

Abstract
Metastatic castration-resistant prostate cancer (mCRPC) accounts for a high percentage of prostate cancer mortality. The proprietary compound galeterone (gal) was designed to inhibit proliferation of androgen/androgen receptor (AR)-dependent prostate cancer cell in vitro and in vivo and is currently in phase III clinical development. Additionally, clinical studies with gal revealed its superb efficacy in four different cohorts of patients with mCRPC, including those expressing splice variant AR-V7. Preclinical studies with gal show that it also exhibits strong anti-proliferative activities against AR-negative prostate cancer cells and tumors through a mechanism involving phosphorylation of eIF2α, which forms an integral component of the eukaryotic mRNA translation complex. Thus, we hypothesized that gal and its new analog, VNPT55, could modulate oncogenic mRNA translation and prostate cancer cell migration and invasion.
We report that gal and VNPT55 profoundly inhibit migration and invasion of prostate cancer cells, possibly by downregulating protein expression of several EMT markers (Snail, Slug, N-Cadherin, Vimentin and MMP-2/-9) via antagonizing the Mnk-eIF4E axis. In addition, gal/VNPT55 inhibited both NF-κB and Twist1 transcriptional activities, downregulating Snail and BMI-1 mRNA expression, respectively. Furthermore, profound up-regulation of E-cadherin mRNA and protein expression may explain the observed significant inhibition of prostate cancer cell migration and invasion. Moreover, expression of self-renewal proteins, β-Catenin, CD44 and Nanog, were markedly depleted. Analysis of gal/VNPT55-treated CWR22Rv1 xenograft tissue sections also revealed that observations in vitro were recapitulated in vivo.
Our results suggest that gal/VNPT55 could become promising agents for the prevention or treatment of all stages of prostate cancer.
Keywords: Prostate cancer, AR degradation agents (ARDAs), Galeterone (Gal)/Gal’s analog (VNPT55), migration and invasion, Mitogen activated protein kinase interacting kinase 1/2 (Mnk1/2), eIF4E phosphorylation
Graphical Abstract
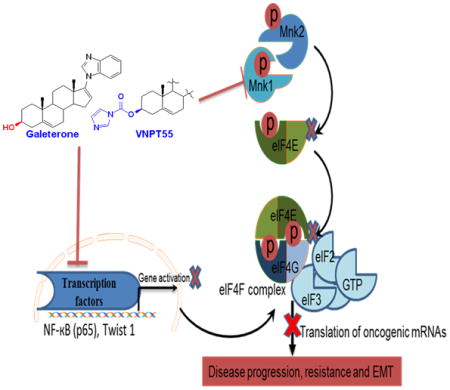
Introduction
Therapy for late-stage prostate cancer (PC) remains very limited and few drugs have shown modest overall survival in patients with metastatic castration-resistant prostate cancer (mCRPC) [1]. PC mortality most frequently results from the metastatic disease which makes it important to design and develop inhibitors to target oncogenes involved in cellular migration and invasion. Epithelial to mesenchymal transition (EMT), a critical process in metastasizing cells [2] is known to be modulated, in part, by the transcriptional activities of NF-κB [3] and the cap-dependent translational complex, mitogen-activated protein kinase (MAPK) kinase interacting kinase1/2 (Mnk1/2)-eukaryotic initiation factor 4E (eIF4E) axis [4, 5].
Substantial studies have revealed the significance of eIF4E phosphorylation (peIF4E) in the progression and metastasis of several cancers including, PC [6]. Interestingly, both the PI3k/Akt/mTOR and Ras/MAPK pathways converge on Mnk1/2-eIF4E axis [7, 8]. Phosphorylation of eIF4E is implicated in EMT and translation of MMP-3 and Snail [9]. Although Mnk-eIF4E axis is reported to be dispensable for normal cell growth and development [10], its role in cancer cell transformation is however very critical [6, 11, 12].
Studies have shown that EMT factors (Snail, Slug, Twist1 and Zeb1/2), can repress the expression of E-cadherin in conjunction with histone deactelyase [13]. BMI-1 (B-cell-specific Moloney murine leukemia virus integration site 1), a putative stem cell factor, is reported to be activated by Twist1 [14]. Several studies have also implicated Twist1 in N-Cadherin and matrix metalloproteinases (MMPs) activation [15, 16]. In a recent study, BMI-1 overexpression was shown to increase expression of Nanog and also enhanced activation of NF-κB thereby increasing stemness and drug resistance [17].
Preclinical and clinical studies suggest that gal possess strong anti-tumor properties against AR-V expressing CRPC models [18] and in men with CRPC [19]. Our previous report on the modulatory effects of gal on endoplasmic reticulum stress response (ERSR) and eIF2α phosphorylation [20] suggests an additional inhibitory activity on translation of mRNAs.
In this study, we observed remarkable gal/VNPT55-induced depletion of Mnk1/2 protein expression which resulted in the suppression of eIF4E phosphorylation. Gal/VNPT55 treatment also resulted in downregulation of several EMT (N-Cadherin, MMPs, Snail and Slug) and putative stem cell factors (β-Catenin, CD44, Nanog, and BMI-1). Knocking down Mnk1 with siRNA showed that gal/VNPT55 mirrored the effects of inhibiting the translation machinery. We also observed a decrease in NF-κB(p65) phosphorylation (p-p65) and RhoA protein expression, implicated in cell migration/invasion and disease progression. Furthermore, both gal and VNPT55 inhibited Twist1 activation of BMI-1 in vitro. We also observed a significant inhibition in PC cell migration and invasion in vitro. Several of these effects were recapitulated in vivo.
Results
Galeterone/VNPT55 inhibit multiple pathways in PC models
Previous studies in PC-3 cells revealed that gal upregulated ERSR genes (CHOP and peIF2α) [20], which are also known to influence protein translation. Following these observations, we evaluated the impact of gal on established PC-3 xenografts. Interestingly, although gal did not prevent tumor formation (Figure 1a), it caused significant inhibition of tumor volumes (Figure 1b) and tumor weights (Figure 1c). The efficacy of gal against PC-3 xenografts in addition to its effects on AR signaling (reviewed in [21]) highlight the multi-target anti-PC activities of gal.
Figure 1.

Efficacy of Gal/VNPT55 on PC-3 xenografts. (a) PC-3 cells were inoculated into the flanks of male SCID mice and treated with either 0.15 mmol/kg gal (12 mice) or vehicle (24 mice) b.i.d. Mice were evaluated daily for the formation of palpable tumor. (b) Male SCID mice were inoculated with PC-3 cells and treated with either vehicle (0.3% hydroxypropyl cellulose, HPC) or 0.15 mmol/kg/b.i.d. d gal. Tumors were measured with calipers as described in materials and methods. (c) Excised PC-3 tumors were weighed following two weeks of treatment. (d) 1 × 106 LNCaP and CWR22Rv1 cells were seeded in 10 cm plates in 5% charcoal dextran supplemented RPMI and subsequently treated with gal (1–5 μM) for duration of 72 h. Immunoblot analysis was utilized to evaluate the expression of ERSR markers. (e) Tumors samples from 4 mice in each treatment group of LAPC4 xenografts were excised and analyzed by western blot for relative expression of ERSR markers, average expression were determined by densitometry (*p<0.05). (f) Cell viability assays were performed in DU145, PC-3 and CWR22Rv1 cells comparing efficacies of gal, VNPT55 and CGP-57380.
Gal’s effects on ERSR genes in PC-3 cells were recapitulated in AR positive cells (LNCaP and CWR22Rv1) in vitro (Figure 1d). However, analysis of peIF2α and BIP expression in AR-positive LAPC4 xenografts [22] revealed no significant difference between vehicle and gal treated groups (Figure 1e). In contrast, cyclin D1 protein expression was significantly down-regulated in vivo (Figure 1e). Since cyclin D1 expression is known to be tightly regulated by the Mnk1/2-eIF4E translation complex [23, 24], this, in addition to the significance of eIF2α in protein translation prompted the hypothesis that gal possibly impacts protein translation, negatively.
To assess the impact/significance of Mnk 1/2 inhibition in PC cells, we compared the anti-proliferative activities of CGP-57380 (Mnk kinase inhibitor) and gal in DU145, PC-3 and CWR22Rv1 cells. Although, cercosporamide inhibits Mnk1/2 with superior activity compared to CGP-57380, it also inhibits a number of kinases (Pim1, GSK3β, ALK4 and Jak3)[25], hence making it unsuitable for selective inhibition of Mnk1/2 as a comparison. Figure 1f shows that whereas the GI50 values of gal and CGP-57380 are comparable, CGP’s efficacy was significantly impaired in PC-3 cells. A study by Bianchini and colleagues reported that PC-3 cells expressed significantly lower levels of peIF4e than DU145 [26], and this could be the reason for CGP’s mediocre efficacy in PC-3 cells.
In response to a suggestion from an astute reviewer, we assessed whether gal/VNPT55’s modulatory effects on both AR and Mnk1/2-eIF4E were partly responsible for their anti-cancer activities. We transfected CWR22Rv1 cells with AR and/or Mnk1 siRNA (Figure 2a), and further analyzed cell viability at 72 h post-treatment with gal/VNPT55. Figure 2b shows that in the absence of AR and/or Mnk1, the GI50 values of gal/VNPT55 were significantly higher in comparison to treated/un-transfected cells. Furthermore, we also transfected CWR22Rv1 cells with Mnk1 and eIF4E plasmids (Figure 2c, left and right panels) [27, 28] and consequently treated them with gal and VNPT55. Interestingly, we observed that overexpressing Mnk1 and/or eIF4E caused an increased expression of markers (MMP-9, Cox-2, Cyclin D1, Slug) known to be regulated by the cap-dependent translation machinery (Figure 2c) and also enhanced the activities of gal and VNPT55, markedly reducing their GI50 values (Figure 2d). This suggests that by silencing AR and/or Mnk1, we eliminated the significant targets of gal/VNPT55, thus minimizing their full impact on cell viability.
Figure 2.

Mnk1-eIF4E overexpression enhances activity of gal/VNPT55 (a) Western blot analysis were conducted on CWR22Rv1 cells transfected with 50 nM of AR and Mnk1 siRNA alone and in combination for 3 days, complexes were washed off and cells lysed after an additional 72 h and analyzed for AR and Mnk1 protein expression. (b) CWR22Rv1 cells treated as in (a) were seeded at 2500 cells/well in a 96 well-plate, allowed to attach overnight and subsequently treated with gal and VNPT55 for an additional 72 h, cell viability was determined as in materials and methods, and GI50 values plotted in a barchart. Results of GI50 values are averages of 3 separate experiments. GI50 values show a significant increase in transfected cells (*p<0.05). (c, left and right panels) CWR22Rv1 cells were transfected with HA tagged eIF4E (HA-eIF4E) and Mnk plasmids for 96 h, western blot was used to analyze protein expression. (d) CWR22Rv1 cells were transfected with Mnk1 and eIF4E plasmids for 16 h, after which cells were counted and seeded at 2500 cells/well in 96-well plates. Cells were subsequently treated with increasing concentrations of gal/VNPT55 for 5 days. GI50 values were computed with graphpad prism and compared to vector-transfected controls (*p<0.05, **p<0.001) (e) CWR22Rv1 and DU145 cells were seeded in 96-well plates and treated with a combination of both gal/VNPT55 and CGP-57380 at a constant ratio of their respective GI50 values. After cell viability was determined with MTT reagent, the fractional effects of the compounds alone and in combination was determined and analyzed by the Calcusyn software to determine the combination indices (CI), to evaluate whether compounds act in synergy (CI < 1), additive (CI = 1) or antagonistic (CI > 1). (f) 1000 cells/well of CWR22Rv1 cells were seeded in 6-well plates and treated with gal/VNPT55 alone or in combination with CGP-57380 (left panel), colonies were stained and counted and presented as bar chart (right graph)
We also evaluated the impact of combining CGP-57380 and gal on PC cell proliferation. Using the calcusyn software, combination indices (CI), in both CWR22Rv1 and DU145 (CI values at ED50 = 10.32393 and 1.58119, respectively) showed antagonistic interaction (Figure 2e). Suggesting that combining Mnk kinase inhibitors with gal may not result in an increased anti-cancer activity. Complementary colony formation studies were also conducted, treating cells with gal, VNPT55, or CGP-57380 and their combinations at ratios of their GI50 values (Figure 2f). This confirmed what was observed in cell viability assays, indicating that Mnk inhibitors may not improve the efficacy of gal/analogs. Although the reasons for these antagonistic interaction [29] needs further in-depth studies, the activity of each agent is not enhanced by the combination.
Galeterone/VNPT55 suppress phosphorylated eIF4E by depleting Mnk1
EIF4E plays a critical role in activating oncogenes implicated in cancer cell survival, proliferation and metastasis [30]. We analyzed expression of peIF4E in LNCaP, CWR22Rv1 and PC-3 cells (Figure 3a–c) post 24 h gal/VNPT55 treatments. Indeed, gal and VNPT55 decreased Mnk1/2 protein expression and markedly downregulated peIF4E. Cell viability assays in Figure 1f (right table), showed that CGP-57380, exhibited mediocre activity with a high GI50 value in PC-3 cells, however, at 5 and 10 μM, CGP-57380 inhibited phosphorylation of eIF4E (Figure 3d). Although gal/VNPT55 do not completely inhibit phosphorylation of eIF4E, the decrease evidently is significantly below the threshold which results in inhibition of translation of oncogenic mRNAs, in agreement with a recent published study [31]. Protein expression analysis of Mnk1/2 in CWR22Rv1 cells demonstrates a dose-dependent effect (Figure 3e). Inhibiting protein translation with cycloheximide with or without gal, shows that gal in combination with cycloheximide enhances the depletion of Mnk1, markedly reducing the half-life of Mnk1 protein expression (Figure 3f, left and right panel). Interestingly Mnk1 depletion by gal was significantly inhibited by MG132 (proteasomal inhibitor), in LNCaP and CWR22Rv1 cells (Figure 3g), implicating the proteasome in gal-induced Mnk1 depletion, similar to our previous report for other agents [32]. We next examined whether ubiquitination preceded gal-induced Mnk1 degradation. Indeed, gal enhanced Mnk1 ubiquitination prior to its degradation (Figure 3h). As expected, the downstream targets of Mnk1-eIF4E (Cox-2, Mcl-1 and Cyclin D1) were also markedly suppressed in LNCaP cells (Figure 3i). Additionally, mRNA analysis of gal/VNPT55 treated PC-3, DU145 and CWR22Rv1 cells (Figure 3j) showed that gal/VNPT55 (5 and 10 μM) significantly decreased Mnk1 mRNA, suggesting a potential effect on transcription factors implicated in Mnk1 gene activation such as SP1[33], ATF (a cAMP-response element also present upstream of the 5′ start site) [34], AP-1[35] and CTF/NF-1 [36].
Figure 3.

Gal and VNPT55 modulate eIF4E phosphorylation by depleting Mnk1/2 expression levels. (a–c) LNCaP, CWR22Rv1 and PC-3 cells were treated with gal and VNPT55 at 10 μM for 24 h. Cells were lysed with RIPA buffer and 50 and 100 μg of total protein used in analyzing total proteins and phosphorylated proteins respectively. (d) PC-3 cells were incubated with 5 and 10μM of CGP-57380 for 24 h and eIF4E phosphorylation analyzed by western blot analysis. (e) CWR22Rv1 cells were treated with increasing dose of gal/VNPT55 and Mnk1/2 protein expression analyzed by western blot with 50 μg total protein. (f) CWR22Rv1 cells were treated with cycloheximide and or gal and Mnk1 protein expression analyzed by western blot analysis. (g) LNCaP and CWR22Rv1 cells incubated with gal alone or in combination with MG132, as in materials and methods to determine the role of the proteasomal enzyme in gal/VNPT55 induced post-translational modulation of Mnk1. (h) LNCaP cells treated with 10 μM for 16 h were incubated with 5 μM MG132 for additional 8h. 500 μg total proteins was separated on a 10% Tris glycine gel, transferred to a Polyvinylidiene fluoride (PVDF) membrane and probed with ubiquitin antibodies. (i) LNCaP cells were treated with 10 μM of gal/VNPT55 and downstream targets of Mnk-eIF4E axis were evaluated. (j) RNA was collected from PC-3, DU145 and CWR22Rv1 cells incubated with 5 and 10 μM of gal/VNPT55 for 24 h. Mnk1 primers were used in quantitative real-time PCR to evaluate effects on mRNA expression levels. Results are represented as means ± S.E.M (*p<0.05).
Galeterone/VNPT55 modulate EMT factors in PC cells
Based on the effects seen on Mnk1/2-eIF4E axis, we hypothesized that gal/VNPT55 may inhibit migration and invasion of PC cells, by downregulating EMT factors. The Mnk-eIF4E axis plays a pivotal role in the expression of mesenchymal markers such as N-cadherin, MMPs and Snail [4, 37, 38]. Thus, we first examined the effects of gal/VNPT55 (5 and 10 μM) on N-Cadherin protein expression in PC-3 and DU145 cells. As shown in Figure 4a, the compounds significantly decreased N-Cadherin protein levels and increased expression of epithelial marker, tight junction protein zona occludens protein, ZO-1 in PC-3 cells. However, effects on N-Cadherin in DU145 were not as significant as observed in PC-3 cells. We also determined whether gal/VNPT55 had any effect on E-cadherin expression levels using fluorescence microscopy. Interestingly, both gal and VNPT55 caused strong up-regulation of E-Cadherin in PC-3 and DU145 cells (Figure 4b), suggesting a reversal in mesenchymal characteristics to a more epithelial non-invasive state.
Figure 4.

Gal/VNPT55 decreases mesenchymal factors and enhances epithelial marker expression. (a) Lysates from PC-3 and DU145 cells treated with 5 and 10 μM were subjected to immunoblot analysis to evaluate N-Cadherin and ZO-1. (b) PC-3 and DU145 cells incubated with gal and VNPT55 at 10 μM for 24 h were stained with E-Cadherin polyclonal antibodies following protocol in materials and methods. Images were taken with Zeiss camera mounted immunofluorescence microscope. (c) PC-3, DU145 and CWR22Rv1 cells were analyzed for MMP-2/-9 protein expression after gal/VNPT55 treatment. (d) LNCaP cells were serum starved for 12 h and treated with 5, 10 and 20 μM gal/VNPT55 for 36 h in serum-free, pen-strep free RPMI media. Culture media was concentrated using Millipore 0.5 ml ultra-centrifugal columns. Conditioned media was separated on 0.1% gelatin Tris/Glycine gel to analyze proteolytic activity of MMPs (top), densitometric analyses (bottom bar chart) shows significant decrease in MMP-2 activity (*p<0.5, **p<0.001). (e) PC-3 cells were treated with gal/VNPT55 at 2.5 and 5 μM for 72 h and media concentrated as in (d) and a zymogram gel used to determine proteolytic activity of MMP-2/-9. (f) DU145 cells were treated as in (e), at 2.5 μM with gal/VNPT55 and analyzed with gelatin gels. Densitometric analysis shows a significant decrease in MMP-2/-9 activity. All zymogram assays were repeated at least three times and represented as means ± S.E.M (*p<0.05).
Dysregulation of MMPs play a critical role in carcinogenesis, tumor invasion and tissue remodeling [39–41]. As shown in Figure 4c, MMP-2 was strongly down-regulated in CWR22Rv1 cells and both MMP-2/-9 were also markedly downregulated in PC-3 and DU145 cells. Media from PC cells incubated with compounds for 36 h (LNCaP) and 72 h (PC-3 and DU145) were analyzed for MMP collagenase activity. Media from LNCaP (Figure 4d), PC-3 and DU145 (Figure 4e and f) cultures show a significant down-regulation of secreted active MMPs.
Galeterone/VNPT55 inhibit migration and invasion of PC cells
We also determined whether gal/VNPT55-induced depletion of EMT markers also subsequently caused inhibition of PC cell migration and invasion. Prior to conducting these assays, we evaluated cell viability with gal/VNPT55 (1–10 μM), at 24 h, to show that doses used did not compromise cell integrity and also that cell number did not increase significantly (Figure 5a). PC-3 and DU145 cells grown to a monolayer and scratched with 200 μl pipette tip, were subsequently treated with gal/VNPT55 (5 μM) over 12 h. Compared to controls gal/VNPT55 significantly inhibited migration of both PC-3 and DU145 cells (Figure 5b and c).
Figure 5.

Gal and VNPT55 inhibit migration and invasion of PC cells in vitro. (a) To evaluate whether PC-3 and DU145 cells used in migration and invasion assays at indicated compound concentrations did not significantly compromise cell numbers and viability, we performed a 24 h MTT cell viability assay with gal/VNPT55 (1–10 μM. (b) PC-3 (top panel) and DU145 (bottom panel) cultured in 24-well plates to a confluent monolayer were scratched with a 200 μl pipette tip and subsequently treated with indicated compounds for 12 h. (c) Wounds were measured before and after the 12 h time point. Distance migrated were quantified by measuring the difference at time 0 and 12 h and normalized to control. (Distance migrated = Distance at time 0 h - distance at 12 h/Distance migrated by control). Experiments were repeated at least 3 times and represented as mean ± S.E.M, shows significant inhibition of cell migration (**p<0.001) (d) PC-3, DU145 and CWR22Rv1 were seeded in BME pre-coated inserts. Cells treated with gal, VNPT55 and CGP at 5 μM in the upper chamber in serum free RPMI media. The bottom chamber was filled with 1ml RPMI media supplemented with 10% FBS. Set-up was placed in 37 °C incubator for 36 h. Cells were fixed in 3.7% paraformaldehyde for 10 minutes and stained with 0.05% crystal violet; cells in upper chamber were wiped off with cotton swabs and invaded cells at the bottom of inserts analyzed by counting. (e) Schematic illustration of invasion assay set-up, with upper and lower chambers. (f) Quantified invaded cells shows a significant inhibition of PC cell invasion (*p < 0.05, **p<0.001). (g and h) PC-3 cells grown to a monolayer and scratched with 200 μl were treated with EGF (10ng/ml) alone and in combination with gal (5 μM), CGP (5 μM) and U0126 (5 μM). Wound healing was analyzed as in (b and c), gal and CGP significantly inhibited migration in the presence of EGF ligand (*p<0.05). All experiments were repeated at least three times and represented as mean ± S.E.M. (i) To evaluate whether activated MMP-9 could be inhibited by galeterone as in Figure 4g, we treated cells with gal, VNPT55 with or without EGF at (gal-5 μM + EGF-10ng/ml) and incubated for 72 conditioned media was concentrated and separated on zymogram gels. It was observed, just as in Figure 5g, that galeterone even in the presence of EGF, was able to inhibit collagenase activity. Experiments were repeated three times (*p<0.05, **p<0.001)
Boyden inserts pre-coated with basement membrane extract (BME) were utilized in invasion assays. 0.75 × 105 PC-3, DU145 and CWR22Rv1 cells were seeded in the upper chamber in phenol red-free RPMI (schematic in Figure 5e) and treated with gal, VNPT55 or CGP-57380. Invading cells after 24 h were stained with 0.5% crystal violet solution (Figure 5d) and quantified by counting cells at the bottom part of the insert, represented in the graph (Figure 5f). Gal, VNPT55 and CGP-57380 significantly inhibited invasion (>95% for gal/VNPT55 vs 35–50% for CGP-57380) of PC cells compared to dimethyl sulfoxide (DMSO) treated controls.
Epidermal growth factor (EGF) family ligands are implicated in cell motility and tumor invasion [42, 43]. The mTORC2-NF-κB pathway, promoting chemoresistance is reported to be activated by epidermal growth factor receptor (EGFR) [44]. Studies have shown that MAPK inhibitor (U0126) retards cell migration and invasion [45]. We evaluated the ability of gal, U0126 and CGP-57380 to inhibit PC cell migration in the presence of EGF (10 ng/ml). We observed that both U0126 and CGP inhibited migration of PC-3 cells (Figure 5g–h). However, in the presence of EGF, only gal and CGP significantly inhibited cell migration. Interestingly, U0126 did not inhibit cell migration in the presence of EGF; probably because the concentration used in the assay (5 μM) was not high enough to overcome the EGF-induced activity. Analysis of MMP-9 collagenase activity in PC-3 cells revealed that gal’s ability to decrease MMP-9 activity was not diminished in the presence of EGF (Figure 5i).
Galeterone/VNPT55 antagonize the NF-κB signaling pathway and Twist1 transcriptional activity
The NF-κB pathway has been implicated in cell proliferation, tumor metastasis, angiogenesis, cell survival [46, 47] and induction of drug resistance. Considering the effects observed on downstream targets of NF-κB signaling, we analyzed effects of gal/VNPT55 on p-p65 and its transcriptional activities. We evaluated expression of p-p65, immunoblot analysis (Figure 6a) revealed a decrease of p-p65 in both PC-3 and DU145 cells. To delineate whether gal/VNPT55 suppression of PC cell migration and invasion resulted from a combined inhibition of both NF-κB and Mnk/eIF4E axis, we analyzed mRNA levels of NF-κB target genes, Snail, Twist1, MMP-9 and E-Cadherin. Interestingly, Snail and MMP-9 mRNA expression were significantly down-regulated with a significant increase in E-Cadherin mRNA (Figure 6b). In contrast, Twist1 mRNA was not significantly affected by gal/VNPT55 (Figure 6b).
Figure 6.

Gal/VNPT55 disrupts NF-κB and Twist1 transcriptional activity. (a) PC-3 and DU145 cells were treated with gal and VNPT55 for 24 h and 50 or 100 μg of total cell lysates separated on a 10% Tris/glycine gel to analyze p65, p52 and phosphorylated p65 (p-p65). (b) RNA collected from treated PC-3 cells were subjected to quantitative real-time PCR to analyze E-Cadherin, MMP-9, Twist1 and Snail mRNA expression (**p<0.001). (c) Protein expression of Snail, Slug, Twist1, RhoA, vimentin and Vascular endothelial growth factor (VEGF) were evaluated by immunoblot analysis after a 24 h treatment in PC-3 and DU145 cells. (d and e) PC-3 and DU145 cells pre-treated with 10 ng TNF-α for 2 h were subsequently treated with gal/VNPT55 at 10 μM for an additional 24 h. Total cell lysates were subjected to cell fractionation. [PC-3-Nu and DU145-Nu are nuclear fractions; PC-3-Cyt and DU145-Cyt are cytosolic fractions]. LSD1 was used as loading controls for nuclear fractions and lactate dehydrogenase (LDH) for cytosolic fraction controls. Twist1, p65 and p-p65 expression levels were analyzed in the different compartments. (f)PC-3 cells were pre-treated with 10 ng TNF-α for 2 h and then treated with gal/VNPT55 for 24 h. Cells were fixed in 3.7% paraformaldehyde and stained with Twist1 mouse monoclonal antibody and images taken
Immunoblot analysis on Snail, Slug, Twist1, Vimentin and VEGF, showed that both gal and VNPT55 significantly depleted protein expression of Snail, Slug, Vimentin and VEGF in both PC-3 and DU145 cells (Figure 6c). However, similar to no observable effects on Twist1 mRNA, Twist1 protein expression was unchanged post-treatment with gal and VNPT55 (Figure 6c). Interestingly, gal/VNPT55 significantly depleted RhoA (Figure 6c), which is involved in cell motility and migration [48–50]. We also evaluated whether decreases in p-p65 prevented p65 nuclear accumulation after gal/VNPT55 treatment. Fractionation of PC-3 and DU145 cells after 24 h incubation with gal/VNPT55 at 10 μM, revealed that p-p65 was significantly decreased in the nuclear compartment, in contrast to no observed change in total p65 (Figure 6d–e). Published work has shown that p65 phosphorylation can occur in the nuclear compartment after translocation [51], possibly gal/VNPT55 effects on p65 phosphorylation occurs in the nuclear compartment. We also observed that Twist1 nuclear translocation was almost completely inhibited (Figure 6d–e). To confirm inhibition of nuclear translocation of Twist1, we stained PC-3 cells with Twist 1 antibody following pre-treatment with 10 ng/ml of TNF-α with or without gal/VNPT55. Our results show that, gal/VNPT55 inhibit Twist1 nuclear translocation (Figure 6f).
We next determined whether decrease in nuclear p-p65 had any consequence on its transcriptional activities, however, it is also important to note that p65-p50 heterodimers that are not phosphorylated on the p65 subunit enhance gene transcription, albeit weakly [52]. Primers spanning the NF-κB consensus binding sequence in Snail promoter region (Figure 7a, upper schematic) were used to amplify cDNA in a chromatin immunoprecipitation (ChIP) assay, immunoprecipitating with NF-κB (p65) antibody. Our data showed that indeed, gal and VNPT55 strongly decreased p65 binding to Snail promoter region (Figure 7a, bottom chart), which may be responsible for the observed decrease in Snail mRNA. We also investigated the transcriptional activities of Twist1 inhibition, on BMI-1 promoter enrichment. QRT-PCR analysis revealed a significant decrease in BMI-1 mRNA in PC-3, DU145 and CWR22Rv1 cells (Figure 7b). ChIP assays showed that Twist1 binding to BMI-1 promoter (Figure 7c) was several folds decreased after gal/VNPT55 treatment (Figure 7d and e, DU145 and CWR22Rv1, respectively). These observations might explain the decrease in BMI-1 and increase in E-Cadherin mRNA levels.
Figure 7.

Gal/VNPT55 inhibits p65 and Twist1 chromatin binding (a) Schematic of NF-κB binding site in Snail promoter region (a, top panel). Gal/VNPT55 decreases p65 binding activity to Snail promoter region in PC-3 cells (a, bottom graph). (b) RNA collected from PC-3, DU145 and CWR22Rv1 cells were analyzed for BMI-1 expression levels after 24 h treatment (*p<0.05, **p<0.001). (c) Schematic representation of Twist1 binding site in BMI-1 promoter region. (d) DU145 and (e) CWR22Rv1 cells were treated with gal/VNPT55 for 36 h at 10 μM and interaction between Twist1 and BMI-1 promoter measured using chromatin immunoprecipitation (ChIP) assay. BMI-1 promoter fold enrichment was normalized to input controls and presented as mean ± SEM (**p<0.001).
Galeterone/VNPT55 depletes stem cell factors and mirrors effects of inhibiting Mnk-eIF4E axis
Since cancer cells undergoing EMT express self-renewal characteristics [53], we evaluated the effects of gal/VNPT55 on putative stem cell factors in two PC cell lines. PC-3 and DU145 cells treated with 10 μM of gal/VNPT55 down-regulated protein expression of Nanog, CD44, β-catenin and BMI-1 (Figure 8a). EZH2, a member of the PRC2, binds to promoters of target genes and methylates lysine 27 of histone H3 (H3K27) [54, 55], methylated H3K27 is recognized by PRC1 (eg. BMI-1) [56], both bind promoter regions of target genes to maintain their suppression. Treatments with gal/VNPT55 significantly decreased expression of EZH2 in PC-3 and DU145 cells (Figure 8a). C-Myc, an oncogene implicated in cell proliferation was also strongly downregulated in both PC-3 and DU145 cells.
Figure 8.

Gal/VNPT55 inhibits PC colony formation and downregulate protein expression of stem cell factors. Mnk1 knockdown exhibited similar effects. (a) PC-3 and DU145 were treated with gal/VNPT55 as indicated and stem cell factors analyzed by immunoblot. (b) 1000 cells/well (PC-3 and DU145), seeded in 6-well plates were treated with indicated concentrations of compounds for a period of 14 days. Media containing compounds were replaced every 3 days. Colonies were stained with 0.05% crystal violet. (c) Colonies from B were counted and presented as bar chart. Results are represented as averages from 3 separate experiments with S.E.M. (*p<0.5, **0.001).
Clonogenic assays to determine the abilities of gal/VNPT55 and CGP (0.5 – 10 μM) to inhibit colony formation (CFU), showed a significant decrease in CFU, further emphasizing their activities on multiple oncogenes. Our data show that, gal/VNPT55 (1 and 2.5 μM) exhibited similar effects as CGP (10 μM) in reducing CFU (Figure 8b and c).
We analyzed whether effects seen on Mnk1/2 may be as a result of gal/VNPT55-induced AR depletion. Thus, we knocked down AR and analyzed Mnk1/2 protein and vice versa. We observed in this study that, depletion of either AR or Mnk1 (with siRNA), had no effect on either targets in both LNCaP and CWR22Rv1 cells (Figure 9a and b, left and right panels).
Figure 9.

Mnk1 knockdown mirrors activity of gal/VNPT55 (a and b) LNCaP (top panel) and CWR22Rv1 (bottom panel) cells were transfected with AR siRNA (25 and 50 nM) for 72 h and Mnk1/2 protein expression analyzed by western blot. LNCaP (top panel) and CWR22Rv1 (bottom panel) cells were transfected with Mnk1 siRNA (25 and 50 nM) for 72 h and full length AR analyzed. (c and d) PC-3 and DU145 cells were transfected with 25 and 50 nM Mnk1 siRNA for 72 h. EMT and stem cell factors were analyzed by western blot.
To show that gal/VNPT55’s effects on Mnk1/2-eIF4E axis resulted in the depletion of multiple oncogenes as observed in this study, we transiently transfected PC-3 and DU145 cells with Mnk1 siRNA. Factors implicated in self-renewal (β-Catenin, EZH2, BMI-1 and Nanog) were down-regulated in PC-3 and DU145 cells (Figure 9c–d). We also analyzed EMT factors (N-Cadherin, MMP-2/-9, Snail, Slug and Twist1); all but Twist1 protein expression were significantly depleted (Figure 9c–d). Previous studies have shown that decreasing peIF4E resulted in a concomitant decrease in MMP-2/-9, Snail, VEGF and β-Catenin protein expression [57]. In summary, these studies clearly show that the effects of silencing Mnk1 (i.e. protein translation) mirrors the effects of gal and VNPT55 in PC cells.
Gal and VNPT55 down-regulate expression of Mnk1/2 and BMI-1 in vivo
Effects seen in vitro are not necessarily observed in vivo, as seen with ERSR markers in our study with LAPC4 xenografts (Figure 1e). It was important to determine whether gal/VNPT55-induced effects on the translation machinery would be observed both in vitro and in vivo. Tumor sections from CWR22Rv1 xenografts, from our recent studies [58], were stained for Mnk1/2, peIF4E, Slug and BMI-1 expression. Representative tumors from gal/VNPT55 treated groups were minced, lysed and used for immunoblot analysis. Gratifyingly, we observed that with the exception of β-Catenin (in gal treated tumors), Mnk1/2, BMI-1 and Oct-4 were markedly down-regulated in vivo in both treated groups compared to vehicle treated controls (Figure 10a–c). Staining of paraffinized sections also confirmed observations from western blot analysis (Figure 10d). Most importantly, both BMI-1 and Slug which are targets of Twist1 were significantly downregulated in vivo (Figure 10e).
Figure 10.

Gal and VNPT55 modulate components of the translational machinery in vivo. (a) Representative tumors-1 and 2 (T1 and T2) from gal and VNPT55 treated groups were excised, minced and lysed. Protein was quantified and western blotting performed to analyze effects on protein expression. Mice (n = 5) were administered with gal (0.15 mmol/kg/twice daily) and VNPT55 (0.15 mmol/kg/twice daily), by intraperitoneal injection, 5 days per week for 34 days. 50 μg of total cell lysate separated on a Tris/glycine gel shows that, in Gal treated xenograft tumors, Protein expression of β-Catenin, Mnk1/2, BMI-1 and Oct-4 are down-regulated. (b) Immunoblot analysis in VNPT55 treated groups also show that effects seen on expression of indicated protein in vitro were also observed in vivo. (c) Densitometry analysis of protein expression from western blot analysis was plotted to quantify the effects seen in vivo. (d) Representative images of Mnk1/2, BMI-1, Slug and peIF4E immunostaining in vehicle and gal/VNPT55 treated groups. Parafinised tumor sections were stained with Mnk1/2 BMI-1, Slug and peIF4E antibodies following protocol in materials and methods. Both gal and VNPT55 show strong depletion effects on Mnk1/2. (e) IHC staining in (e) was quantified using the ImageJ software and represented as a bar chart. (f) Schematic representation of multiple effects of Gal and VNPT55 in inhibiting EMT (1) NF-κB (p65) and p50 heterodimerizes and binds to its cognate sequences in promoter regions of target genes to activate them, gal/VNPT55 in addition to decreasing p65 phosphorylation levels also decrease binding to the chromatin. (2) Twist1 binds to BMI-1 gene to activate its transcription; ChIP analyses shows gal/VNPT55 decreases this interaction. (3) Twist1 also binds to E-Cadherin promoter to repress its transcription, with gal/VNPT55 inhibiting nuclear translocation of Twist1, this significantly inhibits the process and enhances E-Cadherin expression. (4) Mnk1/2 phosphorylation leads to phosphorylation of eIF4E and subsequent formation of the translation complex to translate oncogenic mRNAs. (5) Gal and VNPT55 deplete protein expression of Mnk1/2 and (6) ultimately inhibit the metastatic potential of Mnk-eIF4E axis. Gal and its new analog, VNPT55, decrease transcription and translation of oncogenic mRNA via inhibiting chromatin binding of transcription factors (NF-κB and Twist1) and depleting protein expression of Mnk1/2, respectively. Mnk1/2 depletion culminates in decrease in eIF4E phosphorylation and disruption of cap-dependent mRNA translation, thus inhibiting prostate cancer cell proliferation and epithelial mesenchymal transition (EMT).
Discussion
Multiple pathways and oncogenes are implicated in cancer disease progression, and the great majority of mortality from PC is due to the metastatic disease rather than from the localized disease [59, 60]. The presence of putative cancer stem cells that possess self-renewability and tumor initiating ability enhance resistance to therapeutic agents [61]. This stemness has been implicated with EMT [14]. Activity of BMI-1 has also been implicated in docetaxel resistance in PC [62] and studies have shown that by silencing BMI-1, resistant cells were sensitized to docetaxel therapy. Another axis that increases resistance is the protein kinase c/Twist1 axis which has been reported to be involved in enzalutamide resistance and CRPC [63].
The main objective of this study was to evaluate the efficacy of gal and its analog on pathways involved in PC metastasis, which are also implicated in drug resistance. Here, we report for the first time the inhibitory effects of gal and VNPT55 on Mnk1/2 expression with concomitant decrease in peIF4E. EIF4E activation is reportedly elevated in human PC [64], this promotes tumorigenesis [6] and also enhances EMT [4]. Down-regulation of peIF4E results in a concomitant downregulation of several oncogenic transcription factors implicated in cell invasion, proliferation and resistance, as was observed in this study by silencing Mnk1.
β-Catenin is normally sequestered by E-cadherin, however in metastatic cells, the loss of E-cadherin releases β-Catenin which then forms a complex with T-cell factor/lymphoid enhancer factor (TCF/LEF) to activate the transcription of genes including Snail [65, 66] and VEGF [67]. Our data led us to suggest that gal/VNPT55 may potentially reverse EMT via down-regulation of N-cadherin expression with concomitant increase in E-cadherin.
In addition to effects on the translation machinery, gal and VNPT55 showed inhibitory effects on the NF-κB pathway. The agents also exhibited differential modulation of NF-κB and NF-κB downstream targets. Interestingly, we observed that NF-κB modulation was significantly restricted to the nuclear compartment, which suggests a novel unexplored mechanism of gal/VNPT55-induced NF-κB regulation. Perhaps most significantly, is the inhibition of Twist1 nuclear translocation. Inhibitory effects on Twist1 may have a high clinical impact, as Twist1 is involved in EMT, drug resistance and stem cell renewal. Stem cell factors like CD44 have been implicated in tumorigenicity, cell motility and bone metastases [68, 69], and inhibiting these factors may prove to have favorable clinical implications. Since BMI-1 is upregulated in CRPC and implicated in metastases (mCRPC), its inhibition will greatly impact efficacy of gal and its analogs in PC therapy.
Another important observation was the fact that effects of gal/VNPT55 observed in vitro were recapitulated in vivo. These observations, in addition to their superb toxicity profiles in the laboratory [18] and clinic [19], suggest that, gal and VNPT55 would have significant impact in the clinic. Gal/VNPT55’s effects on multiple signaling pathways based on our in vitro and in vivo results are summarized in Figure 10f.
In summary, our study demonstrates that gal/VNPT55, target oncogenic protein translation, via Mnk1/2 degradation. These two agents markedly attenuate EMT, stem cell characteristics and PC cell migration and invasion. As a consequence of these multiple desirable anti-cancer activities, gal/VNPT55 could become promising agents for the prevention of CRPC and treatment of both early- and late-stage PC. In addition, these agents may be valuable for tackling PC metastasis, a major contributor of PC-associated patient mortality.
Material and Methods
Cell Culture, reagents and immunoblot analysis
Human PC cells lines CWR22Rv1, LNCaP, PC-3 and DU145 PC cells were maintained in RPMI supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. Gal/VNPT55 were synthesized in our laboratory [70] and dissolved in DMSO. Cell culture reagents (FBS, RPMI, and DMEM) were from Invitrogen (Carlsbad, California, USA). U0126, β-actin, Gapdh, Mnk1/2, eIF4E, peIF4E, N-Cadherin, E-Cadherin, Snail, Slug, MMP-2/-9, BMI-1 anti-mouse and anti-rabbit horseradish peroxide (HRP) were purchased from cell signaling (Danvers, Massachusetts, USA). CGP-57380 was purchased from Sigma Aldrich (St. Louis, Missouri, USA). Twist1 polyclonal, EZH2, RhoA, Oct-4 and Nanog antibodies were from Santa Cruz (Dallas, Texas, USA) and the mouse monoclonal antibody from Abcam. β-Catenin, CD44 and VEGF were from BIOSS (Woburn, Massachusetts, USA).
Treated cells were lysed with radioimmunoprecipitation assay (RIPA) buffer (Sigma), supplemented with protease inhibitors (Roche, Indianapolis, Indiana, USA), 1 mM EDTA and 1 mM PMSF (Sigma). Western blotting was done as previously described [58].
Cell viability assays (3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT), colorimetric assay)
MTT assays were performed as described in our previous publications [70]. Briefly, 2500 cells were seeded in 96 well plates overnight. Cells were then treated with indicated concentrations.
Ubiquitination assay
LNCaP treated with gal (20 μM) for 16 h and MG132 (5 μM) for an additional 8 h and lysed with RIPA buffer. 0.5 mg of total cell lysates were pre-cleared with 20 μl of protein A/G sepharose beads (Santa Cruz), and used in immunoprecipitation assays. Polyclonal Mnk1/2 antibodies were used at 1μg to 500 μg total cell lysate.
SiRNA and plasmid transfection
PC-3, DU145, LNCaP and CWR22Rv1 cells were transfected with 25 or 50 nM Mnk1 or AR siRNA (Invitrogen) for 72 h. Scrambled siRNA were transfected as controls. Briefly, 20 μl of lipofectamine RNAiMAX (Invitrogen) reagent was incubated with 25 or 50 nM of siRNA in 1 ml of OPTI-MEM media for 15 minutes at room temperature (r.t). 100 mm plates were subsequently coated with siRNA complexes for an additional 15 minutes. 3 ml of cell suspended in DMEM media without pen/strep were added to plates for 16 h; RNAi complexes were washed off and replaced with regular media. 1 and 2 μg of Mnk1 and HA tagged eIF4E plasmids were transfected into CWR22Rv1 cells, using the Qiagen Effectene transfection kit following manufacturers protocol. 16 h post-transfection, cells were trypsinized, counted and plated in 96-well plates at 2500 cells/well for MTT cell viability assay.
RNA isolation and real-time polymerase chain reaction analysis
Cells were seeded in 6-well plates at 0.3 × 106 cells per well and treated with compounds for 24 h. RNA was isolated with the Qiagen (Valencia, California, USA) RNeasy reagents following manufacturer’s protocol. 1800 ng of RNA were reverse transcribed into cDNA using Qiagen cDNA conversion kit. Relative mRNA levels of Mnk1 (forward: 5′GAGAAGCCAGCCGAGTGGT-3′ and reverse: 5′-TGCCTTTGGTATGCAGGAAGT-3′, MMP-9, and BMI-1 (forward: 5′-AAATGCTGGAGAACTGGAAAG-3′ and reverse: 5′-CTGTGGATGAGGAGACTGC-3′) were quantified with the comparative ΔΔCt using 18S as internal control. Primers for E-cadherin, Twist1, and Snail were as reported in Schech and colleagues [71].
Zymogram Assay
Culture media from LNCaP, PC-3 and DU145 after 72 h treatment was collected and concentrated with 0.5 ml amicon ultra centrifugal columns (Millipore, Bedford, MA). Samples were quantified and 50 μg of conditioned media electrophoresed on a 10% SDS-polyacrylamide gel containing 0.1% gelatin. Gels were briefly washed with de-ionized water and renatured in 1X renaturing buffer for 20 minutes (2X), on a rocker at r.t. Gels were developed in 1X developing buffer for 48 h and stained with page blue and de-stained with de-staining solution (50% methanol, 10% acetic acid and 40% water). Bands representing MMP-2 and MMP-9 were scanned and quantified.
Cell motility (scratch-wound-healing) assay
5 × 105 PC-3 and DU145 cells were seeded per well 24-well plates over 24 h to form a uniform monolayer and maintained in serum-free media for 12 h. Cells were scratched with a 200 μl pipette tip and images taken at 0 h. Treated Cells were incubated at 37 °C for 12 h. Images were taken using the Zeiss microscope.
Invasion assay
Pre-coated Trans-well inserts, 8-μm pore size, (Trevigen) were incubated at 37°C for 2 h. 0.75 × 104 cells were seeded in the top chamber in serum-free media with or without compounds. Bottom chamber was filled with 1 ml of RPMI with 10% FBS. Experimental set-up was placed in a 37 °C incubator for 24 h. Cells at the top chamber of inserts were scraped off with cotton swabs and migrated cells at the bottom were fixed with ice cold methanol and stained in 0.05% crystal violet.
Colony formation assay
0.5 × 103 cells per well were seeded in a 6-well plates and allowed to attach overnight (16h), cells were treated with compounds in RPMI with 10% FBS at the indicated concentrations and replaced every 3rd day for 14 days. Colonies were washed and stained with 0.05% crystal violet for 30 minutes. Colonies were scanned and quantified with ImageJ colony counter. Results are represented as the mean of triplicates.
Nuclear/cytosol fractionation
Fractionation was performed with the active motif nuclear/cytosol kit, following manufacturer’s protocol. Treated cells were washed once with 1X dPBS supplemented with phosphatase inhibitors and scraped into Eppendorf tubes. Cells were pelleted in a swing bucket centrifuge at 500 rpm for 5 minutes and resuspended in 250 μl of hypotonic buffer, incubated on ice for 10 minutes. 25 μl of detergent was added and vortex briefly for 10 seconds. Nuclear was pelleted in a table top micro-centrifuge at 10k rpm for 30 seconds. Supernatant was stored as cytosol fraction and nuclear pellet washed 2X with 1X hypotonic buffer. Pellet was lysed for 30 minutes in 50 μl of complete cell extraction buffer and sonicated twice for 10 seconds. Nuclear debris was pelleted in a cold micro-centrifuge for 20 minutes at 13300 rpm.
Immunofluorescence staining
PC-3 and DU145 cells were seeded and grown to 70% confluence in an 8-chamber slide. Cells were then treated with gal and VNPT55 at 5 and 10 μM in regular media for 24 h. E-Cadherin, Twist1 antibodies were diluted in 2.5% BSA in PBS at 1:1000 and 1:500 respectively. Immunostaining was performed as previously reported [70].
Chromatin immunoprecipitation
Treated cells were cross-linked with 1% formaldehyde at r.t. for 30 minutes and neutralized with glycine (125 nM) for 5 minutes. Washed cells were resuspended in FA lysis buffer. Samples were sonicated 10 sec. for 10 X, clarified and incubated with 5 μg of NF-κB (p65) or 5 μg of Twist1 antibody to 200 μg of quantified lysate and 30 μl of agarose A/G beads for 14 h at 4 °C. Control samples were incubated with 5 μg of normal rabbit immunoglobulin G (IgG) and/or beads alone as negative controls. Complexes were washed 3X with wash buffer and eluted. The qiaquick polymerase chain reaction (PCR) purification kit was used in purifying products resulting from immunoprecipitation. Real-time PCR analysis was conducted on resulting purified DNA. Human BMI-1 promoter primers: forward: 5′-GGTCAAGTACATGTGAC-3′ and reverse: 5′-TCTCCTCTAGCTTGCAG-3′. PCR reaction was amplified for 50 cycles with a 60 °C annealing temperature. Values were normalized to input controls.
Establishment of tumor xenograft and in vivo experiments
All the animal studies were performed according to the guidelines and approval of the Animal Care Committee of the University of Maryland School of Medicine. Male Severe combined immunodeficiency (SCID) mice (4–6weeks old) purchased from the National Cancer Institute-Frederick Cancer Research Center (Frederick, MD) was castrated. PC-3 cells, cultured to sub-confluency were suspended in matrigel (10 mg/ml) at 2.0 × 107 cells/ml. Mice were inoculated with 100 μl of cell suspension in one flank and housed in a pathogen-free environment with a constant room temperature of 24 °C, 12 h dark/light cycles, and free access to food and water. When tumor volumes reached an average size of 90 mm3, mice were put into groups of 10–12 per treatment group. All compounds were formulated in 0.3% hydroxyl propyl cellulose (HPC) in 0.9% saline. Tumors were measured twice weekly and tumor volumes calculated by the formula 4/3π r12 × r2, where r1 is the smaller radius and r2 is the larger radius. Animals were weighed weekly and monitored for general health status and signs of possible toxicity due to treatment. Animal facility reported to researchers any identified animal recommended for sacrifice due to advancement of tumor burden or generalized distress. Mice were euthanized or sacrificed using isoflurane, followed by cervical dislocation.
Immunohistochemical analysis
Specimens were kept in 10% buffered formalin for 24 h and then embedded in paraffin. Sections 4μm thick were mounted on slides for immunostaining. [18]
Statistical analysis
All experiments were repeated at least three times and reported as means with standard error where applicable. Student T-test and Analysis of variance (ANOVA) were used to determine the significance of deviations or lack thereof.
Acknowledgments
This work was supported in part by a grant from NIH and NCI (RO1CA129379), start-up funds from University of Maryland School of Medicine and the Center for Biomolecular Therapeutics (CBT), Baltimore, USA to Professor Vincent C. O. Njar. Andrew K. Kwegyir-Afful was supported in part by University of Maryland School of Medicine Toxicology Program. Dr. Robert D. Bruno was supported in part by National Institute of Environmental Health Safety Training Grant T32 ES007263-16A. The authors thank these agencies for their support.
Abbreviations
- AR
Androgen receptor
- BME
Basement membrane extract
- CI
Combination indices
- EGF
Epidermal growth factor
- EMT
Epithelial-mesenchymal transition
- ERSR
Endoplasmic reticulum stress response
- HPC
hydroxyl propyl cellulose
- VEGF
Vascular endothelial growth factor
Footnotes
Author contributions
This study was designed by VCON, AKKA and RDB. Experiments were performed by AKKA, RDB, PP and FM, while data analyses were performed by VCON, AKKA, RDB, PP and FM. Project supervision was performed by VCON. The manuscript was written by AKKA and VCON with the help of all the authors.
Conflict of interest
Vincent C. O. Njar is the lead inventor of Galeterone (Gal or TOK-001) and VNPT55, patents and technologies thereof owned by the University of Maryland, Baltimore, and licensed to Tokai Pharmaceuticals, Inc. Puranik Purushottamachar and Andrew K. Kwegyir-Afful are co-inventors of VNPT55 and related compounds. A patent application to protect VNPT55 and related novel compounds has been filed. The other authors declare no potential conflict of interest.
References
- 1.Armstrong CM, Gao AC. Drug resistance in castration resistant prostate cancer: resistance mechanisms and emerging treatment strategies. American journal of clinical and experimental urology. 2015;3:64–76. [PMC free article] [PubMed] [Google Scholar]
- 2.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nature reviews Cancer. 2002;2:442–54. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 3.Chua HL, Bhat-Nakshatri P, Clare SE, Morimiya A, Badve S, Nakshatri H. NF-kappaB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2. Oncogene. 2007;26:711–24. doi: 10.1038/sj.onc.1209808. [DOI] [PubMed] [Google Scholar]
- 4.Robichaud N, del Rincon SV, Huor B, Alain T, Petruccelli LA, Hearnden J, Goncalves C, Grotegut S, Spruck CH, Furic L, Larsson O, Muller WJ, Miller WH, Sonenberg N. Phosphorylation of eIF4E promotes EMT and metastasis via translational control of SNAIL and MMP-3. Oncogene. 2015;34:2032–42. doi: 10.1038/onc.2014.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beggs JE, Tian S, Jones GG, Xie J, Iadevaia V, Jenei V, Thomas G, Proud CG. The MAP kinase-interacting kinases regulate cell migration, vimentin expression and eIF4E/CYFIP1 binding. The Biochemical journal. 2015;467:63–76. doi: 10.1042/BJ20141066. [DOI] [PubMed] [Google Scholar]
- 6.Furic L, Rong L, Larsson O, Koumakpayi IH, Yoshida K, Brueschke A, Petroulakis E, Robichaud N, Pollak M, Gaboury LA, Pandolfi PP, Saad F, Sonenberg N. eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:14134–9. doi: 10.1073/pnas.1005320107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haghighat A, Mader S, Pause A, Sonenberg N. Repression of cap-dependent translation by 4E-binding protein 1: competition with p220 for binding to eukaryotic initiation factor-4E. The EMBO journal. 1995;14:5701–9. doi: 10.1002/j.1460-2075.1995.tb00257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hay N. Mnk earmarks eIF4E for cancer therapy. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:13975–6. doi: 10.1073/pnas.1008908107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robichaud N, Del Rincon SV, Huor B, Alain T, Petruccelli LA, Hearnden J, Goncalves C, Grotegut S, Spruck CH, Furic L, Larsson O, Muller WJ, Miller WH, Sonenberg N. Phosphorylation of eIF4E promotes EMT and metastasis via translational control of SNAIL and MMP-3. Oncogene. 2014 doi: 10.1038/onc.2014.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ueda T, Watanabe-Fukunaga R, Fukuyama H, Nagata S, Fukunaga R. Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development. Molecular and cellular biology. 2004;24:6539–49. doi: 10.1128/MCB.24.15.6539-6549.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wendel HG, Silva RL, Malina A, Mills JR, Zhu H, Ueda T, Watanabe-Fukunaga R, Fukunaga R, Teruya-Feldstein J, Pelletier J, Lowe SW. Dissecting eIF4E action in tumorigenesis. Genes & development. 2007;21:3232–7. doi: 10.1101/gad.1604407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ueda T, Sasaki M, Elia AJ, Chio, Hamada K, Fukunaga R, Mak TW. Combined deficiency for MAP kinase-interacting kinase 1 and 2 (Mnk1 and Mnk2) delays tumor development. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:13984–90. doi: 10.1073/pnas.1008136107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nature reviews Cancer. 2007;7:415–28. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 14.Yang MH, Hsu DS, Wang HW, Wang HJ, Lan HY, Yang WH, Huang CH, Kao SY, Tzeng CH, Tai SK, Chang SY, Lee OK, Wu KJ. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nature cell biology. 2010;12:982–92. doi: 10.1038/ncb2099. [DOI] [PubMed] [Google Scholar]
- 15.Hammon M, Herrmann M, Bleiziffer O, Pryymachuk G, Andreoli L, Munoz LE, Amann KU, Mondini M, Gariglio M, Airo P, Schellerer VS, Hatzopoulos AK, Horch RE, Kneser U, Sturzl M, Naschberger E. Role of guanylate binding protein-1 in vascular defects associated with chronic inflammatory diseases. Journal of cellular and molecular medicine. 2011;15:1582–92. doi: 10.1111/j.1582-4934.2010.01146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiss MB, Abel EV, Mayberry MM, Basile KJ, Berger AC, Aplin AE. TWIST1 is an ERK1/2 effector that promotes invasion and regulates MMP-1 expression in human melanoma cells. Cancer research. 2012;72:6382–92. doi: 10.1158/0008-5472.CAN-12-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paranjape AN, Balaji SA, Mandal T, Krushik EV, Nagaraj P, Mukherjee G, Rangarajan A. Bmi1 regulates self-renewal and epithelial to mesenchymal transition in breast cancer cells through Nanog. BMC cancer. 2014;14:785. doi: 10.1186/1471-2407-14-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kwegyir-Afful AK, Ramalingam S, Purushottamachar P, Ramamurthy VP, Njar VC. Galeterone and VNPT55 induce proteasomal degradation of AR/AR-V7, induce significant apoptosis via cytochrome c release and suppress growth of castration resistant prostate cancer xenografts in vivo. Oncotarget. 2015;6:27440–60. doi: 10.18632/oncotarget.4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Montgomery B, Eisenberger MA, Rettig MB, Chu F, Pili R, Stephenson JJ, Vogelzang NJ, Koletsky AJ, Nordquist LT, Edenfield WJ, Mamlouk K, Ferrante KJ, Taplin ME. Androgen Receptor Modulation Optimized for Response (ARMOR) Phase I and II Studies: Galeterone for the Treatment of Castration-Resistant Prostate Cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2016;22:1356–63. doi: 10.1158/1078-0432.CCR-15-1432. [DOI] [PubMed] [Google Scholar]
- 20.Bruno RD, Gover TD, Burger AM, Brodie AM, Njar VC. 17alpha-Hydroxylase/17,20 lyase inhibitor VN/124–1 inhibits growth of androgen-independent prostate cancer cells via induction of the endoplasmic reticulum stress response. Molecular cancer therapeutics. 2008;7:2828–36. doi: 10.1158/1535-7163.MCT-08-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Njar VC, Brodie AM. Discovery and development of Galeterone (TOK-001 or VN/124–1) for the treatment of all stages of prostate cancer. Journal of medicinal chemistry. 2015;58:2077–87. doi: 10.1021/jm501239f. [DOI] [PubMed] [Google Scholar]
- 22.Vasaitis T, Belosay A, Schayowitz A, Khandelwal A, Chopra P, Gediya LK, Guo Z, Fang HB, Njar VC, Brodie AM. Androgen receptor inactivation contributes to antitumor efficacy of 17{alpha}-hydroxylase/17,20-lyase inhibitor 3beta-hydroxy-17-(1H-benzimidazole-1-yl)androsta-5,16-diene in prostate cancer. Molecular cancer therapeutics. 2008;7:2348–57. doi: 10.1158/1535-7163.MCT-08-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosenwald IB, Lazaris-Karatzas A, Sonenberg N, Schmidt EV. Elevated levels of cyclin D1 protein in response to increased expression of eukaryotic initiation factor 4E. Molecular and cellular biology. 1993;13:7358–63. doi: 10.1128/mcb.13.12.7358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosenwald IB, Kaspar R, Rousseau D, Gehrke L, Leboulch P, Chen JJ, Schmidt EV, Sonenberg N, London IM. Eukaryotic translation initiation factor 4E regulates expression of cyclin D1 at transcriptional and post-transcriptional levels. The Journal of biological chemistry. 1995;270:21176–80. doi: 10.1074/jbc.270.36.21176. [DOI] [PubMed] [Google Scholar]
- 25.Konicek BW, Stephens JR, McNulty AM, Robichaud N, Peery RB, Dumstorf CA, Dowless MS, Iversen PW, Parsons S, Ellis KE, McCann DJ, Pelletier J, Furic L, Yingling JM, Stancato LF, Sonenberg N, Graff JR. Therapeutic inhibition of MAP kinase interacting kinase blocks eukaryotic initiation factor 4E phosphorylation and suppresses outgrowth of experimental lung metastases. Cancer research. 2011;71:1849–57. doi: 10.1158/0008-5472.CAN-10-3298. [DOI] [PubMed] [Google Scholar]
- 26.Bianchini A, Loiarro M, Bielli P, Busa R, Paronetto MP, Loreni F, Geremia R, Sette C. Phosphorylation of eIF4E by MNKs supports protein synthesis, cell cycle progression and proliferation in prostate cancer cells. Carcinogenesis. 2008;29:2279–88. doi: 10.1093/carcin/bgn221. [DOI] [PubMed] [Google Scholar]
- 27.Boehm JS, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF, Sjostrom SK, Garraway LA, Weremowicz S, Richardson AL, Greulich H, Stewart CJ, Mulvey LA, Shen RR, Ambrogio L, Hirozane-Kishikawa T, Hill DE, Vidal M, Meyerson M, Grenier JK, Hinkle G, Root DE, Roberts TM, Lander ES, Polyak K, Hahn WC. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007;129:1065–79. doi: 10.1016/j.cell.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 28.Okumura F, Zou W, Zhang DE. ISG15 modification of the eIF4E cognate 4EHP enhances cap structure-binding activity of 4EHP. Genes & development. 2007;21:255–60. doi: 10.1101/gad.1521607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yeh PJ, Hegreness MJ, Aiden AP, Kishony R. Drug interactions and the evolution of antibiotic resistance. Nature reviews Microbiology. 2009;7:460–6. doi: 10.1038/nrmicro2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mamane Y, Petroulakis E, Rong L, Yoshida K, Ler LW, Sonenberg N. eIF4E--from translation to transformation. Oncogene. 2004;23:3172–9. doi: 10.1038/sj.onc.1207549. [DOI] [PubMed] [Google Scholar]
- 31.Avdulov S, Herrera J, Smith K, Peterson M, Gomez-Garcia JR, Beadnell TC, Schwertfeger KL, Benyumov AO, Manivel JC, Li S, Bielinsky AK, Yee D, Bitterman PB, Polunovsky VA. eIF4E threshold levels differ in governing normal and neoplastic expansion of mammary stem and luminal progenitor cells. Cancer research. 2015;75:687–97. doi: 10.1158/0008-5472.CAN-14-2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramalingam S, Gediya L, Kwegyir-Afful AK, Ramamurthy VP, Purushottamachar P, Mbatia H, Njar VC. First MNKs degrading agents block phosphorylation of eIF4E, induce apoptosis, inhibit cell growth, migration and invasion in triple negative and Her2-overexpressing breast cancer cell lines. Oncotarget. 2014;5:530–43. doi: 10.18632/oncotarget.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levinson B, Conant R, Schnur R, Das S, Packman S, Gitschier J. A repeated element in the regulatory region of the MNK gene and its deletion in a patient with occipital horn syndrome. Hum Mol Genet. 1996;5:1737–42. doi: 10.1093/hmg/5.11.1737. [DOI] [PubMed] [Google Scholar]
- 34.Montminy MR, Bilezikjian LM. Binding of a nuclear protein to the cyclic-AMP response element of the somatostatin gene. Nature. 1987;328:175–8. doi: 10.1038/328175a0. [DOI] [PubMed] [Google Scholar]
- 35.Lee W, Mitchell P, Tjian R. Purified transcription factor AP-1 interacts with TPA-inducible enhancer elements. Cell. 1987;49:741–52. doi: 10.1016/0092-8674(87)90612-x. [DOI] [PubMed] [Google Scholar]
- 36.Jones KA, Kadonaga JT, Rosenfeld PJ, Kelly TJ, Tjian R. A cellular DNA-binding protein that activates eukaryotic transcription and DNA replication. Cell. 1987;48:79–89. doi: 10.1016/0092-8674(87)90358-8. [DOI] [PubMed] [Google Scholar]
- 37.De Benedetti A, Graff JR. eIF-4E expression and its role in malignancies and metastases. Oncogene. 2004;23:3189–99. doi: 10.1038/sj.onc.1207545. [DOI] [PubMed] [Google Scholar]
- 38.Graff JR, Konicek BW, Carter JH, Marcusson EG. Targeting the eukaryotic translation initiation factor 4E for cancer therapy. Cancer research. 2008;68:631–4. doi: 10.1158/0008-5472.CAN-07-5635. [DOI] [PubMed] [Google Scholar]
- 39.Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387–92. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 40.Sternlicht MD, Lochter A, Sympson CJ, Huey B, Rougier JP, Gray JW, Pinkel D, Bissell MJ, Werb Z. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell. 1999;98:137–46. doi: 10.1016/s0092-8674(00)81009-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–22. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wells A. EGF receptor. The international journal of biochemistry & cell biology. 1999;31:637–43. doi: 10.1016/s1357-2725(99)00015-1. [DOI] [PubMed] [Google Scholar]
- 43.Wells A, Kassis J, Solava J, Turner T, Lauffenburger DA. Growth factor-induced cell motility in tumor invasion. Acta oncologica. 2002;41:124–30. doi: 10.1080/028418602753669481. [DOI] [PubMed] [Google Scholar]
- 44.Tanaka K, Babic I, Nathanson D, Akhavan D, Guo D, Gini B, Dang J, Zhu S, Yang H, De Jesus J, Amzajerdi AN, Zhang Y, Dibble CC, Dan H, Rinkenbaugh A, Yong WH, Vinters HV, Gera JF, Cavenee WK, Cloughesy TF, Manning BD, Baldwin AS, Mischel PS. Oncogenic EGFR signaling activates an mTORC2-NF-kappaB pathway that promotes chemotherapy resistance. Cancer discovery. 2011;1:524–38. doi: 10.1158/2159-8290.CD-11-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen H, Zhu G, Li Y, Padia RN, Dong Z, Pan ZK, Liu K, Huang S. Extracellular signal-regulated kinase signaling pathway regulates breast cancer cell migration by maintaining slug expression. Cancer research. 2009;69:9228–35. doi: 10.1158/0008-5472.CAN-09-1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nature reviews Immunology. 2005;5:749–59. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 47.Luo JL, Kamata H, Karin M. IKK/NF-kappaB signaling: balancing life and death--a new approach to cancer therapy. The Journal of clinical investigation. 2005;115:2625–32. doi: 10.1172/JCI26322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sahai E, Marshall CJ. RHO-GTPases and cancer. Nature reviews Cancer. 2002;2:133–42. doi: 10.1038/nrc725. [DOI] [PubMed] [Google Scholar]
- 49.Kurokawa K, Matsuda M. Localized RhoA activation as a requirement for the induction of membrane ruffling. Molecular biology of the cell. 2005;16:4294–303. doi: 10.1091/mbc.E04-12-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pertz O, Hodgson L, Klemke RL, Hahn KM. Spatiotemporal dynamics of RhoA activity in migrating cells. Nature. 2006;440:1069–72. doi: 10.1038/nature04665. [DOI] [PubMed] [Google Scholar]
- 51.Reber L, Vermeulen L, Haegeman G, Frossard N. Ser276 phosphorylation of NF-kB p65 by MSK1 controls SCF expression in inflammation. PloS one. 2009;4:e4393. doi: 10.1371/journal.pone.0004393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Richmond A. Nf-kappa B, chemokine gene transcription and tumour growth. Nature reviews Immunology. 2002;2:664–74. doi: 10.1038/nri887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell. 2002;111:185–96. doi: 10.1016/s0092-8674(02)00975-3. [DOI] [PubMed] [Google Scholar]
- 55.Muller J, Hart CM, Francis NJ, Vargas ML, Sengupta A, Wild B, Miller EL, O’Connor MB, Kingston RE, Simon JA. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 2002;111:197–208. doi: 10.1016/s0092-8674(02)00976-5. [DOI] [PubMed] [Google Scholar]
- 56.Min J, Zhang Y, Xu RM. Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes & development. 2003;17:1823–8. doi: 10.1101/gad.269603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lim S, Saw TY, Zhang M, Janes MR, Nacro K, Hill J, Lim AQ, Chang CT, Fruman DA, Rizzieri DA, Tan SY, Fan H, Chuah CT, Ong ST. Targeting of the MNK-eIF4E axis in blast crisis chronic myeloid leukemia inhibits leukemia stem cell function. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E2298–307. doi: 10.1073/pnas.1301838110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kwegyir-Afful AK, Senthilmurugan R, Purushottamachar P, Ramamurthy VP, Njar VC. Galeterone and VNPT55 induce proteasomal degradation of AR/AR-V7, induce significant apoptosis via cytochrome c release and suppress growth of castration resistant prostate cancer xenografts in vivo. Oncotarget. 2015 doi: 10.18632/oncotarget.4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Norgaard M, Jensen AO, Jacobsen JB, Cetin K, Fryzek JP, Sorensen HT. Skeletal related events, bone metastasis and survival of prostate cancer: a population based cohort study in Denmark (1999 to 2007) The Journal of urology. 2010;184:162–7. doi: 10.1016/j.juro.2010.03.034. [DOI] [PubMed] [Google Scholar]
- 60.Sathiakumar N, Delzell E, Morrisey MA, Falkson C, Yong M, Chia V, Blackburn J, Arora T, Kilgore ML. Mortality following bone metastasis and skeletal-related events among men with prostate cancer: a population-based analysis of US Medicare beneficiaries, 1999–2006. Prostate cancer and prostatic diseases. 2011;14:177–83. doi: 10.1038/pcan.2011.7. [DOI] [PubMed] [Google Scholar]
- 61.Li F, Tiede B, Massague J, Kang Y. Beyond tumorigenesis: cancer stem cells in metastasis. Cell research. 2007;17:3–14. doi: 10.1038/sj.cr.7310118. [DOI] [PubMed] [Google Scholar]
- 62.Crea F, Duhagon Serrat MA, Hurt EM, Thomas SB, Danesi R, Farrar WL. BMI1 silencing enhances docetaxel activity and impairs antioxidant response in prostate cancer. International journal of cancer Journal international du cancer. 2011;128:1946–54. doi: 10.1002/ijc.25522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shiota M, Yokomizo A, Takeuchi A, Imada K, Kashiwagi E, Song Y, Inokuchi J, Tatsugami K, Uchiumi T, Naito S. Inhibition of protein kinase C/Twist1 signaling augments anticancer effects of androgen deprivation and enzalutamide in prostate cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:951–61. doi: 10.1158/1078-0432.CCR-13-1809. [DOI] [PubMed] [Google Scholar]
- 64.Graff JR, Konicek BW, Lynch RL, Dumstorf CA, Dowless MS, McNulty AM, Parsons SH, Brail LH, Colligan BM, Koop JW, Hurst BM, Deddens JA, Neubauer BL, Stancato LF, Carter HW, Douglass LE, Carter JH. eIF4E activation is commonly elevated in advanced human prostate cancers and significantly related to reduced patient survival. Cancer research. 2009;69:3866–73. doi: 10.1158/0008-5472.CAN-08-3472. [DOI] [PubMed] [Google Scholar]
- 65.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Developmental cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rao TP, Kuhl M. An updated overview on Wnt signaling pathways: a prelude for more. Circulation research. 2010;106:1798–806. doi: 10.1161/CIRCRESAHA.110.219840. [DOI] [PubMed] [Google Scholar]
- 67.Ceteci F, Ceteci S, Karreman C, Kramer BW, Asan E, Gotz R, Rapp UR. Disruption of tumor cell adhesion promotes angiogenic switch and progression to micrometastasis in RAF-driven murine lung cancer. Cancer cell. 2007;12:145–59. doi: 10.1016/j.ccr.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 68.Hiraga T, Ito S, Nakamura H. Cancer stem-like cell marker CD44 promotes bone metastases by enhancing tumorigenicity, cell motility, and hyaluronan production. Cancer research. 2013;73:4112–22. doi: 10.1158/0008-5472.CAN-12-3801. [DOI] [PubMed] [Google Scholar]
- 69.Gvozdenovic A, Arlt MJ, Campanile C, Brennecke P, Husmann K, Li Y, Born W, Muff R, Fuchs B. CD44 enhances tumor formation and lung metastasis in experimental osteosarcoma and is an additional predictor for poor patient outcome. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2013;28:838–47. doi: 10.1002/jbmr.1817. [DOI] [PubMed] [Google Scholar]
- 70.Purushottamachar P, Godbole AM, Gediya LK, Martin MS, Vasaitis TS, Kwegyir-Afful AK, Ramalingam S, Ates-Alagoz Z, Njar VC. Systematic structure modifications of multitarget prostate cancer drug candidate galeterone to produce novel androgen receptor down-regulating agents as an approach to treatment of advanced prostate cancer. Journal of medicinal chemistry. 2013;56:4880–98. doi: 10.1021/jm400048v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schech AJ, Kazi AA, Gilani RA, Brodie AH. Zoledronic acid reverses the epithelial-mesenchymal transition and inhibits self-renewal of breast cancer cells through inactivation of NF-kappaB. Molecular cancer therapeutics. 2013;12:1356–66. doi: 10.1158/1535-7163.MCT-12-0304. [DOI] [PMC free article] [PubMed] [Google Scholar]