

Abstract
Background and Purpose
Kv7 (Kv7.1–7.5) channels play an important role in the regulation of neuronal excitability and the cardiac action potential. Growing evidence suggests Kv7.4/Kv7.5 channels play a crucial role in regulating vascular smooth muscle contractility. Most of the reported Kv7 openers have shown poor selectivity across these five subtypes. In this study, fasudil – a drug used for cerebral vasospasm – has been found to be a selective opener of Kv7.4/Kv7.5 channels.
Experimental Approach
A perforated whole‐cell patch technique was used to record the currents and membrane potential. Homology modelling and a docking technique were used to investigate the interaction between fasudil and the Kv7.4 channel. An isometric tension recording technique was used to assess the vascular tension.
Key Results
Fasudil selectively and potently enhanced Kv7.4 and Kv7.4/Kv7.5 currents expressed in HEK293 cells, and shifted the voltage‐dependent activation curve in a more negative direction. Fasudil did not affect either Kv7.2 and Kv7.2/Kv7.3 currents expressed in HEK293 cells, the native neuronal M‐type K+ currents, or the resting membrane potential in small rat dorsal root ganglia neurons. The Val248 in S5 and Ile308 in S6 segment of Kv7.4 were critical for this activating effect of fasudil. Fasudil relaxed precontracted rat small arteries in a concentration‐dependent fashion; this effect was antagonized by the Kv7 channel blocker XE991.
Conclusions and Implications
These results suggest that fasudil is a selective Kv7.4/Kv7.5 channel opener and provide a new dimension for developing selective Kv7 modulators and a new prospective for the use, action and mechanism of fasudil.
Abbreviations
- DRG
dorsal root ganglia
- Phe
phenylephrine
- RMP
resting membrane potential
- RTG
retigabine
Tables of Links
| TARGETS | |
|---|---|
| GPCRs a | Voltage‐gated ion channels b |
| α1‐Adrenoceptors | Kv7.1/KCNE1 channel |
| M2 receptor | Kv7.2 channel |
| Kv7.3 channel | |
| Kv7.4 channel | |
| Kv7.5 channel |
| LIGANDS | |
|---|---|
| ACh | Retigabine |
| Fasudil | XE991 |
| Phenylephrine |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a,b).
Introduction
Kv7 (Kv7.1–Kv7.5) is a subfamily of voltage‐gated K+ channels encoded by the KCNQ genes. It plays an important role in the regulation of neuronal excitability (Kv7.2–Kv7.5) and the cardiac action potential (Kv7.1) (Jentsch, 2000; Brown and Passmore, 2009). It has been established that Kv7.2 and Kv7.3 are heterotetramers and are the principal molecular components of the slow voltage‐gated M‐channel, which largely regulates neuronal excitability (Jentsch, 2000; Brown and Passmore, 2009). Recently, Kv7.4 and Kv7.5 channels have also been identified in multiple vascular smooth muscle cells and have been found to play a crucial role in the regulation of vascular smooth muscle contractility and vascular tone (Yeung et al., 2007; Mackie et al., 2008; Greenwood and Ohya, 2009; Jepps et al., 2011; Ng et al., 2011; Brueggemann et al., 2014; Chadha et al., 2014). Kv7 channel modulators provoke significant changes in vascular smooth muscle membrane potential and vascular tone (Yeung and Greenwood, 2005; Joshi et al., 2006; Yeung et al., 2007; Mackie et al., 2008; Yeung et al., 2008; Joshi et al., 2009; Zhong et al., 2010; Jepps et al., 2011; Ng et al., 2011; Jepps et al., 2014). In vascular smooth muscle cells, the physiologically predominant molecular structure is a Kv7.4/Kv7.5 heterotetramer (Brueggemann et al., 2014; Chadha et al., 2014). Thus, the expression of neuronal (Kv7.2/Kv7.3) Kv7 channels can be differentiated from those of vascular (Kv7.4/Kv7.5) Kv7 channels, which provides a selective drug action strategy.
Non‐selective Kv7 modulators may be liable to cause side effects when used clinically. This is certainly true for the first approved Kv7 opener retigabine (RTG), which is used for the treatment of epilepsy [Potiga (Ezogabine)]; comes with side effect warnings for urinary retention [bladder smooth muscle cells are also a target for Kv7 modulators including the openers (Rode et al., 2010; Anderson et al., 2013)]. Most of the reported Kv7 modulators show poor selectivity across the five Kv7 channel subtypes – especially those that affect Kv7.2–Kv7.4 channels. For example, RTG and flupirtine activate all neuronal Kv7 channels (i.e. Kv7.2–Kv7.5) although they do not affect Kv7.1 channels (Tatulian et al., 2001; Schenzer et al., 2005; Wuttke et al., 2005). Thus, compounds that are selective across Kv7 subtypes and also act selectively on either neuronal or vascular Kv7 channels have a significant advantage in terms of therapeutic potential.
Fasudil is the only clinically available RhoA/Rho kinase (ROCK) inhibitor. It is already in use for the treatment of cerebral vasospasm and to improve the cognitive decline seen in stroke victims (Dong et al., 2009; Chen et al., 2013). Its anti‐angina effect has also been confirmed in humans (Vicari et al., 2005). In an earlier preliminary study performed to assess its vasorelaxant effect, we found that fasudil could activate Kv7 channels. Here, we characterized fasudil for its effect on all five Kv7 subtypes and found that fasudil selectively activates Kv7.4/Kv7.5 but does not affect Kv7.2/Kv7.3 channels. The residues Val248 in the S5 and Ile308 in the S6 segment of Kv7.4 were shown to be essential for this effect of fasudil. The Kv7 channel blocker XE991 antagonized the fasudil‐induced vasorelaxation of the small mesenteric artery of the rat.
Methods
cDNA constructs
Plasmids encoding human Kv7.1, Kv7.2, Kv7.3, Kv7.4 and Kv7.5 (GenBank accession numbers: NM000218, AF110020, AF091247, AF105202 and AF249278 respectively) were kindly provided by Diomedes E. Logothetis (Virginia Commonwealth University, Richmond, Virginia, USA) and subcloned into pcDNA3. The Kv7.4(G302T/A304T), Kv7.4(L306I), Kv7.4(I308V) and Kv7.4(L306I/I308V) mutants were produced by Pfu DNA polymerase with a QuickChange kit (Stratagene, USA). The mutants' identity was confirmed with DNA sequencing.
HEK293 cell culture and transfection
HEK293 cells were cultured in DMEM supplemented with 10% FBS (PAA, Austria) and antibiotics in a humidified incubator at 37°C (5% CO2). Cells were seeded on glass coverslips in a 24‐multiwell plate and transfected when 60–70% confluence was reached. For transfection of six wells of cells, a mixture of 2 μg KCNQs (or +2 μg KCNE1) and 2 μg pEGFP (plasmid enhanced green fluorescent protein) cDNAs and 3 μL lipofectamine 2000 reagent (Invitrogen, USA) were prepared in 1.2 mL of DMEM and incubated for 20 min. The mixture was then applied to the cell culture wells and incubated for 4 h. Recordings were made 24 h after cell transfection, and cells were used within 72 h. The transfection rate was ~40–60%. Only cells expressing GFP fluorescence (detected at 510 nM) were used for electrophysiological recordings.
Animals
All animal care and experimental procedures were approved by Animal Care and Ethical Committee of Hebei Medical University (Shijiazhuang, China). All studies are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath and Lilley, 2015). The experiments were performed in the Department of Pharmacology, Hebei Medical University. The animals were housed under controlled conditions at 25°C in a 12 h light/dark cycle with ad libitum access to water and food.
Rat DRG cell culture
Experimental procedures were reviewed and approved by Animal Care and Ethical Committee of Hebei Medical University (Shijiazhuang, China). Rats were anaesthetized with 10% chloral hydrate (3 mL·kg−1, i.p.), throughout the experiment. dorsal root ganglias (DRGs) were isolated from all spinal levels of 1‐week‐old (10–15 g) male Sprague–Dawley rats (obtained from Experimental Animal Centre of Hebei Medical University). Ganglia were placed in modified D‐Hanks' solution, and digested at 37°C with collagenase (2 mg·mL−1, Worthington) for 25 min followed by another 20 min digestion with trypsin (2.5 mg·mL−1, Sigma, USA). They were subsequently suspended and quenched with DMEM medium plus 10% FBS (PAA, Austria) to stop digestion. Ganglia were then dissociated into a suspension of individual cells and plated on poly‐D‐lysine‐coated glass coverslips in 24‐well tissue culture plates. Cells were incubated at 37°C with 5% CO2 and 95% air atmosphere. Electrophysiological recordings were made from small DRG cells (diameter ~20 μm) maintained in culture from day 3 to day 7.
Electrophysiology
Perforated whole‐cell patch recordings were performed on DRG neurons and HEK293 cells. Recordings were made at room temperature (23–25°C). Pipettes were pulled from borosilicate glass capillaries with resistances of 1.5–2.5 MΩ when filled with internal solution. The currents were recorded using an Axon patch 700B amplifier and pClamp 10.0 software (Molecular device, USA). The access resistance in our experiments was measured to be around 7–12 MΩ, and 60–80% series resistance compensation was achieved. Current records were acquired at 5 kHz and filtered at 2 kHz. For perforated patch recording, a pipette was first front‐filled with the standard internal solution and then backfilled with the same internal solution containing amphotericin B (250 μg·mL−1). The external solution used to record rest membrane potential and Kv7 and M currents contained the following (in mM): 160 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 8 glucose at pH 7.4. The internal solution contained the following (in mM): 150 KCl, 5 MgCl2 and 10 HEPES at pH 7.4.
The Kv7 (Kv7.2–7.5) current amplitude was measured from the peak deactivation current (tail current) elicited by a 1.5 s square voltage pulse to −60 mV (−40 mV for Kv7.1/KCNE1) from a holding potential of −30 mV (0 mV for Kv7.1/KCNE1). This was calculated as the difference between the average of a 10‐ms segment taken 20–30 ms into the hyperpolarizing step and the average during the last 50 ms of that step. The current–voltage (I–V) relationships were determined using step pulses between −80 and +30 mV in increments of 10 mV from a holding potential of −80 mV followed by a voltage pulse to −60 mV. Current amplitudes were measured from the peak deactivation current tested at −60 mV.
Homology modelling and docking
Homology models of the Kv7.4 channel used the SWISS‐MODEL server (Arnold et al., 2006; Guex et al., 2009; Biasini et al., 2014). The Kv7.4 sequence (target sequence) was taken from Genbank (http://www.ncbi.nlm.nih.gov/Genbank/). The KcsA channel structure (1BL8) was used as the template structure and was downloaded from the Protein Data Bank (http://www.pdb.org). A three‐dimensional structural model of the S5 to S6 domains of Kv7.4 was constructed based on the homology to KcsA using the crystal structures of the corresponding domains. The global and local model quality was assessed using the QMEAN scoring function. The QMEAN4 score is a composite score consisting of a linear combination of 4 statistical potential terms (Benkert et al., 2011). The QMEAN4 score was −7.36 due to the high sequence homology (about 54% sequence identity to KcsA). The model was compared to its template to verify that the modelling step did not significantly alter the conformation of the backbone or the side chain.
The model was solvated, and K+ and Cl− at ~150 mM were positioned randomly among the solvent to neutralize the system. The system was energy optimized and equilibrated using NAMD2.9 (Phillips et al., 2005) in default settings.
Fasudil was drawn with a Gaussian viewer and manually docked with Autodock 4.0 (Phillips et al., 2005). A grid map was generated for the Kv7.4 channel using CHNOP (i.e. carbon, hydrogen, nitrogen, oxygen and phosphor) elements sampled on a uniform grid containing 120 × 120 × 120 points, 0.375 Å apart. The centre of the grid box was set to the centre of the central cavity of the pore domain of Kv7.4. The AutoDock package docking used the Lamarchian genetic algorithm for ligand conformational search within the grid box. It uses a scoring function based on the orientation, position, conformation and energy of the resulting model, which can rank the docking action. Both the score and the H‐bond match the situation, and 100 docking reference structures were screened to select the optimal combination of conformation. The model with the most likely binding conformation is represented here.
Isometric tension recordings
Isometric tension was recorded in 2 mm segments of the second branch of mesenteric artery suspended on two intraluminal stainless steel wires (40 μm) in a small vessel myography apparatus (DMT, ADInstruments, UK) containing Krebs' solution aerated with 95% O2/5% CO2. After a 30‐min equilibration period, a passive length‐tension curve was constructed in each segment of the mesenteric artery by applying cumulative stretches to the preparation. From this curve, equivalent transmural pressures were estimated, and the vessel was set at a level of tension equivalent to 90% of the diameter of the vessel with a distending pressure of 100 mmHg. After an additional equilibration period, the tissues were bathed in an external solution containing 60 mM KCl for 5 min. This was repeated twice. Changes in the tension were continuously recorded with PowerLab and Chart (version 5, AD Instruments, UK).
Chemicals
Fasudil was purchased from Sigma (St. Louis, USA). RTG and XE991 were synthesized in our department. The stock solutions were made in DMSO and were stored at −20°C. All solutions were freshly prepared from stock solutions before each experiment and kept from light exposure. The final concentration of the DMSO was less than 0.1%.
Data analysis and statistics
The currents were analysed and fitted using Clampfit 10.2 (Molecular device, USA) and Origin 7.5 (Originlab Corporation, USA) software. The concentration–response curve was fitted with logistic equation: y = A 2 + (A 1–A 2)/[1 + (x/x 0)nH], where y is the response, A 1 and A 2 are the maximum and minimum response, respectively, x is the drug concentration, and n H is the Hill coefficient. The current activation curves were generated by plotting the normalized tail current amplitudes against the step potentials and were fitted with a Boltzmann function: y = A/{1 + exp[(Vh – Vm)/k]}. Here, A is the maximal current amplitude, Vh is the voltage for half‐maximal activation, Vm is the test potential and k is the slope factor. All data are given as mean ± SEM. Data statistical analysis was performed using a two sample paired Student's t‐test or one‐way ANOVA. The differences were considered significant at P < 0.05. Where ANOVA was used, a post hoc test (Bonferroni) was followed if F achieved P < 0.05 and there was no significant variance in‐homogeneity.
Blinding was not used in the experiments since all measurements were not subjective but strict quantitative data. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Results
Fasudil selectively activates Kv7.4 and Kv7.4/Kv7.5 currents
We first tested the effects of fasudil (Figure 1A) on Kv7.1/KCNE1, Kv7.2, Kv7.2/Kv7.3, Kv7.4 and Kv7.4/Kv7.5 channels (Figure 1D) expressed in HEK293 cells. The effects of fasudil were compared to those of RTG – a well‐established Kv7 channel opener (Figure 1A) (Tatulian et al., 2001). The Kv7.3 and Kv7.5 homomeric channels were not tested because these channels expressed alone did not give reliable measurable currents. Figure 1D shows the current traces of the Kv7 channels recorded using the voltage protocol shown in Figure 1C. The Kv7 currents were measured in the current amplitude of the deactivating tail currents at −60 mV from preceding voltage steps (from −80 to +30 mV, Figure 1D).
Figure 1.
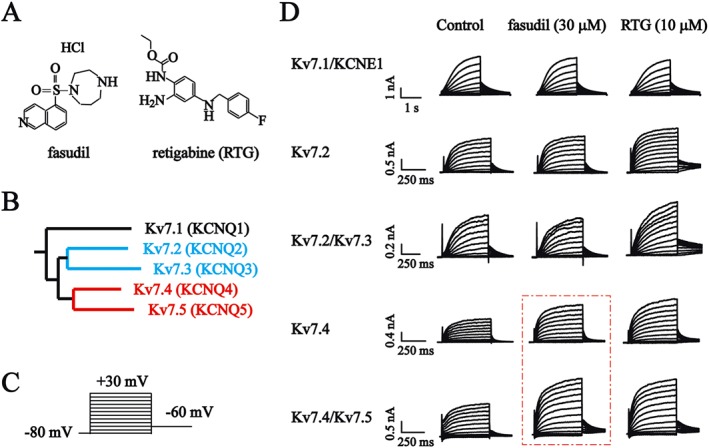
The effects of fasudil and RTG on Kv7 currents in HEK293 cells. (A) Chemical structures of fasudil and RTG. (B) Phylogenetic trees of Kv7 family members. (C) Voltage protocol. Cells were held at −80 mV and stepped to a series of voltages ranging from −80 to +30 mV (in 10 mV increments) for a 500 ms pulse followed by stepping to −60 mV for 500 ms. (D) The whole‐cell currents were recorded from HEK293 cells expressing Kv7 channels. The current traces of Kv7.1/KCNE1, Kv7.2, Kv7.2/Kv7.3, Kv7.4 and Kv7.4/Kv7.5 induced with the voltage protocol are shown in (C). The effects of 30 μM fasudil and 10 μM RTG are also shown.
The effects of fasudil and RTG were quantified by measuring the changes in the peak tail current (measured at −60 mV) amplitudes induced by the drugs. For each cell, the effects of fasudil (30 μM) and RTG (10 μM) were tested on expressed Kv7 currents. The fasudil and RTG were applied for a period of time until the effects were stabilized. This normally took 2–10 min (Figure 2B–E). In most experiments, the effects of fasudil, and RTG could be fully reversed upon drug washout (Figure 2B–E). The summarized results (Figure 2F) show that RTG (10 μM) inhibited Kv7.1/KCNE1 currents and enhanced Kv7.2, Kv7.2/Kv7.3, Kv7.4 and Kv7.4/Kv7.5 currents. The fasudil (30 μM) slightly inhibited Kv7.1/KCNE1 currents and did not affect Kv7.2, Kv7.2/Kv7.3 currents. It enhanced Kv7.4 and Kv7.4/Kv7.5 currents (Figure 2F).
Figure 2.
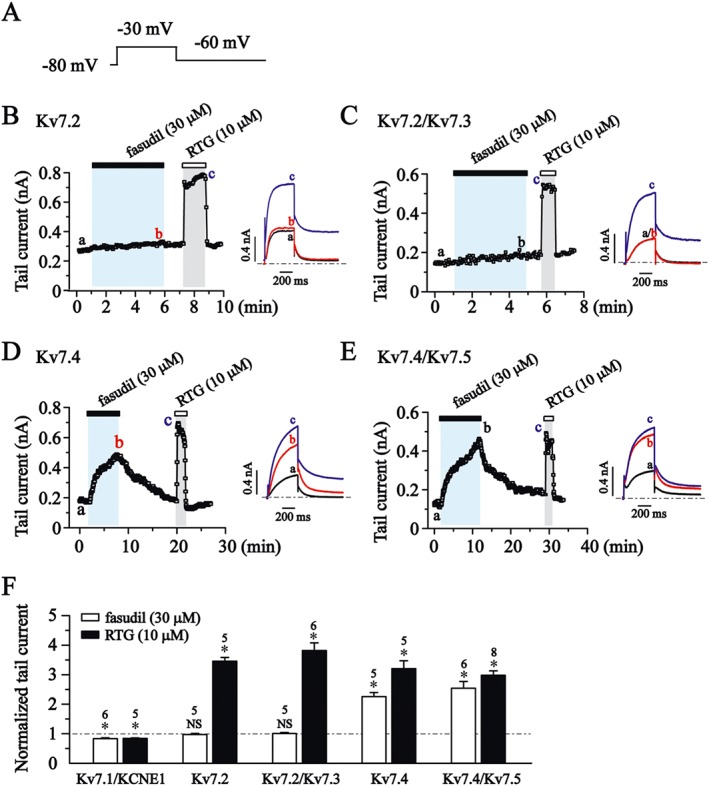
The effects of fasudil and RTG on Kv7 currents in HEK293 cells. (A) Voltage protocol. Cells were held at −80 mV and stepped to +30 mV for a 500 ms pulse followed by stepping to −60 mV for 1000 ms. (B–E) The time course for the effects of fasudil (30 μM), RTG (10 μM) on Kv7.2 (B), Kv7.2/Kv7.3 (C), Kv7.4 (D) and Kv7.4/Kv7.5 (E) currents. The representative current traces under different conditions of the treatments are shown. (F) The effects of fasudil and RTG on Kv7 tail current tested at −60 mV after −30 mV of depolarization. *P < 0.05 compared with the current amplitudes in the absence of drugs; Student's two‐sample paired t‐test; n = 5–8.
The concentration–response relationship of fasudil on Kv7 currents
The concentration–response relationship of fasudil was then established for Kv7.4 and Kv7.4/Kv7.5 currents (Figure 3). For this investigation, the Kv7 peak tail currents measured at −60 mV were measured under the influence of different concentrations of fasudil. The concentration–response curves were constructed from the normalized tail currents over the control and were fitted with a logistic function. As demonstrated in Figure 3A and B, fasudil concentration‐dependently enhanced Kv7.4 and Kv7.4/Kv7.5 currents. The EC50s of fasudil for Kv7.4 and Kv7.4/Kv7.5 were 12.9 ± 2.2 μM and 15.7 ± 1.0 μM respectively. In stark contrast, fasudil at a similar concentration range (1–100 μM) did not affect the current amplitude of heteromeric Kv7.2/Kv7.3 currents (Figure 3C and D). In fact, fasudil at 100 μM slightly inhibited Kv7.2/Kv7.3 currents by 14 ± 4% (P < 0.05, Figure 3D).
Figure 3.
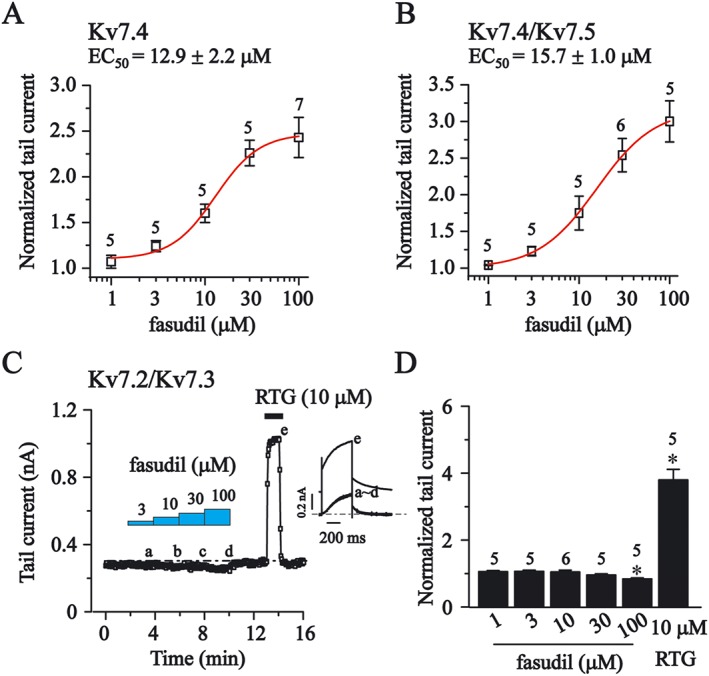
The concentration‐dependent effects of fasudil on Kv7 currents in HEK293 cells. The effects of different concentration of fasudil were tested on Kv7 tail currents, and the protocol used is shown in Figure 2B. (A and B) The concentration–response relationships of fasudil on homomeric Kv7.4 currents and heteromeric Kv7.4/Kv7.5 are shown. The dose–response relationships were fitted with logistic function. The EC50 values are 12.9 ± 2.2 μM (Kv7.4, n = 5–7) and 15.7 ± 1.0 μM (Kv7.4/Kv7.5, n = 5–6). (C) The time course for the effects of fasudil (3–100 μM) and RTG (10 μM) on the currents of Kv7.2/Kv7.3. The representative current traces under different conditions of the treatments are shown in the insets. (D) Summarized data for the effects of fasudil (1–100 μM) and RTG (10 μM) on Kv7.2/Kv7.3 channel. Statistics used Student's paired t‐test; *P< 0.05.
The effects of fasudil on voltage‐dependence of Kv7 activation
The G‐V curves for voltage‐dependent activation of Kv7 currents were established from the tail currents measured at −60 mV, and the half‐activation potential (V1/2) was obtained from the fitting function of the Boltzmann equation as described in the Methods. The currents were recorded when the effects of fasudil (30 μM) and RTG (10 μM) were stabilized. As demonstrated in Figure 4A and C, fasudil (30 μM) and RTG (10 μM) significantly shifted the G‐V curves of Kv7.4 and Kv7.4/Kv7.5 to more negative potentials. More specifically, the V1/2s for Kv7.4 were: control, −21.1 ± 1.1 mV; fasudil, −27.0 ± 0.9 mV; and RTG, −35.8 ± 0.8 mV. The V1/2s for Kv7.4/Kv7.5 were: control, −29.3 ± 0.9 mV; fasudil, −35.9 ± 0.7 mV; and RTG, −45.9 ± 0.5 mV. Similar to its effect on the current amplitude, fasudil did not affect the G‐V curves of Kv7.2 and Kv7.2/Kv7.3 currents (Figure 4E and F), but RTG did significantly shift the G‐V curve of Kv7.2 and Kv7.2/Kv7.3 to more negative potentials. The V1/2s for Kv7.2 were: control, −22.7 ± 1.3 mV; fasudil, −24.6 ± 1.4 mV; and RTG, −75.5 ± 3.5 mV. The V1/2s for Kv7.2/Kv7.3 were: control, −17.1 ± 1.5 mV; fasudil, −19.3 ± 1.0 mV; and RTG, −49.9 ± 1.7 mV.
Figure 4.
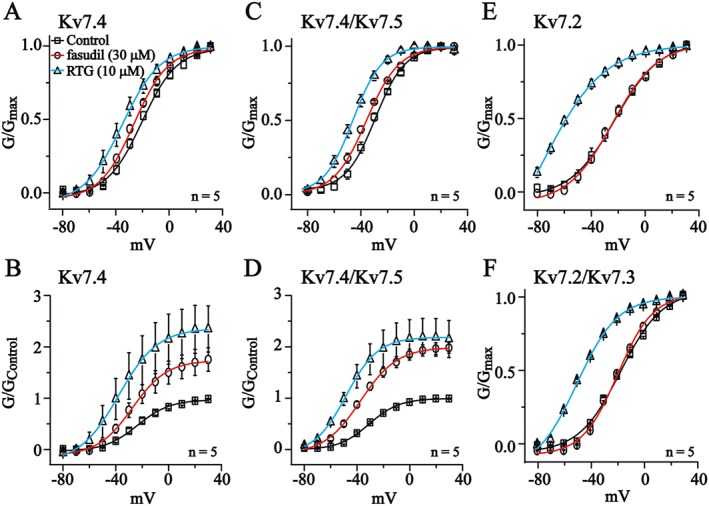
The effects of fasudil on voltage‐dependent activation of Kv7 currents in HEK293 cells. The currents were recorded using the voltage protocol shown at the top of Figure 1C. Voltage‐dependent activation curves were plotted from the tail currents recorded at −60 mV against the preceding voltage step. In (A, C, E and F), the currents (G) were normalized to the maximum value of each group. In (B and D), the currents were normalized to the control maximum value. The data were fitted with a Boltzmann function described in the Methods section; n = 5.
The effects of fasudil and RTG on the maximum channel conductance were also tested. Figure 4B and D show that the tail currents were normalized to the control maximum value, and fasudil and RTG produced a potent increase in the Gmax of Kv7.4 and Kv7.4/Kv7.5 channels (Kv7.4: fasudil, 174 ± 3%; RTG, 236 ± 3%; Kv7.4/Kv7.5: fasudil, 199 ± 2%; and RTG, 218 ± 1%).
The effects of fasudil on M‐type K+ currents in DRG neurons
We next examined whether fasudil could modulate native neuronal Kv7 currents. M‐type K+ currents found in these cells best manifest the Kv7 currents in neuronal cells. The neuronal M‐channels are most likely composed of Kv7.2 and Kv7.3 subunits (Wang et al., 1998; Hadley et al., 2000; Shapiro et al., 2000), which are shown in Figures 1, 2 and 3. These are not affected by fasudil. In our previous study, we observed M‐type K+ currents in the small diameter neuron of the rat DRG (Liu et al., 2010). This M‐type K+ current and the resting membrane potential (RMP) could be significantly modulated by the non‐selective Kv7 opener RTG and the blocker XE991 (Du et al., 2014). Thus, we tested the effect of fasudil on the M‐type K+ currents recorded from rat small DRG neurons using the protocol shown in the inset of Figure 5A. An example of the concentration‐dependent effect of fasudil on the tail currents of M‐channel (recorded at −60 mV) is shown in Figure 5A. The summarized results are shown in Figure 5B.
Figure 5.
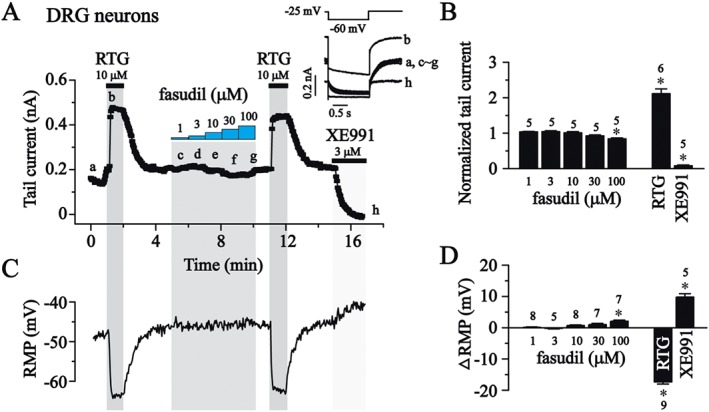
The effects of fasudil on M‐type K+ currents and RMP in small rat DRG neurons. (A) The time course for the effects of fasudil (1–100 μM), RTG (10 μM) and XE991 (3 μM,) on M‐type K+ currents in rat DRG neurons. The representative current traces under different conditions of the treatments were shown in the insets. (B) Summarized data of (A). (C) The time course for the effects of fasudil (1–100 μM), RTG (10 μM) and XE991 (3 μM) on the RMP. (D) Summarized data of change in RMP. Statistics used Student's two sample paired t‐test; *P< 0.05.
Similar to its effect on Kv7.2/Kv7.3, fasudil up to 100 μM did not activate M‐type K+ currents (Figure 3C and D). At 100 μM, it slightly inhibited M‐type K+ currents (by 17 ± 3%, P < 0.05, Figure 5B). In contrast, RTG (10 μM) induced a significant increase in M‐type K+ currents (210 + 10%), which was totally abolished by 3 μM XE991 (Figure 5A and B). Fasudil at lower concentrations (1–30 μM) did not affect the RMP. It slightly depolarized the RMP at 100 μM (2.1 ± 0.4 mV), consistent with an inhibitory effect on the Kv7.2/Kv7.3 currents seen at this concentration (Figure 3D). In contrast, RTG hyperpolarized and XE991 depolarized the RMP significantly (Figure 5C and D). These results suggest that fasudil does not activate the native neuronal M/Kv7 currents (most likely mediated by Kv7.2/Kv7.3 channels); this plays an important role in controlling the RMP.
Key amino acid residues in Kv7.4 channel mediating the effect of fasudil
The differences in the effect of fasudil between Kv7.4/Kv7.5 and Kv7.2/Kv7.3 channels suggests that molecular differences between these channels underpin the selectivity of fasudil. To identify these key amino acid residues in Kv7.4, we first compared the amino acid sequence of S5, pore region and S6 segments of the Kv7 family members including the regions that mediate most of the actions of known Kv7 modulators (Schenzer et al., 2005; Wuttke et al., 2005; Lange et al., 2009). As shown in Figure 5A, residue sequences are highly conserved among the Kv7 family with only a few small differences. We focused on the differences between Kv7.4/Kv7.5 versus Kv7.2/Kv7.3 channels. Residues in the S5, pore region and S6 segment were examined. Thus, Val248, Phe251, Asp262, Ala263, Thr278, Gly302, Ala304, Leu306 and Ile308 are well conserved in Kv7.4 and Kv7.5, but are different in Kv7.2 and Kv7.3.
To identify the binding residues, we performed AutoDock4.0 to determine the docking reference structures by treating the ligand and selected parts of the target as flexible. This approach uses a scoring function based on the orientation, position, conformation and energy of the resulting model and can rank the docking reference structures. The docking simulation results shown in Figure 6B indicate the existence of interactions between fasudil and Ile308 in S6 of the Kv7.4 channel.
Figure 6.
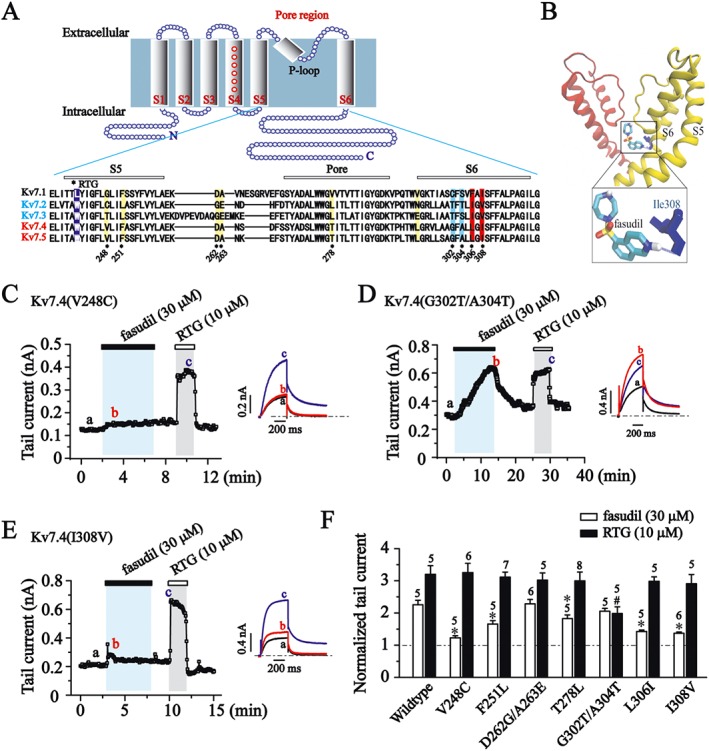
Molecular determinants for fasudil sensitivity. (A) Kv7 cartoon diagram. Transmembrane segments are labelled S1–S6. Alignment of the pore regions of Kv7.1 to Kv7.5 (KCNQ1–5). RTG binding sites are labelled with the violet band, and residues that are different between Kv7.4/Kv7.5 versus Kv7.2/Kv7.3 in the S5 segment and pore region are labelled with a yellow band. Residues Gly302, Ala304, Leu306 and Ile308 in the S6 segment are labelled with blue or red band. (B) Docking result for the binding of fasudil within the pore domain of Kv7.4 (KCNQ4) channel. Two subunits are shown (yellow and red) of energy‐optimized homology models of the Kv7.4 pore domain derived from the crystal structures of KcsA. The hydrogen bond between fasudil and Ile308 in Kv7.4 (KCNQ4) channel is shown as a blue dotted line. Residue I308 is shown in blue. (C–E) The time courses for the effects of fasudil (30 μM) and RTG (10 μM) on Kv7.4 point mutants recorded at −60 mV. Each mutation site was indicated in (A). (F) Histogram shows the effect of fasudil (30 μM) and RTG (10 μM) on the tail currents of Kv7.4 point mutants (−60 mV). *P < 0.05 compared with the Kv7.4 wildtype currents in the presence of fasudil, #P < 0.05 compared with the Kv7.4 wildtype currents in the presence of RTG, n = 5–8 (one‐way ANOVA, Bonferroni post hoc test).
The responses of Kv7.4(V248C) and Kv7.4(I308V) mutant channels to fasudil were greatly attenuated (P < 0.05), but the responses of these mutant channels to RTG were maintained (Figure 6C, E and F, P > 0.05). These data indicate that the Val248 in S5 and Ile308 in S6 of Kv7.4 are critical for the selective potentiation effect of fasudil. In contrast, fasudil (30 μM) potentiated the Kv7.4(G302 T/A304 T) currents similar to the Kv7.4 wildtype (P>0.05; Figure 6D and F). However, the potentiation effect of RTG (10 μM) on these mutant channels were greatly reduced (P <0.05).
The role of Kv7 activation in fasudil‐induced vasorelaxation of rat mesenteric artery
Activation of Kv7.4 and Kv7.5 channels induces vasorelaxation in many vascular tissues (Ohya et al., 2003; Yeung et al., 2007; Greenwood and Ohya, 2009), and fasudil is a potent vasodilator for treatment of cerebral vasospasm (Dong et al., 2009). Here, we used a classic Kv7 blocker XE991 to investigate whether activation of Kv7.4, and Kv7.5 channels play a role in fasudil‐induced vascular relaxation.
The second branch of rat mesenteric artery segments was used, and the integrity of functional endothelial cells was tested using ACh (1 μM) for each vessel ring. Only the vessel rings that could be fully relaxed by ACh (1 μM) were used (Figure 7A). The effect of fasudil on the phenylephrine (Phe)‐induced contraction was tested first. The Phe‐induced contraction shows limited relaxation at 30 min (3.52 ± 2.49%, n = 5, P > 0.05, Supporting Information Figure S2), and we studied the concentration‐dependent effect of fasudil at this point. Fasudil relaxed the second branch of the rat mesenteric artery segments precontracted by Phe (10 μM) in a concentration‐dependent fashion with an EC50 of 0.83 ± 0.25 μM (Figure 7B and E). In the presence of 10 μM XE991, which blocks Kv7.4/Kv7.5 currents (Supporting Information Figure S3), the dose–response curve of fasudil was significantly right‐shifted with an EC50 of 5.04 ± 0.96 μM (fasudil + XE991) (Figure 7C and E). We then tested the effect of fasudil on the high KCl‐induced vascular contraction. Fasudil also relaxed the KCl‐induced contraction, but with a much high EC50 (5.73 ± 0.26 μM; Figure 7D and E). These data indicate a possible involvement of Kv7.4 and Kv7.5 channels in the fasudil‐induced vasorelaxation.
Figure 7.
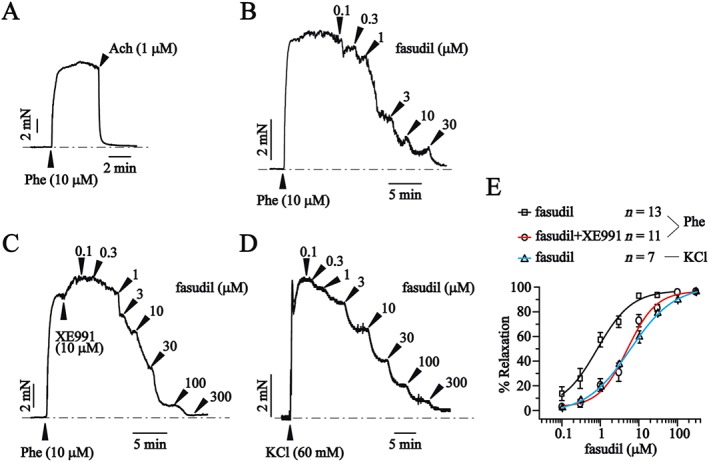
Kv7 blocker XE991 antagonizes the vasorelaxant effects of fasudil on precontracted rat mesenteric artery segments. (A) Representative tension trace of the effect of ACh (1 μM) on a mesenteric artery segment precontracted with phenylephrine (Phe; 10 μM). (B) Representative tension trace of the effect of fasudil (0.1–100 μM) on a mesenteric artery segment precontracted with Phe (10 μM). (C) Representative tension trace of the effect of fasudil (0.1–300 μM) + XE991 (10 μM) on a mesenteric artery segment precontracted with Phe (10 μM). (D) Representative tension trace of the effect of fasudil (0.1–300 μM) on a mesenteric artery segment precontracted with KCl (60 mM). (E) Concentration‐response relationships for fasudil. Data are fitted with the logistic equation. The EC50s are 0.83 ± 0.25 μM (fasudil/Phe, n = 13) and 5.04 ± 0.96 μM (fasudil + XE991/Phe, n = 11) and 5.73 ± 0.26 μM (fasudil/KCl, n = 7).
Discussion and conclusions
To our knowledge, this is the first report to describe a highly selective opener for Kv7.2–Kv7.5 channels, and more specifically for vascular Kv7.4/Kv7.5 rather than neuronal Kv7.2/Kv7.3 channels. It is highly selective because fasudil did not activate Kv7.2/Kv7.3 channels in any concentrations tested here, but it did significantly enhance the effects of Kv7.4/Kv7.5 channels. The reported Kv7 openers so far have either low selectivity or no selectivity among the Kv7.2–Kv7.5 channels. For example, RTG and flupirtine show no significant selectivity among Kv7.2–Kv7.5 channels (Lange et al., 2009); acrylamide (S)‐1 enhances the maximal current amplitude of Kv7.4 and Kv7.5 at all potentials, but it inhibits/activates Kv7.2 and Kv7.2/Kv7.3, which are strongly voltage‐dependent (Bentzen et al., 2006). ICA‐27243 is more potent at activating heteromeric Kv7.2/Kv7.3 channels (EC50 of 0.4 μM) than homomeric Kv7.4 channels (EC50 of 9.7 μM) and heteromeric Kv7.3/Kv7.5 channels [EC50 >10 μM; (Wickenden et al., 2008; Blom et al., 2010)]. Diclofenac enhances the maximum current amplitudes and shifts the voltage‐dependent activation of Kv7.4 channels to more negative potentials, but it inhibited the maximum current amplitudes and shifts the voltage‐dependent activation of Kv7.5 to more negative potentials (Brueggemann et al., 2011). Celecoxib inhibits Kv7.1 currents and enhances Kv7.2, Kv7.4 and Kv7.3/Kv7.5 currents (Du et al., 2011); Znpy and QO‐58 enhances all Kv7.1–Kv7.5 currents (Xiong et al., 2007; Zhang et al., 2013).
A single tryptophan residue at the cytoplasmic end of S5 [Trp236 (Kv7.2) or Trp242 (Kv7.4)] is critical for the effect of RTG on Kv7 channels (Schenzer et al., 2005; Wuttke et al., 2005) and is crucial for the enhancement effect of many reported Kv7 openers such as acrylamide (S)‐2 (Blom et al., 2009), acrylamide (S)‐1 and BMS204352 (Bentzen et al., 2006), celecoxib (Du et al., 2011). Mutation of this tryptophan diminishes the enhancing effects of these compounds. This tryptophan is conserved among the Kv7.2–Kv7.5, and this may explain why the reported Kv7 openers show either low selectivity or no selectivity among the Kv7 family. In this study, the mutagenesis results suggest that Val248 in S5 and Ile308 in the S6 segment of Kv7.4 are crucial for the selective activation by fasudil. This is supported by the docking simulation study shown in Figure 6. Importantly, mutation of these two amino acids did not affect the activation of Kv7.4 by RTG. These results provide a strategy for further development of a selective modulator of the Kv7 channels.
The selective effect of fasudil on the vascular (Kv7.4/Kv7.5) versus neuronal Kv7 (Kv7.2/Kv7.3) was further strengthened by the selective relaxation effect of fasudil on the mesenteric artery and the lack of effect on M‐type K+ currents as well as the related membrane hyperpolarization on DRG neurons. This proves that fasudil not only selectively activates the expressed vascular Kv7.4/Kv7.5 channels but also selectively activates the naturally expressed Kv7.4/Kv7.5 channels. However, while the vascular Kv7 could be limited to Kv7.4/Kv7.5 (Jepps et al., 2013), the neuronal Kv7 includes all Kv7.2–Kv7.5, although the Kv7.2/Kv7.3 may be predominant (Jentsch, 2000; Brown and Passmore, 2009). Nevertheless, it seems that fasudil will affect a cell (tissue, vascular smooth muscle) that expresses Kv7.4/Kv7.5 but not a cell (tissue, e.g. DRG) that expresses Kv7.2/Kv7.3 channels. The significance of this finding could be perceived via three aspects: it provides an experimental foundation for selective targeting of neuronal versus vascular Kv7 channels and their related physiological functions; it provides a new impetus and new information for developing new selective Kv7 modulators; and it provides a new mechanistic explanation for the vasodilator effect of fasudil.
The Kv7 channels are probably not the sole mechanism responsible for fasudil‐induced vascular relaxation because fasudil is a ROCK inhibitor. This – in combination with other mechanisms such as NO production – is likely to contribute to the relaxation (Disli et al., 2009). This NO could then activate Kv7.4/Kv7.5 channels and contribute to the vasodilator effect of fasudil that underpins its clinical use against vasospasm. The limited in vitro results prevent a firm conclusion from being reached. However, there are a few points that should be considered. (1) The XE991 antagonized the fasudil‐induced, concentration‐dependent relaxation of mesenteric arteries suggesting that Kv7 is indeed involved in fasudil‐induced vasorelaxation. (2) Fasudil induced a relaxation of KCl‐induced contraction with an EC50 of 5.73 ± 0.26 μM, which is much higher (seven times) than that of fasudil on Phe‐induced contraction (Figure 7E) and is similar to Phe‐induced relaxation in the presence of XE991 (5.04 ± 0.96 μM) and strongly suggests the involvement of K+ channels. (3) Fasudil activates Kv7.4/Kv7.5 with a EC50 of 15.7 ± 1.0 μM. (4) Fasudil is used clinically to treat vasospasm at a dose of 30 mg, which gives rise to plasma concentrations Cmax of 188 mg·L−1 (573 μM) (Shibuya et al., 1992; Chen et al., 2010). We also tested the effects of another ρ kinase inhibitor Y‐27 632, which is structurally different from fasudil –Y‐27632 (30 μM) did not affect the Kv7.4/Kv7.5 currents (Supporting Information Figure S1) suggesting that ρ kinase inhibition is not related to the fasudil‐induced activation of Kv7.4/Kv7.5 channels.
In summary, our results suggest that fasudil is a selective Kv7.4 and Kv7.5 channel opener and may provide a new dimension for developing selective Kv7 modulators and a new prospective for the use, action and mechanism of fasudil. These findings and the increased understanding of the molecular determinants of selective Kv7 modulators are crucial to discovering more potent and specific Kv7 channel modulators with potential clinical utility.
Author contributions
X.Z., H.A., J.L., Y.Z., Y.L. and Z.J. performed the research. X.Z., W.Z., L.C. and H.Z. designed the research study. X.Z. analysed the data. X.Z., L.C. and H.Z. wrote the paper.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 The effect of Y‐27632 on Kv7.4/7.5 current.
Supporting info item
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31270882 to HZ, 31401003 to XZ and 81573416 to WZ), the National Basic Research Program of China (2013CB531302 to HZ), Natural Science Foundation of Hebei Province (H2015423014 to XZ), Project Funded by China Postdoctoral Science Foundation (58th to XZ), the Ministry of Education (Young Thousand Talent Program) to WZ, High Talent Science Research Project of Education Bureau Hebei Province (GCC2014015 to WZ) and by the Natural Science Fund for Distinguished Young Scholars of the Hebei Province of China (C2015202340 to HA).
Zhang, X. , An, H. , Li, J. , Zhang, Y. , Liu, Y. , Jia, Z. , Zhang, W. , Chu, L. , and Zhang, H. (2016) Selective activation of vascular Kv7.4/Kv7.5 K+ channels by fasudil contributes to its vasorelaxant effect. British Journal of Pharmacology, 173: 3480–3491. doi: 10.1111/bph.13639.
Contributor Information
Li Chu, Email: chuli0614@126.com.
Hailin Zhang, Email: zhanghl@hebmu.edu.cn.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson UA, Carson C, Johnston L, Joshi S, Gurney AM, McCloskey KD (2013). Functional expression of KCNQ (Kv7) channels in guinea pig bladder smooth muscle and their contribution to spontaneous activity. Br J Pharmacol 169: 1290–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K, Bordoli L, Kopp J, Schwede T (2006). The SWISS‐MODEL workspace: a web‐based environment for protein structure homology modelling. Bioinformatics 22: 195–201. [DOI] [PubMed] [Google Scholar]
- Benkert P, Biasini M, Schwede T (2011). Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27: 343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentzen BH, Schmitt N, Calloe K, Dalby Brown W, Grunnet M, Olesen SP (2006). The acrylamide (S)‐1 differentially affects Kv7 (KCNQ) potassium channels. Neuropharmacology 51: 1068–1077. [DOI] [PubMed] [Google Scholar]
- Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T et al. (2014). SWISS‐MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42 (Web Server issue): W252–W258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom SM, Schmitt N, Jensen HS (2009). The acrylamide (S)‐2 as a positive and negative modulator of Kv7 channels expressed in Xenopus laevis oocytes. PLoS One 4: e8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom SM, Schmitt N, Jensen HS (2010). Differential effects of ICA‐27243 on cloned K(V)7 channels. Pharmacology 86: 174–181. [DOI] [PubMed] [Google Scholar]
- Brown DA, Passmore GM (2009). Neural KCNQ (Kv7) channels. Br J Pharmacol 156: 1185–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brueggemann LI, Mackie AR, Martin JL, Cribbs LL, Byron KL (2011). Diclofenac distinguishes among homomeric and heteromeric potassium channels composed of KCNQ4 and KCNQ5 subunits. Mol Pharmacol 79: 10–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brueggemann LI, Mackie AR, Cribbs LL, Freda J, Tripathi A, Majetschak M et al. (2014). Differential protein kinase C‐dependent modulation of Kv7.4 and Kv7.5 subunits of vascular Kv7 channels. J Biol Chem 289: 2099–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadha PS, Jepps TA, Carr G, Stott JB, Zhu HL, Cole WC et al. (2014). Contribution of kv7.4/kv7.5 heteromers to intrinsic and calcitonin gene‐related peptide‐induced cerebral reactivity. Arterioscler Thromb Vasc Biol 34: 887–893. [DOI] [PubMed] [Google Scholar]
- Chen H, Lin Y, Han M, Bai S, Wen S (2010). Simultaneous quantitative analysis of fasudil and its active metabolite in human plasma by liquid chromatography electro‐spray tandem mass spectrometry. J Pharm Biomed Anal 52: 242–248. [DOI] [PubMed] [Google Scholar]
- Chen M, Liu A, Ouyang Y, Huang Y, Chao X, Pi R (2013). Fasudil and its analogs: a new powerful weapon in the long war against central nervous system disorders? Expert Opin Investig Drugs 22: 537–550. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disli OM, Ozdemir E, Berkan O, Bagcivan I, Durmus N, Parlak A (2009). Rho‐kinase inhibitors Y‐27632 and fasudil prevent agonist‐induced vasospasm in human radial artery. Can J Physiol Pharmacol 87: 595–601. [DOI] [PubMed] [Google Scholar]
- Dong M, Yan BP, Yu CM (2009). Current status of rho‐associated kinases (ROCKs) in coronary atherosclerosis and vasospasm. Cardiovasc Hematol Agents Med Chem 7: 322–330. [DOI] [PubMed] [Google Scholar]
- Du XN, Zhang X, Qi JL, An HL, Li JW, Wan YM et al. (2011). Characteristics and molecular basis of celecoxib modulation on K(v)7 potassium channels. Br J Pharmacol 164: 1722–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Hao H, Gigout S, Huang D, Yang Y, Li L et al. (2014). Control of somatic membrane potential in nociceptive neurons and its implications for peripheral nociceptive transmission. Pain 155: 2306–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood IA, Ohya S (2009). New tricks for old dogs: KCNQ expression and role in smooth muscle. Br J Pharmacol 156: 1196–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex N, Peitsch MC, Schwede T (2009). Automated comparative protein structure modeling with SWISS‐MODEL and Swiss‐PdbViewer: a historical perspective. Electrophoresis 30 (Suppl 1): S162–S173. [DOI] [PubMed] [Google Scholar]
- Hadley JK, Noda M, Selyanko AA, Wood IC, Abogadie FC, Brown DA (2000). Differential tetraethylammonium sensitivity of KCNQ1‐4 potassium channels. Br J Pharmacol 129: 413–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch TJ (2000). Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci 1: 21–30. [DOI] [PubMed] [Google Scholar]
- Jepps TA, Chadha PS, Davis AJ, Harhun MI, Cockerill GW, Olesen SP et al. (2011). Downregulation of Kv7.4 channel activity in primary and secondary hypertension. Circulation 124: 602–611. [DOI] [PubMed] [Google Scholar]
- Jepps TA, Olesen SP, Greenwood IA (2013). One man's side effect is another man's therapeutic opportunity: targeting Kv7 channels in smooth muscle disorders. Br J Pharmacol 168: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepps TA, Bentzen BH, Stott JB, Povstyan OV, Sivaloganathan K, Dalby‐Brown W et al. (2014). Vasorelaxant effects of novel Kv 7.4 channel enhancers ML213 and NS15370. Br J Pharmacol 171: 4413–4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi S, Balan P, Gurney AM (2006). Pulmonary vasoconstrictor action of KCNQ potassium channel blockers. Respir Res 7: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi S, Sedivy V, Hodyc D, Herget J, Gurney AM (2009). KCNQ modulators reveal a key role for KCNQ potassium channels in regulating the tone of rat pulmonary artery smooth muscle. J Pharmacol Exp Ther 329: 368–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange W, Geissendorfer J, Schenzer A, Grotzinger J, Seebohm G, Friedrich T et al. (2009). Refinement of the binding site and mode of action of the anticonvulsant Retigabine on KCNQ K+ channels. Mol Pharmacol 75: 272–280. [DOI] [PubMed] [Google Scholar]
- Liu B, Linley JE, Du X, Zhang X, Ooi L, Zhang H et al. (2010). The acute nociceptive signals induced by bradykinin in rat sensory neurons are mediated by inhibition of M‐type K+ channels and activation of Ca2 + −activated Cl‐ channels. J Clin Invest 120: 1240–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie AR, Brueggemann LI, Henderson KK, Shiels AJ, Cribbs LL, Scrogin KE et al. (2008). Vascular KCNQ potassium channels as novel targets for the control of mesenteric artery constriction by vasopressin, based on studies in single cells, pressurized arteries, and in vivo measurements of mesenteric vascular resistance. J Pharmacol Exp Ther 325: 475–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng FL, Davis AJ, Jepps TA, Harhun MI, Yeung SY, Wan A et al. (2011). Expression and function of the K+ channel KCNQ genes in human arteries. Br J Pharmacol 162: 42–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohya S, Sergeant GP, Greenwood IA, Horowitz B (2003). Molecular variants of KCNQ channels expressed in murine portal vein myocytes: a role in delayed rectifier current. Circ Res 92: 1016–1023. [DOI] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E et al. (2005). Scalable molecular dynamics with NAMD. J Comput Chem 26: 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rode F, Svalo J, Sheykhzade M, Ronn LC (2010). Functional effects of the KCNQ modulators retigabine and XE991 in the rat urinary bladder. Eur J Pharmacol 638: 121–127. [DOI] [PubMed] [Google Scholar]
- Schenzer A, Friedrich T, Pusch M, Saftig P, Jentsch TJ, Grotzinger J et al. (2005). Molecular determinants of KCNQ (Kv7) K+ channel sensitivity to the anticonvulsant retigabine. J Neurosci 25: 5051–5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MS, Roche JP, Kaftan EJ, Cruzblanca H, Mackie K, Hille B (2000). Reconstitution of muscarinic modulation of the KCNQ2/KCNQ3 K(+) channels that underlie the neuronal M current. J Neurosci 20: 1710–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya M, Suzuki Y, Sugita K, Saito I, Sasaki T, Takakura K et al. (1992). Effect of AT877 on cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Results of a prospective placebo‐controlled double‐blind trial. J Neurosurg 76: 571–577. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatulian L, Delmas P, Abogadie FC, Brown DA (2001). Activation of expressed KCNQ potassium currents and native neuronal M‐type potassium currents by the anti‐convulsant drug retigabine. J Neurosci 21: 5535–5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicari RM, Chaitman B, Keefe D, Smith WB, Chrysant SG, Tonkon MJ et al. (2005). Efficacy and safety of fasudil in patients with stable angina: a double‐blind, placebo‐controlled, phase 2 trial. J Am Coll Cardiol 46: 1803–1811. [DOI] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS et al. (1998). KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M‐channel. Science 282: 1890–1893. [DOI] [PubMed] [Google Scholar]
- Wickenden AD, Krajewski JL, London B, Wagoner PK, Wilson WA, Clark S et al. (2008). N‐(6‐chloro‐pyridin‐3‐yl)‐3,4‐difluoro‐benzamide (ICA‐27243): a novel, selective KCNQ2/Q3 potassium channel activator. Mol Pharmacol 73: 977–986. [DOI] [PubMed] [Google Scholar]
- Wuttke TV, Seebohm G, Bail S, Maljevic S, Lerche H (2005). The new anticonvulsant retigabine favors voltage‐dependent opening of the Kv7.2 (KCNQ2) channel by binding to its activation gate. Mol Pharmacol 67: 1009–1017. [DOI] [PubMed] [Google Scholar]
- Xiong Q, Sun H, Li M (2007). Zinc pyrithione‐mediated activation of voltage‐gated KCNQ potassium channels rescues epileptogenic mutants. Nat Chem Biol 3: 287–296. [DOI] [PubMed] [Google Scholar]
- Yeung SY, Greenwood IA (2005). Electrophysiological and functional effects of the KCNQ channel blocker XE991 on murine portal vein smooth muscle cells. Br J Pharmacol 146: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung SY, Pucovsky V, Moffatt JD, Saldanha L, Schwake M, Ohya S et al. (2007). Molecular expression and pharmacological identification of a role for K(v)7 channels in murine vascular reactivity. Br J Pharmacol 151: 758–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung S, Schwake M, Pucovsky V, Greenwood I (2008). Bimodal effects of the Kv7 channel activator retigabine on vascular K+ currents. Br J Pharmacol 155: 62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Liu Y, Xu J, Zhang F, Liang H, Du X (2013). Membrane microdomain determines the specificity of receptor‐mediated modulation of Kv7/M potassium currents. Neuroscience 254: 70–79. [DOI] [PubMed] [Google Scholar]
- Zhong XZ, Harhun MI, Olesen SP, Ohya S, Moffatt JD, Cole WC et al. (2010). Participation of KCNQ (Kv7) potassium channels in myogenic control of cerebral arterial diameter. J Physiol 588 (Pt 17): 3277–3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 The effect of Y‐27632 on Kv7.4/7.5 current.
Supporting info item