a different chromosome number is also presented. Self-pollinated seeds from F1 plants with 44 or 45 chromosomes were selected (as indicated by the arrows on the bottom) to produce offspring. (B) The segregation of plants with different chromosome numbers at the F2 generation, which originated separately from F1 plants with 44 or 45 chromosomes. Plants with 36 to 38 chromosomes were selected to produce offspring. (C) The segregation of the plants with different chromosome number in the F3 generation, 50 to 80% of plants derived from F2 plants with 38, 37, or 36 chromosomes were identified as having 36 chromosomes; only plants with 36 chromosomes were selected as new-type B. juncea. (D) The majority of the plants in the F4 generation inherited 36 chromosomes. The arrows indicating the selected plants were filled with colors and grains that were the same as the columns in the picture in the next generation. The cytological photographs illustrate the correct chromosome numbers of 45, 36, 36, and 36 in the pentaploids, target F2 plants, and new-type B. juncea (F3 and F4), respectively.
Approximately 1000 F2 plants were obtained, and approximately one-third of the plants exhibited abundant pollen (as observed by eye); they were selected as candidate tetraploid plants for chromosomal counting. In the F2 population descended from the F1 specimens with 45 chromosomes, only 2.69% of the plants exhibited 36 chromosomes; most of the plants had extra chromosomes (Figure 3B). To obtain more euploid progeny (Ar/jAr/jBc/jBc/j, 2n = 36) in the next generation, F2 plants with 37 or 38 chromosomes (which may be aneuploid with one or two extra chromosomes that could be lost in the next cycle of meiosis) were selected together with plants with 36 chromosomes to produce the F3 generation via self-pollination.
More than 2000 F3 plants were obtained from various 24 F2 plants derived from different pentaploids with 36, 37, or 38 chromosomes, which were established as 24 derived F2:3 families according to the origin of the pentaploid cross in the F2 generation. The chromosome numbers were assessed for those fertile plants that generated abundant pollen at flowering and produced a large number of seeds. Of the fertile plants, 70% (119 of 170) were regarded as euploid (2n = 36), 85% of the inferred euploid specimens were derived from 13 F2 plants with 36 chromosomes (Table 1, Figure 3C), and 5 of the 13 F2 families constituted 69% of the inferred F3 euploid plants. The plants derived from 37- or 38-chromosome F2 plants accounted for 15% of the inferred euploid specimens in the F3 generation (Supplementary Tables 4, 5).
Table 1.
The number of plants and families investigated and identified in the F3 generation.
| Chromosome number in the F2 generation | Total F3 plants/ F2:3 families grown to maturity | No. of F3 plants/ showing plentiful pollen | No. of fertile F3 plants*/ F2:3 families checked | No. of F3 plants/ F2:3 families with 36 chromosomes |
|---|---|---|---|---|
| 36 | 1200/17 | 465/14 | 139/13 | 101/13 |
| 37 | 450/23 | 344/21 | 17/9 | 12/7 |
| 38 | 550/37 | 276/29 | 14/9 | 6/4 |
| Total | 2200/77 | 1085/64 | 170/31 | 119/24 |
A plant with ≥ 8 seeds/silique was regarded as a fertile plant.
Approximately 2500 F4 plants derived from 87 F3 plants with 36 chromosomes were obtained. Overall, 94% of the plants, counted from 116 F4 individuals, were euploid (2n = 36) (Figure 3D, Supplementary Table 6). To verify that the “inferred euploid” specimens with 36 chromosomes exhibited AABB euploidy, the chromosome composition of five F4 plants with 36 chromosomes was investigated by cytological observation. All of the plants exhibited normal behavior at meiosis in pollen mother cells with 20 chromosomes from the A genome and 16 chromosomes from the B genome (Figure 4). We further counted the chromosome numbers of 78 F6 plants with normal fertility derived from 21 F4 families and found that 96.2% of the F6 plants had 36 chromosomes (Supplementary Tables 6, 7), and all of the plants descended from seven 36-chromosome F3/F4 families (25 of the 78 plants) consistently exhibited 36 chromosomes as well. It indicated that the lines of new-type B. juncea were genetically stable, at least, from the F6 generation.
Figure 4.

Demonstration of the chromosome composition of new-type B. juncea lines via cytological observations. A pollen mother cell in meiotic anaphase I depicting the chromosome composition of a new-type B. juncea plant, C18J23-1-2-8, at the F4 generation (A–C) compared with traditional B. juncea accession, J56 (D–F), as revealed by in situ hybridization. (A,D) Merged images of DAPI staining (blue) and a B genome DNA probe (bluish green). Sixteen chromosomes from the B genome (bluish green) were distinguished from 36 chromosomes in total, and separated equally into two nuclei. Twenty chromosomes from the A genome (pure blue) were observed. (B,E) Images of a B-genome probe (which appears green without DAPI interference); only 16 chromosomes from the B genome were distinguished. (C,F) Images of a C-genome probe (red); no obvious red signals were found, suggesting that the 20 chromosomes in (A,D) with a blue color were from the A genome and not the C genome.
The consequences of selection on plant fertility
The seed set of plants in the F1 generation varied widely among individually bagged plants, ranging from no seed set to close to a normal level. One-fifth of the F1 plants were seedless, while approximately half of the plants yielded more than 100 self-pollinated seeds, and a small proportion of plants (approximately 4%) produced more than 500 seeds (Figure 5A).
Figure 5.

Plant fertility in different generations after interspecific crossing. (A) The categories of plants exhibiting different fertility and levels of seed set in the F1 generation. The levels of seed set varied markedly from zero to hundreds of seeds produced per plant. (B) The seed set rate (seeds/silique) was determined in the F2, F3, and F4 generations by an analysis of 90, 55, and 72 plants, respectively. The horizontal arrows at the bottom indicate the consequences of chromosome transmission from generation to generation, and the vertical arrows above the columns indicate the maximal number of seeds per silique in one plant. The red bars in the column of the F3 generation indicate the mean value of seeds per silique counted from 675 plants; chromosomes were not counted in most of these plants. Six cultivars of traditional B. juncea were used as a control.
Plants with 36, 37, and 38 chromosomes were used to investigate the fertility of the F2 generation. The plants with 36 chromosomes exhibited the highest rate of seed set (6.04 seeds/silique), which was significantly higher (P < 0.05) than the seed set rate of plants with 37 or 38 chromosomes (Supplementary Table 5). Some of the 36-chromosome F2 plants borne more than 10 seeds per silique (Figure 5B).
The majority of F3 plants exhibited good fertility and flowered normally with abundant pollen (Table 1, Supplementary Figure 2). However, the F3 progeny derived from the 36-chromosome F2 plants had higher rates of seed set than the offspring derived from 37- or 38-chromosome F2 plants, as judged by the number of seeds per silique and the seed yield per plant (Figure 5B, Supplementary Table 8). Compared with the F2 generation, the number of seeds per silique in the plants with 36 chromosomes increased in the F3 generation regardless of whether they were derived from 36-, 37-, or 38-chromosome F2 plants.
The number of seeds per silique increased again in the F4 generation, and the best F3-derived lines with 36 chromosomes had a good seed set rate that was comparable to that of traditional B. juncea lines (Table 2). The results of cross-pollination with traditional B. juncea accessions showed that the lines of new-type B. juncea were not sexually isolated from but were instead compatible with traditional cultivars of B. Juncea (Supplementary Figure 2).
Table 2.
Extent of trait variation in the ‘new-type B. juncea lines’ (F4-F6) and traditional accessions of B. juncea.
| Agronomic trait* | New-type B. juncea lines | Traditional B. juncea accessions | |||
|---|---|---|---|---|---|
| Generation | Mean ±SD | Range | Mean ±SD | Range | |
| Plant height (cm) | 179.04±18.90 | 132~232 | 154.10±24.95 | 101.71~187.00 | |
| Branch number | 9.47±3.01 | 5.75~18.50 | 8.76±1.98 | 6.08~11.10 | |
| Silique number per plant | F4* | 400±156.40 | 172.89~948.51 | 401.32±177.91 | 173.67~767.53 |
| Kilo-seed weight (g) | 2.01±0.39 | 1.35~3.40 | 1.71±0.46 | 1.22~2.48 | |
| Seed number (per silique) | 11.46±1.96 | 7.69~15.54 | 12.41±2.09 | 9.61~16.72 | |
| Seed yield (g/plant) | 9.34±3.42 | 3.31~22.80 | 7.52±2.55 | 2.13~7.79 | |
| Theoretical seed yield (g/plant)** | 9.21 | 8.52 | |||
| Kilo-seed weight (g) | F6*** | 2.00±0.37 | 1.38~2.57 | 1.88±0.42 | 1.27~2.64 |
| Seed number (per silique) | 12.81±2.70 | 8.39~20.34 | 12.40±1.43 | 8.98~13.98 | |
Data were calculated from 445 F4 plants from 72 F3:4 families of new-type B. juncea and compared with the accessions of traditional B. juncea.
Calculated from Silique number × Kilo-seed weight × Seed number/1,000. The kilo-seed weight is the weight of 1000 seeds, therefore, we divided 1000 to calculate the seed yield per plant.
Data were calculated from 81 F6 plants from 20 F5:6 families of new-type B. juncea compared with ten accessions of traditional B. juncea.
Genetic diversity in a population of new-type B. juncea compared with traditional B. juncea accessions
A set of 978 SSR markers was used to analyze the genetic diversity of the new-type B. juncea lines compared with their parental species. Of them, 44.2, 17.5, and 9.6% markers could be assigned to A, B, C genome according to the origin of the alleles amplified in different parental species, respectively. However, 28.7% of the markers could not be distinguished because of multiple amplified products in different genomes. For the plants from the same family, we estimated the percentage of the introgression according to the origin of the alleles from both parents (Supplementary Table 9). Those alleles were not originated from both of the parents, but newly appeared or disappeared in the new-type B. juncea lines were noted as novel allelic variation. Averagely 39.2, 25.5, and 35.4% of the genome of new-type B. juncea were constituted with the introgression of the traditional B. juncea parents, hexaploid parents and novel allelic variation (Supplementary Table 9). For the introgression from hexaploid parents, 55.5 and 15.1% of the alleles were originated from the A and B genome, respectively, but 29.4% of them could not be assigned. The new-type B. juncea lines exhibited substantial genetic distance from their hexaploid parents, as assayed via principal ordination analysis (Figure 6A). The F2:3 family, C34J27-1, along with most of other F2:3 families, exhibited clear genetic differences relative to their tetraploid parents and traditional B. juncea accessions. Most of the F2:3 families of new-type B. juncea, especially C18J05-2, also exhibited marked polymorphisms within their families and demonstrated increased genetic variation as a whole compared with traditional B. juncea accessions. An estimate of the gene diversity index between the different populations indicated that the new-type B. juncea lines exhibited the richest genetic diversity and the highest Shannon Index (0.49), which was considerably higher than that of traditional B. juncea accessions (0.31). A phylogenetic tree revealed that the new-type B. juncea was obviously separated from traditional B. juncea and had a greater genetic distance from other Brassica species than traditional B. juncea (Figure 6B). Population genetic structure analyses indicated that three apparent genetic clusters existed among the investigated lines and their parents (Figure 6C, Supplementary Figure 3). The new-type B. juncea lines inherited major genetic components from both parents (the hexaploids and traditional B. juncea).
Figure 6.

Genetic divergence between the new-type B. juncea lines and relative species. (A) The genetic differences between the new-type B. juncea lines in the F3 generation and their parents were revealed by principle ordination analysis with multi-dimensional scaling. We draw three ovals to outline the three genetic clusters of the new-type B. juncea, traditional B. juncea and hexaploid, respectively. The logos for different lines of new-type B. juncea were designed to have the same color as their hexaploid parents (shown in cycles with different colors) and the same shape as their B. juncea parents (shown with different geometric shapes). The arrows at the upper right indicate two plants from F2:3 family C18J0502 with remarkable genetic distance from traditional B. juncea. The key to the symbols is shown on the right, and the Shannon Index for the new-type B. juncea and their parental species is shown in the brackets. (B) Phylogenetic tree of eight families of new-type B. juncea, eight accessions of traditional B. juncea, two accessions of B. carinata, five hexaploid lines and two accessions of B. napus. The new-type B. juncea formed a group that could be distinguished from traditional B. juncea and other species, including B. napus (AACC), another tetraploid species in the genus Brassica. The length of the branches represents Nei's genetic distance, and the numerals above the branches indicate the reliability by bootstrap analysis. (C) The population structure of new-type B. juncea and its parental species at K = 3 (Supplementary Figure 3). The blue color represents the genetic structure of hexaploids, while the red and green color represent the genetic structure of traditional B. juncea. The genetic structure of new-type B. juncea reflected the membership fractions from both parental species (hexaploid and traditional B. juncea).
Potential for trait variation and inter-subgenomic heterosis in the early generations of the new-type B. juncea lines
In total, 119 of the best F3 plants with marked variation in morphology and agronomic traits were selected from 24 F2:3 families, which represent 14 combinations involving five hexaploid lines (each synthesized from different accessions of B. carinata and B. rapa) and 11 accessions of traditional B. juncea (Supplementary Table 10). After testing the seed yield and yield components, seeds from 76 F3 plants representing all of the founder parents and cross combinations were ultimately sown in the field to generate the F4 generation, which resulted in approximately 2500 plants that exhibited distinct differences from their parental species and presented comparable agronomic traits to traditional B. juncea (Table 2, Supplementary Figures 4A–D). The agronomic and morphological traits of the new-type B. juncea lines were further improved at the F6 generation through selection, resulting in an obviously increased seed number per pod.
We estimated the heterosis potential for the seed yield of the inter-subgenomic hybrids between the new-type B. juncea lines in different early generations (F3, F4, and F5) and traditional B. juncea accessions. The seed yield of the hybrids was significantly increased compared with parental accessions when grown as a spring crop in the northwest of China and as winter crop rapeseeds in central China (Figure 7, Supplementary Table 11 and Supplementary Figure 4E). The average over superior parent heteoris and mid-parent heterosis of the tested hybrids across environments were 60 and 96%, respectively. However, the heterosis potential of different lines also varied extensively.
Figure 7.
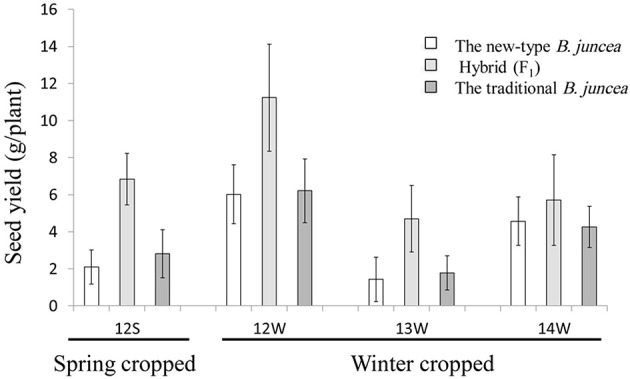
Seed yield of new-type B. juncea lines, traditional B. juncea accessions and the hybrids between them. The spring crop data were collected from nine F3 lines of new-type B. juncea, eight cultivars of traditional B. juncea, and six hybrids between the new-type B. juncea lines and traditional B. juncea accessions at the spring-type growing environment in 2012 (environment abbreviation as 12S), and the winter crop data were collected from 10 F3 lines of new-type B. juncea, five cultivars of traditional B. juncea, and 10 hybrids in 2012–2013 at the winter-type environment (environment abbreviation as 12W). The winter crop data for 2013–2014 and 2014–2015 in the winter-type environment (environment abbreviation as 13 and 14W) were successively collected from eight F4 and 10 F5 lines of new-type B. juncea, five and seven cultivars of traditional B. juncea, and 13 and 21 derived hybrids, respectively (for more detail, see Supplementary Table 10).
Discussion
To broaden the genetic base of traditional B. juncea, we demonstrated an approach to generate new-type B. juncea lines with introgression of subgenomes from its related oilseed species, B. rapa and B. carinata, via interspecific crossing and intensive selection. We performed direct selection on the hybrid offspring obtained from interspecific crosses between artificial allohexaploid (ArArBcBcCcCc) lines and traditional B. juncea (AjAjBjBj) accessions. Subsequently, we conducted intensive phenotypic selection and cytological screening in the offspring of the pentaploids under self-pollination over three successive generations (Figure 2). As a result, a population of new-type B. juncea founded with multiple parents was developed (Figures 4–6). The new-type B. juncea was stable in terms of the chromosome composition up to the F6 generation (Supplementary Tables 6, 7), presented rich genetic diversity within the population, and diverged from, but was not isolated from traditional B. juncea (Figure 6). The traits of the new-type B. juncea lines, were comparable to the traditional parental B. juncea (Table 2, Supplementary Figure 4). Strong seed yield heterosis was observed in the hybrids between new-type B. juncea and traditional B. juncea accessions from different environments (Figure 7, Supplementary Table 11). The novel germplasm may be a valuable resource to bring the old Brassica oilseed crop into the world of modern breeding, especially due to its unique favorable traits for global climate changing such as drought-resistance. This work helps to elucidate how plant crops may receive genome-wide benefits when bred with related species.
Our strategy also demonstrated the efficiency of direct selection for introducing considerable exotic genomic components to the donor species. The analysis of genetic structure showed that each of the investigated lines of new-type B. juncea exhibited a large membership fraction from the hexaploid parents synthesized from B. carinata and B. rapa. One-third of the total estimated alleles, according to estimation by a total of 978 molecular markers (Supplementary Table 9), showed novel variation in the new-type B. juncea lines compared with the parents, including newly appearing alleles and missing alleles compared with the parents. Novel allelic variation has been frequently observed in the progeny of interspecific hybrids in plants (Pontes et al., 2004; Han et al., 2005; Wang et al., 2010; Zou et al., 2011). The appearance of novel alleles, including new alleles and missing alleles, suggests extensive genome reshuffling and recombination between the three Brassica genomes in these lines. The genome was altered by thousands of genes via exotic introgression and induced novel variation. Therefore, it is not surprising that the new-type B. juncea diverges from traditional B. juncea as well as relative species, as revealed by population genetic analysis of a phylogenetic tree and assessment of genetic diversity through principal ordination analysis (Figure 6). Genetic diversity was achieved by using multiple founder parents (five different hexaploids and 11 cultivars of traditional B. juncea) and maintaining a relatively large population, as more than 100 plants were selected from thousands of plants grown in each generation, in contrast to a few offspring obtained from a few combinations that are routinely used for addressing the problems caused by reproductive barriers in distant crosses (Kovach and McCouch, 2008; Jena, 2010; Camadro et al., 2012). Further studies to analyze the novel genomic variation of the new-type B. juncea lines would provide more information and insights to understand the genomic changes.
To create a novel germplasm for a given species, a balance should be sought between genetic diversity, fertility and chromosome stability. Considering that the gametes originating from the hexaploid parents would be abnormal (Plummer et al., 2010; Tian et al., 2010; Chen et al., 2011) and that errors may occur when counting a large number of chromosomes in each cell, we adopted F1 plants with 44 to 45 chromosomes as candidates during the process of screening for pentaploids (2n = 45). Actually, approximately two-thirds of the plants that met our selection criteria on chromosome number (2n = 36) were derived from “pseudo-pentaploid” specimens with 44 chromosomes. But we also selected F2 plants with 37 and 38 chromosomes (15% of the plants with features of interest to the new-type B. juncea) and only retained those plants with 36 chromosomes at the F3 generation. At F3 generation, we firstly checked the pollen amount, then investigated the seed set of the plants with good pollen. After selection on the pollen amount and seed set, we performed chromosome counting for the plants with good pollen and seed set, and selected those plants with 36 chromosomes to next generation. A significantly increased percentage of plants with a stable chromosome number (2n = 36) was observed from the F2 generation to F4 generation. This implies that the new-type B. juncea was chromosomally stable after intensive selection in a few generations (Supplementary Table 4). Selecting relatively fertile plants to assess chromosome number increased the efficiency of chromosome counting and also improved the most important agronomic trait for the new-type B. juncea population. This strategy might provide a reference for improving the efficiency of generating novel Brassica synthetics.
In early generations, the new-type B. juncea lines presented comparable agronomic traits to traditional B. juncea accessions (Table 2). These traits could be further improved via recurrent selection, which has been demonstrated to be an efficient means of population improvement in crops (Hallauer and Carena, 2012; de Morais et al., 2015; Shelton and Tracy, 2015). Additionally, favorable traits from two different groups of traditional B. juncea (Pradhan et al., 1993; Chen et al., 2013) and exotic genome components from B. napus (AnAnCnCn) (which has been intensively improved by modern breeding and is most widely grown as an oilseed Brassica crop) can be introgressed into the pool via recurrent selection. The latter strategy has been demonstrated as a good approach by a group of Indian scientists who developed lines of new-type B. juncea with an AnAnBcBc constitution from octoploid AnAnBcBcCcCcCnCn plants derived from interspecific crosses between B. napus (AnAnCnCn) and B. carinata (Gupta et al., 2015).
The size of the B genome of B. nigra has been reported to be either 632 Mb (larger than that of B. rapa, at 529 Mb; Johnston et al., 2005) or close to that of B. rapa (Navabi et al., 2013). The results of a genome sequencing project revealed that there are 41,174 protein-coding genes in the Ar genome of B. rapa (Wang et al., 2011); therefore, it can be estimated that B. juncea harboring the AB genome can exhibit approximately 80,000 genes. It was estimated that a considerable percentage (average 25.5% per line and 31.3 % in the total population) of Ar/Bc components were introgressed into the genome of the new-type B. juncea (Supplementary Table 9), corresponding to approximately 20,400 genes per line and 25,040 genes in the total population. This massive introgression of alien genes, including numerous favorable alleles, would comprehensively alter the old genome of B. juncea and broaden the genetic base of this species. However, abundant space remains for thousands of alien genes to be further introduced in the genome of new-type B. juncea.
An obvious heterosis potential for seed yield is observed in the inter-subgenomic hybrids between the new-type B. juncea lines and traditional B. juncea accessions (Figure 7), which can be explained by the clear genetic distance between the new-type B. juncea and traditional B. juncea. Strong heterosis was also observed for other traits, such as seed weight (Supplementary Figure 4E). Further estimates of the trait variation and heterosis potential of the new-type B. juncea might be evaluated more precisely based on field traits, assessed in large blocks and multiple locations in late generations. The potential for inter-subgenomic heterosis would be strengthened by pyramiding higher introgression, by intercrossing new-type B. juncea plants and selection based on genomic components and agronomic traits, as shown in the new-type B. napus (Zou et al., 2010). However, in further estimation of the hybrid heterosis for seed yield, the possibility of introducing a male-sterile system into the population of new-type B. juncea should be considered.
In modern breeding systems, more importance is given to the sustainability of plant breeding, which urgently requires a high level of genetic diversity among breeding lines and elite cultivars with resource-conserving and environmentally friendly characteristics (Falk, 2010). This study may provide insights into the creation of novel germplasm pools for crops by exploring the genetic resources from related species, especially species in which subgenomes or subspecies already exist, such as wheat, rice, cotton and oil palm. We anticipate that with the recent rapid development of genomic technologies, such as high-throughput genotyping, genome sequencing, genome-wide association studies, and genomic selection, it has become possible for novel germplasms with massive exotic introgression to be appropriately evaluated and effectively used in crop breeding (Mahmood et al., 2007; Navabi et al., 2011; Nakaya and Isobe, 2012).
Author contributions
ZW and JZ performed the research, analyzed the data and wrote the manuscript. MW and SC performed field experiments and cytological observations. MW and CW screened the molecular markers. PL analyzed the molecular marker data. JZ and JM designed the research, revised the manuscript, and funded the project. All authors read and approved the final manuscript.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors gratefully acknowledge Prof. Zhongsong Liu (Hunan Agriculture University) for providing pollen from traditional B. juncea, Dr. Xianhong Ge and Prof. Zaiyun Li for providing probes for genomes B and C, and Dr. Xiuqiang Huang for selecting the SSR markers. We also acknowledge DuPont Pioneer of Caledon, Ontario, Canada for their support of the molecular marker analysis in this study. This work was supported by the National Key Research and Development Program of China (No. 2016YFD0101300), the Special Fund for Agro-scientific Research in the Public Interest of China (201203026-6) and the Natural Science Foundation of Hubei Province Key Program (2014CFA008).
Supplementary material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.01677/full#supplementary-material
References
- Ahmed N. U., Park J. I., Jung H. J., Seo M. S., Kumar T. S., Lee I. H., et al. (2012). Identification and characterization of stress resistance related genes of Brassica rapa. Biotechnol. Lett. 34, 979–987. 10.1007/s10529-012-0860-4 [DOI] [PubMed] [Google Scholar]
- Allard R. W. (1960). Principles of Plant Breeding. New York, NY: John Wiley and Sons. Inc. [Google Scholar]
- Amanullah H. M., Hassan M., Malhi S. S. (2011). Phenology and seed quality response of rape (B. napus) versus mustard (B. juncea) to sulfur and potassium fertilization in Northwest Pakistan. J. Plant Nutr. 34, 1175–1185. 10.1080/01904167.2011.558160 [DOI] [Google Scholar]
- Bennett R. A., Seguin-Swartz G., Rahman H. (2012). Broadening genetic diversity in canola using the C-genome species Brassica oleracea L. Crop Sci. 52, 2030–2039. 10.2135/cropsci2011.11.0580 [DOI] [Google Scholar]
- Burton W. A., Ripley V. L., Potts D. A., Salisbury P. A. (2004). Assessment of genetic diversity in selected breeding lines and cultivars of canola quality Brassica juncea and their implications for canola breeding. Euphytica 136, 181–192. 10.1023/B:EUPH.0000030672.56206.f0 [DOI] [Google Scholar]
- Camadro E. L., Erazzú L. E., Maune J. F., Bedogni M. C. (2012). A genetic approach to the species problem in wild potato. Plant Biol. 14, 543–554. 10.1111/j.1438-8677.2012.00563.x [DOI] [PubMed] [Google Scholar]
- Chalhoub B., Denoeud F., Liu S., Parkin I. A., Tang H., Wang X., et al. (2014). Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345, 950–953. 10.1126/science.1253435 [DOI] [PubMed] [Google Scholar]
- Chauhan J. S., Singh K. H., Singh V. V., Satyanshu K. (2011). Hundred years of rapeseed-mustard breeding in India: accomplishments and future strategies. Indian J. Agr. Sci. 81, 1093–1109. [Google Scholar]
- Chen S., Nelson M. N., Chèvre A., Jenczewski E., Li Z., Mason A. S., et al. (2011). Trigenomic bridges for Brassica improvement. Crit. Rev. Plant Sci. 30, 524–547. 10.1080/07352689.2011.615700 [DOI] [Google Scholar]
- Chen S., Wan Z., Nelson M. N., Chauhan J. S., Redden R., Burton W. A., et al. (2013). Evidence from genome-wide simple sequence repeat markers for a polyphyletic origin and secondary centers of genetic diversity of Brassica juncea in China and India. J. Hered. 104, 416–427. 10.1093/jhered/est015 [DOI] [PubMed] [Google Scholar]
- Chen S., Zou J., Cowling W. A., Meng J. (2010). Allelic diversity in a novel gene pool of canola-quality Brassica napus enriched with alleles from B. Rapa and B. Carinata. Crop Pasture Sci. 61, 483–492. 10.1071/CP09327 [DOI] [Google Scholar]
- Cheung W., Friesen L., Rakow G., Seguin-Swartz G., Landry B. (1997). A RFLP-based linkage map of mustard [Brassica juncea (L.) Czern. And Coss.]. Theor. Appl. Genet. 94, 841–851. [Google Scholar]
- Clarke K., Gorley R. (2006). PRIMER v6: User Manual/Tutorial. Plymouth, UK: PRIMER-E Ltd. [Google Scholar]
- Cui C., Ge X., Gautam M., Kang L., Li Z. (2012). Cytoplasmic and genomic effects on meiotic pairing in Brassica hybrids and allotetraploids from pair crosses of three cultivated diploids. Genetics 191, 725–738. 10.1534/genetics.112.140780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Morais O. P., Jr, Melo P. G. S., de Morais O. P., de Castro A. P., Breseghello F., Utumi M. M., et al. (2015). Genetic progress after cycles of upland rice recurrent selection. Sci. Agric. 72, 297–305. 10.1590/0103-9016-2014-0137 [DOI] [Google Scholar]
- Doyle J. J. (1990). Isolation of plant DNA from fresh tissue. Focus 12, 13–15. [Google Scholar]
- Evanno G., Regnaut S., Goudet J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620. 10.1111/j.1365-294X.2005.02553.x [DOI] [PubMed] [Google Scholar]
- Falk D. E. (2010). Generating and maintaining diversity at the elite level in crop breeding. Genome 53, 982–991. 10.1139/G10-081 [DOI] [PubMed] [Google Scholar]
- Falush D., Stephens M., Pritchard J. K. (2003). Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164, 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks S. J. (2011). Plasticity and evolution in drought avoidance and escape in the annual plant Brassica rapa. New Phytol. 190, 249–257. 10.1111/j.1469-8137.2010.03603.x [DOI] [PubMed] [Google Scholar]
- Ge X. H., Li Z. Y. (2007). Intra- and intergenomic homology of B-genome chromosomes in trigenomic combinations of the cultivated Brassica species revealed by GISH analysis. Chromosome Res. 15, 849–861. 10.1007/s10577-007-1168-4 [DOI] [PubMed] [Google Scholar]
- Girke A., Schierholt A., Becker H. C. (2012a). Extending the rapeseed genepool with resynthesized Brassica napus L. I: Genetic diversity. Genet. Resour. Crop Evol. 59, 1441–1447. 10.1007/s00122-011-1765-7 [DOI] [Google Scholar]
- Girke A., Schierholt A., Becker H. C. (2012b). Extending the rapeseed gene pool with resynthesized Brassica napus II: Heterosis. Theor. Appl. Genet. 124, 1017–1026. 10.1007/s00122-011-1765-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta M., Gupta S., Kumar H., Kumar N., Banga S. S. (2015). Population structure and breeding value of a new type of Brassica juncea created by combining A and B genomes from related allotetraploids. Theor. Appl. Genet. 128, 221–234. 10.1007/s00122-014-2423-7 [DOI] [PubMed] [Google Scholar]
- Hallauer A. R., Carena M. J. (2012). Recurrent selection methods to improve germplasm in maize. Maydica 57, 266–283. [Google Scholar]
- Han F., Fedak G., Guo W., Liu B. (2005). Rapid and repeatable elimination of a parental genome-specific DNA repeat (pGc1R-1a) in newly synthesized wheat allopolyploids. Genetics 170, 1239–1245. 10.1534/genetics.104.039263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu C., Song X., Qiang S. (2009). ISSR variation within and among wild Brassica juncea populations: implication for herbicide resistance evolution. Genet. Resour. Crop Evol. 56, 913–924. 10.1007/s10722-009-9410-x [DOI] [Google Scholar]
- Jena K. K. (2010). The species of the genus Oryza and transfer of useful genes from wild species into cultivated rice, O. sativa. Breed. Sci. 60, 518–523. 10.1270/jsbbs.60.518 [DOI] [Google Scholar]
- Johnston J. S., Pepper A. E., Hall A. E., Chen Z. J., Hodnett G., Drabek J., et al. (2005). Evolution of genome size in Brassicaceae. Ann. Bot. 95, 229–235. 10.1093/aob/mci016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovach M. J., McCouch S. R. (2008). Leveraging natural diversity: back through the bottleneck. Curr. Opin. Plant Biol. 11, 193–200. 10.1016/j.pbi.2007.12.006 [DOI] [PubMed] [Google Scholar]
- Li M., Qian W., Meng J., Li Z. (2004). Construction of novel Brassica napus genotypes through chromosomal substitution and elimination using interploid species hybridization. Chromosome Res. 12, 417–426. 10.1023/B:CHRO.0000034722.66981.94 [DOI] [PubMed] [Google Scholar]
- Li Q., Zhou Q., Mei J., Zhang Y., Li J., Li Z., et al. (2014). Improvement of Brassica napus via interspecific hybridization between B. napus and B. oleracea. Mol. Breed. 34, 1955–1963. 10.1007/s11032-014-0153-9 [DOI] [Google Scholar]
- Mahmood T., Rahman M. H., Stringam G. R., Yeh F., Good A. G. (2007). Quantitative trait loci for early maturity and their potential in breeding for earliness in Brassica juncea. Euphytica 154, 101–111. 10.1007/s10681-006-9276-3 [DOI] [Google Scholar]
- Malik R. S. (1990). Prospects for Brassica carinata as an oilseed crop in India. Exp. Agric. 26, 125–129. 10.1017/S0014479700015465 [DOI] [Google Scholar]
- Mason A. S., Batley J. (2015). Creating new interspecific hybrid and polyploid crops. Trends Biotechnol. 33, 436–441. 10.1016/j.tibtech.2015.06.004 [DOI] [PubMed] [Google Scholar]
- Meng J., Shi S., Gan L., Li Z., Qu X. (1998). The production of yellow-seeded Brassica napus (AACC) through crossing inter-specific hybrids of B. campestris (AA) and B. Carinata (BBCC) with B. napus. Euphytica 103, 329–333. 10.1023/A:1018646223643 [DOI] [Google Scholar]
- Nakaya A., Isobe S. N. (2012). Will genomic selection be a practical method for plant breeding? Ann. Bot. 110, 1303–1316. 10.1093/aob/mcs109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navabi Z. K., Huebert T., Sharpe A. G., O'Neill C. M., Bancroft I., Parkin I. A. (2013). Conserved microstructure of the Brassica B genome of Brassica nigra in relation to homologous regions of Arabidopsis thaliana, B. rapa and B. oleracea. BMC Genomics 14:250. 10.1186/1471-2164-14-250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navabi Z. K., Stead K. E., Pires J. C., Xiong Z., Sharpe A. G., Parkin I. A., et al. (2011). Analysis of B-genome chromosome introgression in interspecific hybrids of Brassica napus x B. Carinata. Genetics 187, 659–673. 10.1534/genetics.110.124925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M., Tajima F., Tateno Y. (1983). Accuracy of estimated phylogenetic trees from molecular data. II. Gene frequency data. J. Mol. Evol. 19, 153–170. 10.1007/BF02300753 [DOI] [PubMed] [Google Scholar]
- Plummer J. A., Pradhan A., Si P., Mason A., Nelson M., Cowling W., et al. (2010). Development for tri-genomic hexaploid Brassica populations. Hort. Sci. 45, S156–S156. [Google Scholar]
- Pontes O., Neves N., Silva M., Lewis M. S., Madlung A., Comai L., et al. (2004). Chromosomal locus rearrangements are a rapid response to formation of the allotetraploid Arabidopsis suecica genome. Proc. Natl. Acad. Sci. U.S.A. 101, 18240–18245. 10.1073/pnas.0407258102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan A. K., Pental D. (2011). Genetics of Brassica juncea, in Genetics and Genomics of the Brassicaceae. New York, NY: Springer Verlag. [Google Scholar]
- Pradhan A. K., Sodhi Y. S., Mukhopadhyay A., Pental D. (1993). Heterosis breeding in Indian mustard (Brassica juncea L. Czern & Coss): analysis of component characters contributing to heterosis for yield. Euphytica 69, 219–229. 10.1007/BF00022368 [DOI] [Google Scholar]
- Prakash S., Raut R. N. (1983). Artifcial synthesis of Brassica napus and its prospects as an oilseed crop in India. Indian J. Genet. 43, 191–283. [Google Scholar]
- Pritchard J. K., Stephens M., Donnelly P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J. P., Dickson M. H., Earle E. D. (2000). Improved resistance to bacterial soft root by protoplast fusion between Brassica rapa and B. oleracea. Theor. Appl. Genet. 100, 810–819. 10.1007/s,001220051356 [DOI] [Google Scholar]
- SAS Institute (1999). SAS Online Doc (R), version 8.0. Cary, NC: SAS Institute. [Google Scholar]
- Shelton A., Tracy W. (2015). Recurrent selection and participatory plant breeding for improvement of two organic open-pollinated sweet corn (Zea mays L.) populations. Sustainability 7, 5139–5152. 10.3390/su7055139 [DOI] [Google Scholar]
- Takezaki N., Nei M., Tamura K. (2010). POPTREE2: software for constructing population trees from allele frequency data and computing other population statistics with windows interface. Mol. Biol. Evol. 27, 747–752. 10.1093/molbev/msp312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian E., Jiang Y., Chen L., Zou J., Liu F., Meng J. (2010). Synthesis of a Brassica trigenomic allohexaploid (B. Carinata x B. Rapa) de novo and its stability in subsequent generations. Theor. Appl. Genet. 121, 1431–1440. 10.1007/s00122-010-1399-1 [DOI] [PubMed] [Google Scholar]
- UN (1935). Genomic analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. Jap. J. Bot. 7, 389–452. [Google Scholar]
- Waalen W. M., Tanino K. K., Olsen J. E., Eltun R., Rognli O. A., Gusta L. V. (2011). Freezing tolerance of winter canola cultivars is best revealed by a prolonged freeze test. Crop Sci. 51, 1988–1996. 10.2135/cropsci2011.02.0098 [DOI] [Google Scholar]
- Wang H. Y., Tian Q., Ma Y. Q., Wu Y., Miao G. J., Ma Y., et al. (2010). Transpositional reactivation of two LTR retrotransposons in rice-Zizania recombinant inbred lines (RILs). Hereditas 147, 264–277. 10.1111/j.1601-5223.2010.02181.x [DOI] [PubMed] [Google Scholar]
- Wang X., Wang H., Wang J., Sun R., Wu J., Liu S., et al. (2011). The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43, 1035–1039. 10.1038/ng.919 [DOI] [PubMed] [Google Scholar]
- Warwick S. I. (2011). Brassicaceae in Agriculture, in Genetics and Genomics of the Brassicaceae, eds Schmidt R., Bancroft I. (New York, NY: Springer Verlag; ), 33–65. [Google Scholar]
- Woods D. L., Capcara J. J., Downey R. K. (1991). The potential of mustard (Brassica juncea (L.) Coss) as an edible oil crop on the Canadian prairies. Can. J. Plant Sci. 71, 195–198. 10.4141/cjps91-025 [DOI] [Google Scholar]
- Xiao Y., Chen L., Zou J., Tian E., Xia W., Meng J. (2010). Development of a population for substantial new type Brassica napus diversified at both A/C genomes. Theor. Appl. Genet. 121, 1141–1150. 10.1007/s00122-010-1378-6 [DOI] [PubMed] [Google Scholar]
- Yang J., Liu D., Wang X., Ji C., Cheng F., Liu B., et al. (2016). The genome sequence of allopolyploid Brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat. Genet. 48, 1225–1232. 10.1038/ng.3657 [DOI] [PubMed] [Google Scholar]
- Yeh F., Yang R., Boyle T. (1999). Microsoft Windows-Based Free Ware for Population Genetic Analysis (Release 1.31). Edmonton: University of Alberta. [Google Scholar]
- Yu F., Lydiate D. J., Gugel R. K., Sharpe A. G., Rimmer S. R. (2012). Introgression of Brassica rapa subsp. Sylvestris blackleg resistance into B. napus. Mol. Breed. 30, 1495–1506. 10.1007/s11032-012-9735-6 [DOI] [Google Scholar]
- Zamir D. (2001). Improving plant breeding with exotic genetic libraries. Nat. Rev. Genet. 2, 983–989. 10.1038/35103590 [DOI] [PubMed] [Google Scholar]
- Zhou J., Tan C., Cui C., Ge X., Li Z. (2016). Distinct subgenome stabilities in synthesized Brassica allohexaploids. Theor. Appl. Genet. 129, 1257–1271. 10.1007/s00122-016-2701-7 [DOI] [PubMed] [Google Scholar]
- Zou J., Fu D., Gong H., Qian W., Xia W., Pires J. C., et al. (2011). De novo genetic variation associated with retrotransposon activation, genomic rearrangements and trait variation in a recombinant inbred line population of Brassica napus derived from interspecific hybridization with Brassica rapa. Plant J. 68, 212–224. 10.1111/j.1365-313X.2011.04679.x [DOI] [PubMed] [Google Scholar]
- Zou J., Raman H., Guo S., Hu D., Wei Z., Luo Z., et al. (2014). Constructing a dense genetic linkage map and mapping QTL for the traits of flower development in Brassica carinata. Theor. Appl. Genet. 127, 1593–1605. 10.1007/s00122-014-2321-z [DOI] [PubMed] [Google Scholar]
- Zou J., Zhu J., Huang S., Tian E., Xiao Y., Fu D., et al. (2010). Broadening the avenue of intersubgenomic heterosis in oilseed Brassica. Theor. Appl. Genet. 120, 283–290. 10.1007/s00122-009-1201-4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.