

CONSPECTUS
RNA polymerase II (Pol II) is an essential enzyme that catalyzes transcription with high efficiency and fidelity in eukaryotic cells. During transcription elongation, Pol II catalyzes the nucleotide addition cycle (NAC) to synthesize messenger RNA using DNA as the template. The transitions between the states of the NAC require conformational changes of both the protein and nucleotides. Although X-ray structures are available for most of these states, the dynamics of the transitions between states are largely unknown. Molecular dynamics (MD) simulations can predict structure-based molecular details and shed light on the mechanisms of these dynamic transitions. However, the employment of MD simulations on a macromolecule (tens to hundreds of nanoseconds) such as Pol II is challenging due to the difficulty of reaching biologically relevant timescales (tens of microseconds or even longer). To overcome this challenge, kinetic network models (KNMs) such as Markov State Models (MSMs) have become a popular approach to assess long-timescale conformational changes using many short MD simulations.
We describe here our application of KNMs to characterize the molecular mechanisms of the NAC of Pol II. First, we introduce the general background of MSMs and further explain the procedures for the construction and validation of MSMs by providing some technical details. Next, we give an outline of our previous studies in which we applied MSMs to investigate the individual steps of the NAC, including translocation and pyrophosphate ion release. We make a summary of the major findings for each of these MSM applications. Furthermore, we describe in detail how to build the structural models, the procedures to generate conformations for seeding MD simulations and the parameters used to construct MSMs for each of the application we present. Finally, in order to study the overall NAC, we combine the individual steps of the NAC into a five-state KNM based on a non-branched Brownian ratchet scheme to explain the single-molecule optical tweezers experimental data. In the description of the KNM application, we explicitly write out the underlying assumptions of the five-state KNM and discuss the open questions and future studies that can help us refine the KNMs. The studies we discuss in the review complement experimental observations and provide molecular mechanisms for the transcription elongation cycle. In the long term, incorporation of sequence-dependent kinetic parameters into KNMs has a great potential for identifying error-prone sequences and predicting transcription dynamics in genome-wide transcriptomes.
Graphical abstract
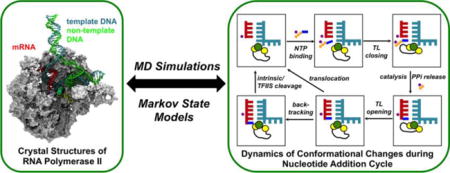
1. INTRODUCTION
RNA polymerase II (Pol II) is the eukaryotic enzyme that is responsible for transcribing the genetic information encoded in DNA into messenger RNA (mRNA). With the assistance of various transcription factors, Pol II can catalyze gene transcription efficiently and accurately1–3. In the stable Pol II elongation complex (EC), the incoming double-stranded DNA (dsDNA) unwinds in the downstream region, and the template DNA strand enters the active site to form a hybrid with the nascent mRNA; the non-template DNA strand makes a ~90° turn near the active site, and the unwound DNA region forms the transcription bubble. The non-template DNA strand re-anneals with the template DNA strand to form the exiting dsDNA in the upstream region1,2 (Figure 1a).
Figure 1. Structure of the Pol II elongation complex and the NAC.
(a) Left: cut-view of Pol II EC. Right: close-up of the active site in the post-translocation state. (b) Schematic representation of the six states of the Pol II NAC.
During transcription elongation, the Pol II EC catalyzes the nucleotide addition cycle (NAC) to add nucleoside triphosphate (NTP) to the growing mRNA strand1,4 (Figure 1b). In general, the NAC can be described by six states, most of which have been captured by X-ray crystallographic studies5–12. Initially, Pol II is in the post-translocation state, which is characterized by an empty active site and open trigger loop (TL) (State I)5. The substrate NTP diffuses into the enzyme and binds in a non-catalytic preinsertion state (State II)6,7,13. If the incoming NTP matches the template DNA, the TL closes beneath the active site (State III)8. Next, Pol II catalyzes RNA incorporation, and the pyrophosphate ion (PPi) exits the enzyme (State IV). The TL then opens, and the EC enters the pre-translocation state (State V)9. Finally, to begin another round of the NAC, the EC translocates by one base pair from the pre- to post-translocation state to free the active site1,2. During elongation, RNA misincorporation occasionally occurs, and to maintain fidelity, Pol II backtracks (State VI) to remove the misincorporated nucleotide either by intrinsic cleavage or with the aid of transcription factor IIS (TFIIS)10–12. Although X-ray crystallographic studies are essential to understanding the structural basis of transcription, crystal structures are static and thus do not reveal the underlying dynamics.
Molecular dynamics (MD) simulation is an essential tool for modeling the dynamics of biomolecules by considering actual atomic interactions. Therefore, MD has great potential to elucidate the molecular mechanism of Pol II transcription14–17. For example, we performed MD simulations that provided molecular insight into the role of TL in nucleotide selection by identifying the atomic interactions between NTP and the closed TL14. Moreover, we analyzed the MD conformations of Pol II with a complete transcription bubble and predicted that the secondary channel is the major route for NTP diffusion into the enzyme15.
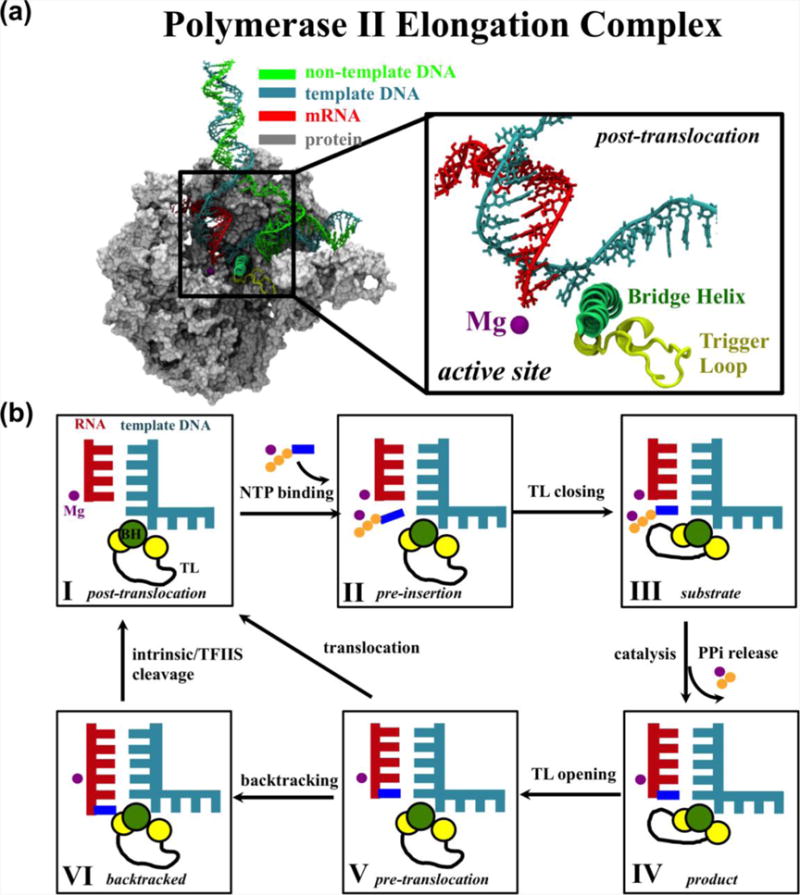
Although MD simulations have great capacity for elucidating structure-based molecular details, one major challenge to their application to large systems, such as Pol II, is achieving biologically relevant timescales (Figure 2a). MD simulations for Pol II are usually limited to tens to hundreds of nanoseconds; however, the transition between states in the NAC occurs on the order of tens of microseconds or even longer. Thus, there exists a timescale gap between MD simulations and essential conformational changes. The Markov State Model (MSM), a type of kinetic network model (KNM), has recently become a popular approach to bridge the timescale gap18–31. Our group has successfully applied MSM to study various biological systems32–41.
Figure 2. General background of MSMs.
(a) Timescale gap between atomistic MD simulations and experiments. (b) Sampling of the free energy landscape by conventional MD simulations initiated from different conformations, with each individual simulation labeled by a line (figure adapted with permission from ref.24. Copyright (2009) National Academy of Sciences, USA.). (c) “Splitting and lumping” algorithm to construct MSMs (figure adapted with permission from ref.28. Copyright (2009) Elsevier.). (d) Implied timescale plot (figure adapted with permission from ref.38. Copyright (2015) Jiang et al.). (e) Validation of MSMs by testing the residence probability (figure adapted with permission from ref.38. Copyright (2015) Jiang et al.).
Here, we describe our application of KNMs to characterize the molecular mechanism underlying Pol II transcription elongation. First, we introduce the basic concepts underlying MSMs, followed by an illustration of the construction and validation of MSMs. Then, we review our previous work on the elucidation of the individual steps of the NAC as well as the overall NAC transcription dynamics39–42. Finally, we provide a discussion and future perspectives.
2. SIMULATING LONG-TIMESCALE DYNAMICS OF BIOMOLECULES
2.1. Markov State Model Theory
The construction of MSMs using automatic algorithms24–28,37,43 can facilitate the examination of long-timescale dynamic processes via a number of short MD simulations that reach local equilibrium (Figure 2b)18–31. The basic concept of MSM is to partition the conformational space into a number of metastable states, and fast motions are integrated out by discretizing time in units of τ (lag time) (Figures 2b–c). If τ is long enough to allow full relaxation within each state, the model becomes Markovian, i.e., the probability for the system to visit a given state at time t+τ depends only on its current position at t. Under this condition, the long-timescale dynamics can be obtained by propagating the transition probability matrix,
| (1) |
where is the state population vector at time , and , the element of , denotes the transition probability from state i to state j after a lag time of τ. Specifically, can be generated by counting transitions between pairs of states at τ from many short MD simulations.
2.2. Construction and validation of Markov State Models
To construct MSMs, we first divide MD conformations into a large number of clusters or microstates according to their structural similarity (Figure 2c). This step is often performed by geometric clustering algorithms26,27,44 such as K-centers and K-means. Root mean square deviation (RMSD) between pairs of conformations is a popular choice of the distance metric for clustering. In practice, one may need some physical understanding of the system in order to choose an appropriate set of atoms to be included in the RMSD calculations. More recently, new algorithms29–31 such as the time-structure based Independent Component Analysis (tICA)31, have been developed to identify a set of key tICs that can sufficiently describe slowest dynamics of the system, and subsequently distances between pairs of conformations can also be computed in the reduced dimensional space containing these tICs. The tICA method provides a promising approach to automatically choose metrics that can best describe the conformational dynamics of interest. When constructing MSMs based on microstates, one assumes that conformations within each microstate are structurally similar and thus also kinetically similar. The interplay between length of lag time (τ) and number of states is critical to achieve a Markovian model, because shorter lag times always require more states to ensure that the system can lose memory within each state. To build Markovian models for biomolecules, one often needs thousands of microstates, because the lag time is limited by the length of individual short MD simulations44. However, when the number of states is too big, statistical errors may become dominant. Under this condition, many states only contain one or few conformations each, and this may result in substantial uncertainties in the estimated transition probabilities between states. Nevertheless, microstate-MSMs are particularly useful for quantitative comparisons with experiments44.
As microstate-MSMs containing thousands of states are usually too complicated for gaining mechanistic insights, microstates are often coarse grained into fewer macrostates based on their kinetic proximity (Figure 2c). Several algorithms such as Hierarchical Nyström method25 developed in our group, Perron-cluster cluster analysis (PCCA)45 and its improved version (PCCA+)46, Bayesian agglomerative clustering engine47, and the most probable paths algorithm48 can perform this task. Number of macrostates can be determined by searching for the leading and stable gap in the implied timescales, while physical insights sometimes may also help for complicated biological systems. Macrostate-models (containing a few states) are often non-Markovian and thus are not suitable for computing any quantitative properties. In this regard, all reported quantitative properties of macrostates should be computed based on the validated microstate-MSMs39,49. Even so, the applications of macrostates can still greatly help the visualization of conformational dynamics, and facilitate the interpretation of molecular mechanisms of biological processes.
To choose an appropriate lag time that can render the model Markovian, one often examines the implied timescale , which can be calculated as
| (2) |
where is the kth eigenvalue of the transition matrix at lag time . Each implied timescale describes an aggregate transition between two subsets of states. When the model is Markovian, its predicted implied timescales should become invariant with the lag time 50.
MSMs can be validated by the Chapman-Kolmogorov test20, which examines if time evolutions of state populations predicted by an MSM (Equation (1)) are consistent with those directly obtained from MD simulations. A derivation of this test, where one examines probabilities for the system to remain in a certain state rather than state populations, is also widely adopted in validating MSMs20 (Figure 2e).
2.3 Computing Thermodynamic and Kinetic Properties from Markov State Models
Useful quantities could be acquired from validated MSMs such as equilibrium populations of states, ensemble averages of particular observables, mean first passage times (MFPTs) between pairs of states, and dominant transition pathways from the initial to the final state (obtained from the transition path theory51).
3. APPLICATIONS
The characterization of the dynamic transition between functional states is essential to understanding Pol II transcription elongation. We have used MSMs to study the individual steps in the NAC (Figure 1b): translocation39 and PPi release40. Furthermore, we have combined the individual steps to construct a five-state KNM to study the overall NAC42. In this section, we present these results as examples of the application of KNMs to predict the dynamics of Pol II transcription elongation.
3.1 Dynamics of translocation
Pol II translocation describes the reversible dynamic process by which Pol II moves by one register from the pre- to the post-translocation state to free the active site or the reverse process (Figure 1b). Translocation is also a necessary step to establish Pol II at a new stage to allow the next round of the NAC. We used MSMs based on MD simulations to investigate the underlying molecular mechanism of Pol II translocation39. In general, the result supports a Brownian ratchet model: Pol II can oscillate between pre- and post-translocation state via a Brownian motion driven by thermal energy. On one hand, NTP binding and incorporation can act as the pawl in this Brownian ratchet that biases Pol II movement to favor the forward (pre- to post-translocation) direction. On the other hand, RNA transcript hydrolysis and pyrophosphorolysis may work as the pawl to bias movement of Pol II in the reverse direction. Moreover, two intermediates were identified, and the bridge helix (BH) was shown to interact with nucleotides to facilitate the process.
We built the structural models of Pol II in the pre- and post-translocation states based on available crystal structures (see reference39 for details). Initial pathways connecting pre- and post-translocation states were generated using the modified Climber algorithm39,52. We note that the string method53 is another method of providing good initial pathways. Representative conformations were selected from these initial pathways to perform two rounds of unbiased MD simulations at 310K using the amber03 force field54. The aggregated simulation time reaches ~2,500ns, and the second round of MD simulations (1,600ns in total) were used to build MSMs. In particular, we split MD conformations into microstates by performing two independent k-center clusterings using RMSD as the distance metric. As the motion of transition nucleotide (TN) is directly related to translocation, the first clustering was performed based on the RMSD of TN after the alignment of the whole Pol II. To capture how Pol II responds to the translocation of RNA/DNA, we also performed a second clustering based on the RMSDs of nucleotides as well as BH and TL. We then combined these sets of clusters, and removed the empty ones to obtain the final model containing 976 microstates. For visualizing the translocation mechanisms, we further lumped these microstates into four metastable states.
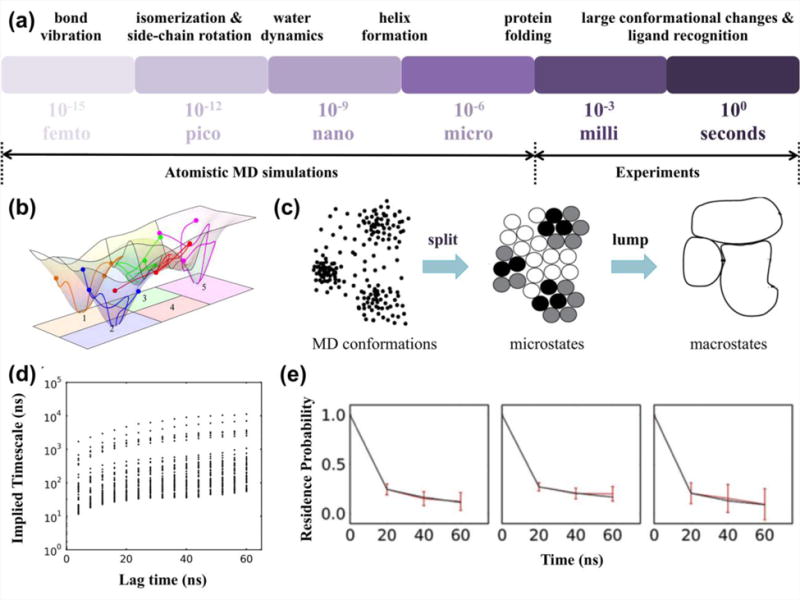
Our MSMs revealed that Pol II oscillates between the pre- (S1) and post-translocation (S4) states, through two intermediate states (S2 and S3) (Figure 3a). MFPT calculations demonstrated that the transition between S1 and S2 is the rate-limiting step (~20 μs) and states S2~S4 could form a wide flat energy basin (Figure 3). We also found pre- and post-translocation states coexist and both of them are populated, which is consistent with experimental observations55,56.
Figure 3. Pol II translocation elucidated by MSMs.
(a) Four metastable states identified by MSMs. The TN (orange), Y836 (purple) and T831 (grey) are shown. (b) Schematic free energy profile of translocation. The transition between S1 and S2 has the highest energy barrier (rate-limiting step, as shown in (a)). The cartoon representations of the states indicate that the translocation of the RNA:DNA hybrid (red and blue) is asynchronous with the translocation of the TN (orange). The population of each state is also displayed.
Structural analysis of the four metastable states provided the molecular details of Pol II translocation. In the intermediate state S2, the backbone of the RNA:DNA hybrid has reached its final position, as in the post-translocation (S4) state. However, the TN lags behind and forms key stacking interactions with the BH residue Y836. The π-stacking interaction between the TN and Y836 can lower the energy barrier of translocation. In the intermediate state S3, the TN crosses over the BH and approaches its final canonical i+1 position. These intermediates are consistent with the X-ray crystal structures of intermediates trapped by α-amanitin or DNA damage57–60.
We also observed that the central region of the BH is very flexible and it can bend as much as 10Å toward the active site to facilitate translocation (Figures 2a–b in reference39). For the transition from S1 to S2, BH bending aids the translocation of the RNA:DNA hybrid. In S2 and S3, the BH residues Y836 and T831 facilitate TN translocation. Our MSM suggests that Pol II can oscillate between the pre- and post-translocation conformations until the incoming NTP stabilizes the post-translocation state. These results provide atomic details about translocation and support a Brownian ratchet mechanism.
3.2 Dynamics of PPi release
PPi is the byproduct of NTP incorporation, and the release of PPi is a prerequisite for the continuation of the NAC (Figures 1b and 4a). We constructed MSMs based on MD simulations, suggesting that PPi release along the secondary channel follows a four-state hopping model in Pol II (Figures 4b–c)40. This PPi release process is further coupled with the TL tip motion.
Figure 4. Pol II PPi release elucidated by MSMs.
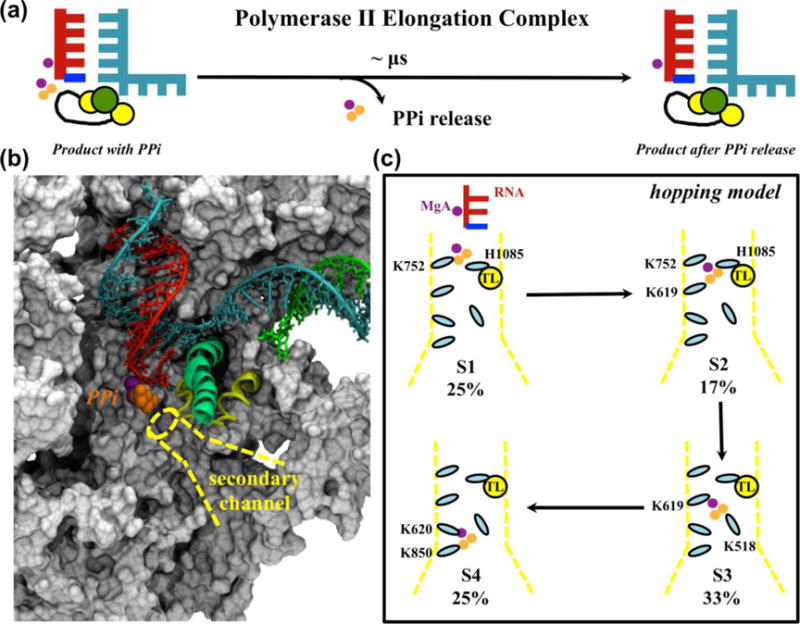
(a) Schematic representation of the PPi release process. (b) Pol II structural model used to study PPi (orange spheres) release along the secondary channel (yellow dotted lines). (c) PPi release follows a four-state hopping model. The residues interacting with PPi at each hopping site are labeled. The population of each metastable state is shown as a percentage.
The simulation model for Pol II in complex with PPi was built based on the crystal structures of Pol II bound to NTP (see reference40 for details) (Figure 4b). Initial pathways for PPi release were first generated from steered MD simulations. Afterwards, representative conformations along the pathways were selected to seed unbiased MD simulations at 310K and 1bar using amber03 force field54 with regenerating the partial charges of (Mg-PPi)2−. MD simulations with an aggregated time of >700ns were used to build a 274 microstate MSM via a k-center clustering based on the RMSD of the PPi after the structural alignment of BH. To facilitate the comprehension of the release mechanism, we further applied the PCCA+ algorithm46 to lump these microstates into 4 macrostates.
Our MSMs demonstrate that PPi adopts a hopping model to jump from the active site to the funnel. Within each hopping site, several positively charged amino acids form stable interactions with PPi and assist its release (Figure 4c). Interestingly, four of the positively charged residues are conserved among eukaryotes, and two are conserved between both eukaryotes and prokaryotes (Figure 3c in reference40), indicating the biological significance of these residues during evolution. This study also demonstrated the correlation between TL motion and PPi release. The dynamics of the TL tip help PPi exit from the active site via the interaction between TL and PPi; once PPi is outside the active site, its release can assist the initial opening of TL by inducing conformational changes of TL tip.
In addition to Pol II, we have also studied PPi release in bacterial RNA polymerase41, and a simpler two-state model was elucidated. The different number of states between yeast and bacteria are due to differences in protein surface layout: positively charged residues are located separately in yeast but are continuously distributed in bacteria. Also different from Pol II, we found the PPi release in a bacterial RNA polymerase is only coupled with the side chain rotation of a TL residue R1239, but cannot induce any substantial conformational changes of the TL backbone. In this regard, the application of MSMs has provided deeper insights into the structural features that influence the dynamics of PPi release in different systems41.
3.3 Five-state KNM of the overall Pol II NAC
In addition to focusing on a single step of the NAC, we have recently built a KNM that describes the overall NAC by combining all the transitions among states I~V42 (Figure 5a). After fitting to the single-molecule experiments61, our KNM provides explanations to two slow steps during Pol II elongation, as identified by the single-molecule experiment61 (Figure 5a).
Figure 5. Elucidation of the overall dynamics of the NAC by KNM.
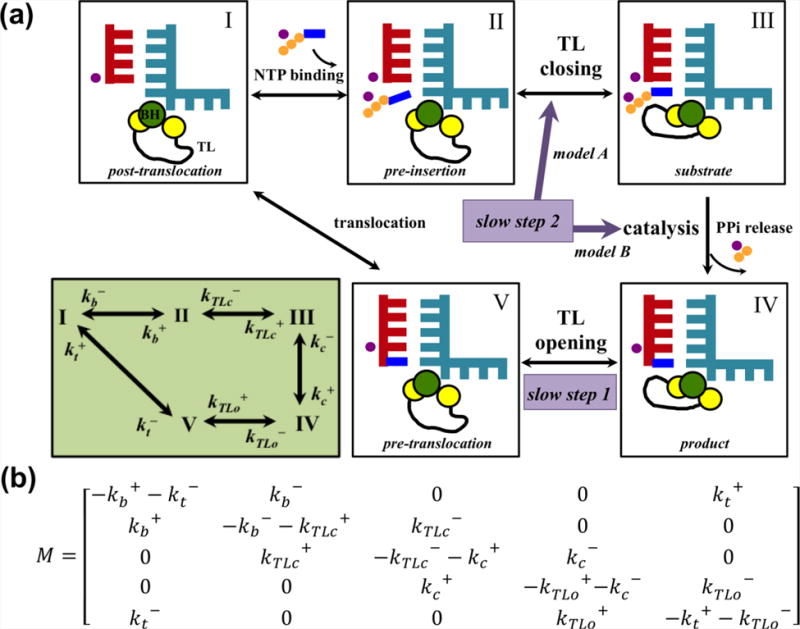
(a) A five-state KNM provides explanations to the two slow steps in transcriptional elongation: slow step 1 is assigned to the TL opening, whereas two scenarios are possible for the slow step 2: TL closing (model A: II to III) or pre-catalytic conformational rearrangement (model B: III to IV). The lower left panel shows the transition rates between states. The state definition here is consistent with that in Figure 1, but the backtracking state is not included in this model. (b) The transition rate matrix for the five-state KNM.
In this work, we included five states in the KNM and assigned individual rates for each of the forward and backward transitions (Figure 5a). Based on the five-state scheme with the population vector defined as Π=(PIPIIPIIIPIVPV)T, we formulated the following master equation:
| (3) |
with the transition rate matrix M shown in Figure 5b. By solving Equation (3) at the steady state under42, we derived the elongation rate. Fitting the elongation rate to single-molecule optical tweezers experimental data in the pause-free region61 yielded the transition rates. These results suggested that one slow step corresponds to the TL opening prior to translocation, while the other slow step may be explained by two possible scenarios: TL closing upon NTP binding or a pre-catalytic conformational adjustment of the active site (Figure 5a). Accurate determination of the intrinsic properties of NTP binding could improve the understanding of elongation dynamics.
This work provides a critical link between the overall transcription elongation process and the individual steps of the NAC, thus enabling investigations of how the local structural and dynamic perturbations that occur in a single transition affect the overall elongation kinetics.
4. DISCUSSION AND PERSPECTIVES
We will conduct further studies of other critical conformational changes during the NAC, such as the TL motions and backtracking. TL undergoes conformational changes and plays important roles in Pol II transcription elongation8,14 (Figure 1b). We aim to construct MSMs to elucidate the mechanisms of TL motions. Also of interest is backtracking, which is a crucial process for maintaining the high accuracy of transcription by Pol II11. There are two hypotheses for backtracking: the concerted model (RNA backtracking is concurrent with the movement of DNA)5 and the stepwise model (RNA fraying occurs prior to the DNA movement)12. We will apply MSMs to decipher the backtracking mechanism and reveal how Pol II can detect mismatched nucleotides to promote backtracking. Moreover, the effects of DNA damage or backbone heterogeneity on Pol II transcription in molecular detail will be of interest59,60,62–65. In addition, it will be interesting to investigate the thermodynamics and kinetics of NTP loading, and how NTP loading couples with translocation. Our previous work on translocation was conducted using a minimum scaffold of the Pol II elongation complex39. Recently, the crystal structure with a full transcription bubble was resolved66,67. This structure could provide a structural basis for investigating Pol II translocation and backtracking by considering the effect of the base pairs that break and reform at the two edges of the transcription bubble. Finally, we also plan to incorporate the backtracking state in our KNM so that it can take into account the proofreading process and its impact on transcriptional dynamics.
Recent studies have demonstrated that transcription errors do not occur at random sites but occur preferentially in specific sequence motifs68,69. Therefore, in the long term, a KNM containing DNA sequence dependent transition rates could be applied to pinpoint hot-spot sequence motifs of Pol II transcription, thus providing a mechanistic link between the structural-mechanics of Pol II fidelity and error-prone sequences of the transcriptome. These studies could open up a new perspective in understanding human diseases and aging problems related to transcription fidelity.
Acknowledgments
This work was supported by the Hong Kong Research Grant Council [grant numbers HKUST C6009-15G, 16302214, 609813, AoE/M-09/12, M-HKUST601/13, and T13-607/12R to X.H.]; the National Science Foundation of China [grant number 21273188 to X.H.]; the National Institutes of Health [grant number GM102362 to D.W.]; the Hong Kong PhD Fellowship Scheme [grant number PF10-17123 to F.P.A.] and the Consejo Nacional de Ciencia y Technología Fellowship [grant number 215482 to F.P.A.].
Biographies
Lu Zhang obtained her Ph.D. in Chemistry at Hong Kong University of Science and Technology (HKUST) in 2013. She is now a research associate at HKUST. Her research interests include RNA polymerase transcription and energy transfer in photosynthesis.
Fátima Pardo-Avila earned her Ph.D. in Chemistry at HKUST in 2015. Her research focuses on elucidating the atomic mechanism of enzymes involved in gene expression.
Ilona Christy Unarta graduated from HKUST in 2013 with a B.S. in Chemistry. She is now a second-year M.Phil. student in Bioengineering at HKUST. Her research interest is describing biological mechanisms at the molecular level through computational methods.
Peter Pak-Hang Cheung completed his Ph.D. in Molecular Virology at the University of Hong Kong in 2012. He is now a postdoctoral fellow at HKUST. His research interest involves the use of molecular biology, biophysics, and bioinformatics approaches to understand eukaryotic and viral polymerases.
Guo Wang received her B.S. from Sun Yat-sen University in 2013. She is now a second-year M.Phil. student in Chemistry at HKUST. Her research interest is mechanistic studies of biological systems using computational methods.
Dong Wang is an Associate Professor at the University of California, San Diego. He obtained his Ph.D. with Prof. Lippard at MIT in 2004. He then joined Prof. Kornberg’s group at Stanford University as a postdoctoral fellow (2004–2009). His research focuses on understanding the mechanisms of transcription and epigenetic regulation, chromatin remodeling, and DNA damage and repair.
Xuhui Huang is an Associate Professor at HKUST. He earned his Ph.D. with Prof. Bruce J. Berne at Columbia University in 2006. He subsequently worked with Profs. Michael Levitt and Vijay S. Pande as a postdoctoral fellow (2006–2008) and then as a research associate (2008–2009) at Stanford University. His research focuses on understanding the functional conformational changes of complex biological systems by developing and applying novel computational tools that bridge the gap between experiments and simulations.
Footnotes
Notes
The authors declare no competing financial interests.
References
- 1.Kornberg RD. The molecular basis of eukaryotic transcription. Proc Natl Acad Sci U S A. 2007;104:12955–12961. doi: 10.1073/pnas.0704138104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheung ACM, Cramer P. A Movie of RNA Polymerase II Transcription. Cell. 2012;149:1431–1437. doi: 10.1016/j.cell.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Klein BJ, Bose D, Baker KJ, Yusoff ZM, Zhang XD, Murakami KS. RNA polymerase and transcription elongation factor Spt4/5 complex structure. Proc Natl Acad Sci U S A. 2011;108:546–550. doi: 10.1073/pnas.1013828108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brueckner F, Ortiz J, Cramer P. A movie of the RNA polymerase nucleotide addition cycle. Curr Opin Struct Biol. 2009;19:294–299. doi: 10.1016/j.sbi.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 5.Westover KD, Bushnell Da, Kornberg RD. Structural basis of transcription: Separation of RNA from DNA by RNA polymerase II. Science. 2004;303:1014–1016. doi: 10.1126/science.1090839. [DOI] [PubMed] [Google Scholar]
- 6.Westover KD, Bushnell Da, Kornberg RD. Structural basis of transcription: nucleotide selection by rotation in the RNA polymerase II active center. Cell. 2004;119:481–489. doi: 10.1016/j.cell.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 7.Kettenberger H, Armache KJ, Cramer P. Complete RNA polymerase II elongation complex structure and its interactions with NTP and TFIIS. Mol Cell. 2004;16:955–965. doi: 10.1016/j.molcel.2004.11.040. [DOI] [PubMed] [Google Scholar]
- 8.Wang D, Bushnell Da, Westover KD, Kaplan CD, Kornberg RD. Structural basis of transcription: role of the trigger loop in substrate specificity and catalysis. Cell. 2006;127:941–954. doi: 10.1016/j.cell.2006.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gnatt AL, Cramer P, Fu JH, Bushnell DA, Kornberg RD. Structural basis of transcription: An RNA polymerase II elongation complex at 3.3 angstrom resolution. Science. 2001;292:1876–1882. doi: 10.1126/science.1059495. [DOI] [PubMed] [Google Scholar]
- 10.Cheung ACM, Cramer P. Structural basis of RNA polymerase II backtracking, arrest and reactivation. Nature. 2011;471:249–253. doi: 10.1038/nature09785. [DOI] [PubMed] [Google Scholar]
- 11.Wang D, Bushnell Da, Huang X, Westover KD, Levitt M, Kornberg RD. Structural basis of transcription: backtracked RNA polymerase II at 3.4 angstrom resolution. Science. 2009;324:1203–1206. doi: 10.1126/science.1168729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sydow JF, Brueckner F, Cheung ACM, Damsma GE, Dengl S, Lehmann E, Vassylyev D, Cramer P. Structural basis of transcription: mismatch-specific fidelity mechanisms and paused RNA polymerase II with frayed RNA. Mol Cell. 2009;34:710–721. doi: 10.1016/j.molcel.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 13.Landick R. NTP-entry routes in multi-subunit RNA polymerases. Trends Biochem Sci. 2005;30:651–654. doi: 10.1016/j.tibs.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 14.Huang X, Wang D, Weiss DR, Bushnell Da, Kornberg RD, Levitt M. RNA polymerase II trigger loop residues stabilize and position the incoming nucleotide triphosphate in transcription. Proc Natl Acad Sci U S A. 2010;107:15745–15750. doi: 10.1073/pnas.1009898107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang L, Silva D-A, Pardo-Avila F, Wang D, Huang X. Structural Model of RNA Polymerase II Elongation Complex with Complete Transcription Bubble Reveals NTP Entry Routes. PLoS Comput Biol. 2015;11:e1004354. doi: 10.1371/journal.pcbi.1004354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang B, Feig M, Cukier RI, Burton ZF. Computational simulation strategies for analysis of multisubunit RNA polymerases. Chem Rev. 2013;113:8546–8566. doi: 10.1021/cr400046x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang B, Opron K, Burton ZF, Cukier RI, Feig M. Five checkpoints maintaining the fidelity of transcription by RNA polymerases in structural and energetic details. Nucleic Acids Res. 2014;43:1133–1146. doi: 10.1093/nar/gku1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bowman GR, Voelz VA, Pande VS. Taming the complexity of protein folding. Curr Opin Struct Biol. 2011;21:4–11. doi: 10.1016/j.sbi.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chodera JD, Noe F. Markov state models of biomolecular conformational dynamics. Curr Opin Struct Biol. 2014;25:135–144. doi: 10.1016/j.sbi.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prinz JH, Wu H, Sarich M, Keller B, Senne M, Held M, Chodera JD, Schutte C, Noe F. Markov models of molecular kinetics: Generation and validation. J Chem Phys. 2011;134:174105. doi: 10.1063/1.3565032. [DOI] [PubMed] [Google Scholar]
- 21.Buchete NV, Hummer G. Coarse master equations for peptide folding dynamics. J Phys Chem B. 2008;112:6057–6069. doi: 10.1021/jp0761665. [DOI] [PubMed] [Google Scholar]
- 22.Pan AC, Roux B. Building Markov state models along pathways to determine free energies and rates of transitions. J Chem Phys. 2008;129:064107. doi: 10.1063/1.2959573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malmstrom RD, Lee CT, Van Wart AT, Amaro RE. Application of Molecular-Dynamics Based Markov State Models to Functional Proteins. J Chem Theory Comput. 2014;10:2648–2657. doi: 10.1021/ct5002363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang X, Bowman GR, Bacallado S, Pande VS. Rapid equilibrium sampling initiated from nonequilibrium data. Proc Natl Acad Sci U S A. 2009;106:19765–19769. doi: 10.1073/pnas.0909088106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yao Y, Cui RZ, Bowman GR, Silva D-A, Sun J, Huang X. Hierarchical Nystrom methods for constructing Markov state models for conformational dynamics. J Chem Phys. 2013;138:174106. doi: 10.1063/1.4802007. [DOI] [PubMed] [Google Scholar]
- 26.Sheong FK, Silva D-A, Meng L, Zhao Y, Huang X. Automatic State Partitioning for Multibody Systems (APM): An Efficient Algorithm for Constructing Markov State Models To Elucidate Conformational Dynamics of Multibody Systems. J Chem Theory Comput. 2015;11:17–27. doi: 10.1021/ct5007168. [DOI] [PubMed] [Google Scholar]
- 27.Zhao Y, Sheong FK, Sun J, Sander P, Huang X. A fast parallel clustering algorithm for molecular simulation trajectories. J Comput Chem. 2013;34:95–104. doi: 10.1002/jcc.23110. [DOI] [PubMed] [Google Scholar]
- 28.Bowman GR, Huang X, Pande VS. Using generalized ensemble simulations and Markov state models to identify conformational states. Methods. 2009;49:197–201. doi: 10.1016/j.ymeth.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nuske F, Keller BG, Perez-Hernandez G, Mey ASJS, Noe F. Variational Approach to Molecular Kinetics. J Chem Theory Comput. 2014;10:1739–1752. doi: 10.1021/ct4009156. [DOI] [PubMed] [Google Scholar]
- 30.Perez-Hernandez G, Paul F, Giorgino T, De Fabritiis G, Noe F. Identification of slow molecular order parameters for Markov model construction. J Chem Phys. 2013;139:015102. doi: 10.1063/1.4811489. [DOI] [PubMed] [Google Scholar]
- 31.Schwantes CR, Pande VS. Improvements in Markov State Model Construction Reveal Many Non-Native Interactions in the Folding of NTL9. J Chem Theory Comput. 2013;9:2000–2009. doi: 10.1021/ct300878a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cui RZ, Silva D-A, Song J, Bowman GR, Zhuang W, Huang X. Bridging the Gap Between Optical Spectroscopic Experiments and Computer Simulations for Fast Protein Folding Dynamics. Curr Phys Chem. 2012;2:45–58. [Google Scholar]
- 33.Gu S, Silva DA, Meng LM, Yue A, Huang XH. Quantitatively Characterizing the Ligand Binding Mechanisms of Choline Binding Protein Using Markov State Model Analysis. PLoS Comput Biol. 2014;10:e1003767. doi: 10.1371/journal.pcbi.1003767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qiao Q, Bowman GR, Huang X. Dynamics of an Intrinsically Disordered Protein Reveal Metastable Conformations That Potentially Seed Aggregation. J Am Chem Soc. 2013;135:16092–16101. doi: 10.1021/ja403147m. [DOI] [PubMed] [Google Scholar]
- 35.Silva D-A, Bowman GR, Sosa-Peinado A, Huang X. A Role for Both Conformational Selection and Induced Fit in Ligand Binding by the LAO Protein. PLoS Comput Biol. 2011;7:e1002054. doi: 10.1371/journal.pcbi.1002054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhuang W, Cui RZ, Silva D-A, Huang X. Simulating the T-jump-triggered unfolding dynamics of trpzip2 peptide and its time-resolved IR and two-dimensional IR signals using the Markov state model approach. J Phys Chem B. 2011;115:5415–5424. doi: 10.1021/jp109592b. [DOI] [PubMed] [Google Scholar]
- 37.Huang X, Yao Y, Bowman GR, Sun J, Guibas LJ, Carlsson G, Pande VS. Constructing multi-resolution Markov State Models (MSMs) to elucidate RNA hairpin folding mechanisms. Pac Symp Biocomput. 2010:228–239. doi: 10.1142/9789814295291_0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang H, Sheong FK, Zhu L, Gao X, Bernauer J, Huang X. Markov State Models Reveal a Two-Step Mechanism of miRNA Loading into the Human Argonaute Protein: Selective Binding followed by Structural Re-arrangement. PLoS Comput Biol. 2015;11:e1004404. doi: 10.1371/journal.pcbi.1004404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Silva DA, Weiss DR, Pardo-Avila F, Da LT, Levitt M, Wang D, Huang XH. Millisecond dynamics of RNA polymerase II translocation at atomic resolution. Proc Natl Acad Sci U S A. 2014;111:7665–7670. doi: 10.1073/pnas.1315751111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Da LT, Wang D, Huang X. Dynamics of pyrophosphate ion release and its coupled trigger loop motion from closed to open state in RNA polymerase II. J Am Chem Soc. 2012;134:2399–2406. doi: 10.1021/ja210656k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Da LT, Pardo-Avila F, Wang D, Huang X. A Two-State Model for the Dynamics of the Pyrophosphate Ion Release in Bacterial RNA Polymerase. PLoS Comput Biol. 2013;9:e1003020. doi: 10.1371/journal.pcbi.1003020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu J, Da LT, Huang XH. Constructing kinetic models to elucidate structural dynamics of a complete RNA polymerase II elongation cycle. Phys Biol. 2015;12:016004. doi: 10.1088/1478-3975/12/1/016004. [DOI] [PubMed] [Google Scholar]
- 43.Bowman GR, Meng L, Huang X. Quantitative comparison of alternative methods for coarse-graining biological networks. J Chem Phys. 2013;139:121905. doi: 10.1063/1.4812768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bowman GR, Beauchamp KA, Boxer G, Pande VS. Progress and challenges in the automated construction of Markov state models for full protein systems. J Chem Phys. 2009;131:124101. doi: 10.1063/1.3216567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deuflhard P, Huisinga W, Fischer A, Schutte C. Identification of almost invariant aggregates in reversible nearly uncoupled Markov chains. Linear Algebra Appl. 2000;315:39–59. [Google Scholar]
- 46.Deuflhard P, Weber M. Robust Perron cluster analysis in conformation dynamics. Linear Algebra Appl. 2005;398:161–184. [Google Scholar]
- 47.Bowman GR. Improved coarse-graining of Markov state models via explicit consideration of statistical uncertainty. J Chem Phys. 2012;137:134111. doi: 10.1063/1.4755751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jain A, Stock G. Identifying Metastable States of Folding Proteins. J Chem Theory Comput. 2012;8:3810–3819. doi: 10.1021/ct300077q. [DOI] [PubMed] [Google Scholar]
- 49.Da LT, E C, Duan B, Zhang C, Zhou X, Yu J. A Jump-from-Cavity Pyrophosphate Ion Release Assisted by a Key Lysine Residue in T7 RNA Polymerase Transcription Elongation. PLoS Comput Biol. 2015;11:e1004624. doi: 10.1371/journal.pcbi.1004624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Swope WC, Pitera JW, Suits F. Describing protein folding kinetics by molecular dynamics simulations. 1. Theory. J Phys Chem B. 2004;108:6571–6581. [Google Scholar]
- 51.E WN, Vanden-Eijnden E. Transition-Path Theory and Path-Finding Algorithms for the Study of Rare Events. Annu Rev Phys Chem. 2010;61:391–420. doi: 10.1146/annurev.physchem.040808.090412. [DOI] [PubMed] [Google Scholar]
- 52.Weiss DR, Levitt M. Can Morphing Methods Predict Intermediate Structures? J Mol Biol. 2009;385:665–674. doi: 10.1016/j.jmb.2008.10.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pan AC, Sezer D, Roux B. Finding transition pathways using the string method with swarms of trajectories. J Phys Chem B. 2008;112:3432–3440. doi: 10.1021/jp0777059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong GM, Zhang W, Yang R, Cieplak P, Luo R, Lee T, Caldwell J, Wang JM, Kollman P. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 55.Abbondanzieri Ea, Greenleaf WJ, Shaevitz JW, Landick R, Block SM. Direct observation of base-pair stepping by RNA polymerase. Nature. 2005;438:460–465. doi: 10.1038/nature04268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hein PP, Palangat M, Landick R. RNA Transcript 3′-Proximal Sequence Affects Translocation Bias of RNA Polymerase. Biochemistry. 2011;50:7002–7014. doi: 10.1021/bi200437q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brueckner F, Cramer P. Structural basis of transcription inhibition by alpha-amanitin and implications for RNA polymerase II translocation. Nat Struct Mol Biol. 2008;15:811–818. doi: 10.1038/nsmb.1458. [DOI] [PubMed] [Google Scholar]
- 58.Wang D, Zhu GY, Huang X, Lippard SJ. X-ray structure and mechanism of RNA polymerase II stalled at an antineoplastic monofunctional platinum-DNA adduct. Proc Natl Acad Sci U S A. 2010;107:9584–9589. doi: 10.1073/pnas.1002565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walmacq C, Wang L, Chong J, Scibelli K, Lubkowska L, Gnatt A, Brooks PJ, Wang D, Kashlev M. Mechanism of RNA polymerase II bypass of oxidative cyclopurine DNA lesions. Proc Natl Acad Sci U S A. 2015;112:E410–419. doi: 10.1073/pnas.1415186112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang LF, Zhou Y, Xu L, Xiao R, Lu XY, Chen L, Chong J, Li HR, He C, Fu XD, Wang D. Molecular basis for 5-carboxycytosine recognition by RNA polymerase II elongation complex. Nature. 2015;523:621–625. doi: 10.1038/nature14482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dangkulwanich M, Ishibashi T, Liu SX, Kireeva ML, Lubkowska L, Kashlev M, Bustamante CJ. Complete dissection of transcription elongation reveals slow translocation of RNA polymerase II in a linear ratchet mechanism. Elife. 2013;2:e00971. doi: 10.7554/eLife.00971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kellinger MW, Ulrich S, Chong J, Kool ET, Wang D. Dissecting chemical interactions governing RNA polymerase II transcriptional fidelity. J Am Chem Soc. 2012;134:8231–8240. doi: 10.1021/ja302077d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu L, Zhang L, Chong J, Xu J, Huangb XH, Wang D. Strand-specific (asymmetric) contribution of phosphodiester linkages on RNA polymerase II transcriptional efficiency and fidelity. Proc Natl Acad Sci U S A. 2014;111:E3269–E3276. doi: 10.1073/pnas.1406234111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kellinger MW, Song C-X, Chong J, Lu X-Y, He C, Wang D. 5-formylcytosine and 5-carboxylcytosine reduce the rate and substrate specificity of RNA polymerase II transcription. Nat Struct Mol Biol. 2012;19:831–833. doi: 10.1038/nsmb.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu L, Wang W, Zhang L, Chong J, Huang X, Wang D. Impact of template backbone heterogeneity on RNA polymerase II transcription. Nucleic Acids Res. 2015;43:2232–2241. doi: 10.1093/nar/gkv059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Barnes Christopher O, Calero M, Malik I, Graham Brian W, Spahr H, Lin G, Cohen Aina E, Brown Ian S, Zhang Q, Pullara F, Trakselis Michael A, Kaplan Craig D, Calero G. Crystal Structure of a Transcribing RNA Polymerase II Complex Reveals a Complete Transcription Bubble. Mol Cell. 2015;59:258–269. doi: 10.1016/j.molcel.2015.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bernecky C, Herzog F, Baumeister W, Plitzko JM, Cramer P. Structure of transcribing mammalian RNA polymerase II. Nature. 2016;529:551–554. doi: 10.1038/nature16482. [DOI] [PubMed] [Google Scholar]
- 68.Imashimizu M, Oshima T, Lubkowska L, Kashlev M. Direct assessment of transcription fidelity by high-resolution RNA sequencing. Nucleic Acids Res. 2013;41:9090–9104. doi: 10.1093/nar/gkt698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gout JF, Thomas WK, Smith Z, Okamoto K, Lynch M. Large-scale detection of in vivo transcription errors. Proc Natl Acad Sci U S A. 2013;110:18584–18589. doi: 10.1073/pnas.1309843110. [DOI] [PMC free article] [PubMed] [Google Scholar]