

ABSTRACT
A variety of missense mutations in LMNA (the gene for lamin C and prelamin A) cause familial partial lipodystrophy (FPLD), a disease associated with reduced adipose tissue, particularly in the limbs. Several studies have reported that fibroblasts from FPLD subjects have an accumulation of prelamin A. Those findings were intriguing but also perplexing because many of the LMNA missense mutations associated with lipodystrophy are located in sequences distant from the sequences required for the farnesylation of prelamin A and ZMPSTE24-mediated conversion of prelamin A to mature lamin A. Here, we revisited the issue of prelamin A accumulation in the setting of FPLD mutations. We used western blots with lamin A/C antibodies and prelamin A–specific monoclonal antibodies to assess prelamin A levels in wild-type fibroblasts and fibroblasts carrying LMNA mutations associated with lipodystrophy (R482W, I299V, C591F, T528M). None of the mutant fibroblasts exhibited an accumulation of prelamin A. Also, the amount of prelamin A accumulation in response to lopinavir (an inhibitor of ZMPSTE24) was similar in wild-type and mutant fibroblasts. Thus, the LMNA lipodystrophy mutations that we examined did not lead to prelamin A accumulation, nor did they render those cells more susceptible to prelamin A accumulation when ZMPSTE24 was inhibited by lopinavir.
KEYWORDS: lamin A/C, laminopathy, lipodystrophy, progeria, prelamin A
Introduction
A variety of LMNA missense mutations cause familial partial lipodystrophy (FPLD2 or FPLD, Dunnigan Type), a disease associated with partial loss of adipose tissue from the limbs and trunk. Cao and Hegele1 reported in 2000 that a heterozygous LMNA mutation, R482Q, causes FPLD2 and predisposes to hyperlipidemia, diabetes mellitus, and atherosclerotic heart disease. In the same year, Shackleton et al.2 showed that R482W and R482L mutations cause FPLD2. Speckman et al.3 studied 15 FPLD families and found the R482W mutation in 7 families, a R482Q mutation in 5 families, and a G465D mutation in one family. Later, other LMNA mutations were identified in association with lipodystrophy (e.g., D230N, R399C, I299V, D47Y, L92F, L387V, R399H, L421P).4-6 Most of the mutations are located in sequences shared by lamin C and lamin A.
Loss of adipose tissue clearly occurs in the setting of “lamin A related” progeroid syndromes. Children with Hutchinson-Gilford progeria syndrome (HGPS) exhibit a striking loss of subcutaneous adipose tissue.7 HGPS is caused by a mutation that alters LMNA splicing and results in an in-frame deletion of 50 amino acids in prelamin A.8 This deletion eliminates the ZMPSTE24 cleavage site and therefore prevents the processing step that would normally convert farnesyl–prelamin A to mature lamin A;9 consequently, a large amount of an internally truncated farnesyl–prelamin A (progerin) accumulates in cells. Progerin is toxic to cells and is responsible for all HGPS disease phenotypes, including the loss of adipose tissue. More recently, Wang et al.10 uncovered a subject with a LMNA missense mutation (L647R) that eliminates prelamin A's ZMPSTE24 cleavage site and thus causes an accumulation of farnesyl–prelamin A; that subject had multiple progeria-like disease phenotypes, including reduced adipose tissue. ZMPSTE24 missense mutations that reduce ZMPSTE24 catalytic activity also cause an accumulation of farnesyl–prelamin A, resulting in a progeroid syndrome with reduced adipose tissue.11,12 In the setting of these progeroid syndromes, the prelamin A that accumulates is farnesylated, as judged by metabolic labeling experiments.13-15
Farnesyl–prelamin A accumulates in fibroblasts treated with lopinavir, an HIV-protease inhibitor that binds to ZMPSTE24 and blocks its catalytic activity.14,16 The inhibition of ZMPSTE24 by lopinavir might be relevant to the loss of adipose tissue that occurs in lopinavir-treated HIV patients.17-19 Of note, the farnesyl–prelamin A that accumulates in lopinavir-treated cells is distinct from the prelamin A that accumulates with a protein farnesyltransferase inhibitor or an inhibitor of HMG-CoA reductase (i.e., a statin); those drugs block protein farnesylation and lead to an accumulation of nonfarnesylated prelamin A.20,21
The fact that “progeroid syndrome” mutations that block the processing of farnesyl–prelamin A are accompanied by reduced adipose tissue inevitably raised the question of whether LMNA missense mutations causing FPLD also result in prelamin A accumulation. In recent years, several groups performed protein gel blots on FPLD fibroblasts with commercial antibodies and concluded that FPLD mutations (including the classic R482W mutation) are associated with an accumulation of prelamin A.6,22-24 These reports were intriguing, but we reasoned that caution was warranted. First, it was unclear why the LMNA mutations causing FPLD would affect prelamin A processing, given that the responsible amino acid substitutions are widely dispersed within lamin A/C and most are not located in sequences important for prelamin A processing. Second, we have learned to be cautious about commercial antibodies against lamin A/C and prelamin A because some lots of those antibodies bind nonspecifically to other proteins. For that reason, we generated 2 monoclonal antibodies against prelamin A; both are specific and sensitive in western blot and immunocytochemistry studies.25-28
In the current studies, we used a polyclonal antibody against lamin A/C and the prelamin A monoclonal antibodies to determine if prelamin A accumulates in FPLD fibroblasts. We also tested whether FPLD fibroblasts would manifest an exaggerated accumulation of prelamin A in the presence of lopinavir.
Methods
Fibroblasts from FPLD subjects
Fibroblasts were isolated by 2 of the authors (D.A.-V. and S.S.-I.) from skin biopsies of normal subjects and FPLD subjects with one of 4 LMNA mutations (R482W, C591F, T528M, I299V). This study was approved by the Ethics Review Panel of the Conselleria de Sanidade (Xunta de Galicia, Spain) and carried out according to the ethical guidelines of the Declaration of Helsinki. All of the patients provided informed consent for participation in the study and publication of their clinical, and genetic information. The identity of the fibroblasts was confirmed at UCLA by DNA sequencing. Fibroblasts were cultured in 5% CO2 at 37°C in Dulbecco's modified Eagle's medium (Gibco, Life Technologies) containing 10% fetal bovine serum (FBS; HyClone, Logan, UT), 2 mM glutamine, 10 units/ml penicillin, and 10 μg/ml streptomycin (Gibco). Most studies were performed on fibroblasts that had been seeded into 12-well plates. In some studies, the cells were treated with a protein farnesyltransferase inhibitor (FTI) (lonafarnib, 0.5 µM final concentration); an HIV protease inhibitor (lopinavir, 20 to 30 µM final concentration); or vehicle (DMSO) alone at 37°C for 72 h. Lopinavir was obtained from the National Institutes of Health AIDS Research and Reference Reagent Program; lonafarnib was provided by Schering-Plough, Kenilworth, NJ. In some studies, fibroblasts were grown in medium containing lopinavir (25 µM) and in medium containing from 0 to 50% FBS. In other studies, fibroblasts were grown in medium containing bovine serum albumin (BSA; 1–30 mg/ml), 3% FBS, and either an FTI (0.5 µM) or lopinavir (25 µM).
Western blots
Urea-soluble extracts of fibroblasts were prepared as described.29 Proteins were size-fractionated on 4–12% gradient polyacrylamide Bis-Tris gels (Invitrogen) and transferred to a nitrocellulose membrane. Western blots were performed with a goat polyclonal antibody against lamin A/C (1:400; sc-6215, Santa Cruz Biotechnology); a goat polyclonal antibody against actin (1:2,000; sc-1616, Santa Cruz Biotechnology); a rat monoclonal antibody (7G11) that binds to nonfarnesylated prelamin A; or a rat monoclonal antibody (3C8) that binds to both farnesylated and nonfarnesylated prelamin A. Antibody 3C8 was labeled directly with an infrared dye with the DyLight Antibody Labeling Kit 53026 (Thermo Scientific). Binding of polyclonal antibodies and antibody 7G11 was detected with a secondary antibody labeled with an infrared dye (Rockland Immunochemicals). The intensity of prelamin A bands, relative to lamin C or actin, was quantified with an Odyssey infrared scanner (Li-Cor).
We also assessed the effects of lopinavir on prelamin A accumulation in primary embryonic fibroblasts from wild-type mice and 2 littermates that were heterozygous for a knockout mutation in Zmpste24 (Zmpste24+/−).
Quantitative RT-PCR
RNA was isolated with RNeasy Mini Kit (QIAGEN) and treated with DNase I (Ambion, Life Technologies); it was then reverse transcribed with oligo(dT), random primers, and SuperScript III (Invitrogen). Quantitative RT-PCR (qPCR) reactions were performed on a 7900HT Fast Real-Time PCR system (Applied Biosystems) with SYBR Green PCR Master Mix (Bioline). Transcript levels were determined by the comparative cycle threshold method and normalized to GAPDH for human samples and cyclophilin A for mouse samples using the 2-ΔΔ CT method.30 Primers for human LMNA were 5′-ATGAGGATGGAGATGACCTGC-3′ and 5′-AGGCAGAAGAGCCAGAGGAGA-3′ (exons 10 and 11, respectively); primers for human ZMPSTE24 were 5′-GGAAGTTGGGACATACAGTCAAA-3′ and 5′-TCCTTTCGACCAATTAATACAGC-3′ (exons 8 and 9, respectively); another set of primers for human ZMPSTE24 was 5′-TATGAGATCACTCAGTCCCTGGT-3′ and 5′-AGTCTGTTGATTGAAGCCATGTT-3′ (exons 3/4 and 4/5, respectively). Primers for mouse Lmna were 5′-GGTTGAGGACAATGAGGATGA-3′ and 5′-TGAGCGCAGGTTGTACTCAG-3′ (exons 10 and 11, respectively). Primers for mouse Zmpste24 were 5′-ACTGGAAGTTGGGACACACAGTA-3′ and 5′- TTCCCTTCGACCAATTAATACAG-3′ (exons 8 and 9, respectively).
Results
We examined prelamin A levels in wild-type fibroblasts and fibroblasts from FPLD subjects carrying one of 4 LMNA mutations (R482W, C591F, T528M, I299V). The R482W mutation is associated with classic FPLD2 and has been encountered by multiple investigators.2,3,22,31 The C591F mutation causes an atypical FPLD accompanied by hypertrophic cardiomyopathy.32 A T528M mutation was encountered in 2 sisters with typical FPLD2; this mutation was identified previously in FPLD subjects (compound heterozygotes who also carried an S583L mutation)33 and in a subject with a progeroid syndrome (a compound heterozygote who also carried an M540T mutation).34 The I299V mutation was identified in an atypical FPLD subject (severe insulin resistance, an absence of adipose tissue in limbs and buttocks but increased subcutaneous adipose tissue and increased adipose tissue in the trunk).5
After verifying the identity of R482W, I299V, C591F, and T528M fibroblasts by DNA sequencing (Fig. 1), we used protein gel blots to determine if fibroblasts from 2 R482W FPLD subjects (L8, L160) contained more prelamin A than fibroblasts from a wild-type subject (C9). In the absence of lopinavir or a farnesyltransferase inhibitor (FTI; lonafarnib), prelamin A could not be detected in either wild-type or R482W fibroblasts, as judged by western blots with a polyclonal lamin A/C antibody (Fig. 2A–B) or 2 different prelamin A–specific monoclonal antibodies (3C8, 7G11) (Fig. 2C–D). When lopinavir was added to the medium, the amount of farnesyl–prelamin A was similar in wild-type and R482W fibroblasts (Fig. 2A–C). Antibody 7G11 is specific for nonfarnesylated prelamin A; consequently, it did not bind to prelamin A in lopinavir-treated fibroblasts (Fig. 2D). As expected, the electrophoretic mobility of the farnesyl–prelamin A in lopinavir-treated cells was slightly faster than that of nonfarnesylated prelamin A in FTI-treated cells (Fig. 2A–C).
Figure 1.
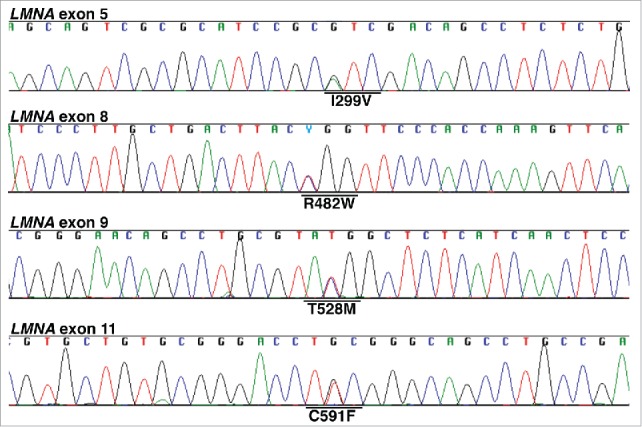
DNA sequencing chromatograms to verify the identity of R482W, T528M, C591F, and I299V fibroblasts. Sequencing chromatograms were obtained on cDNA amplicons generated from fibroblast RNA. All fibroblasts were heterozygous for the indicated LMNA mutation.
Figure 2.
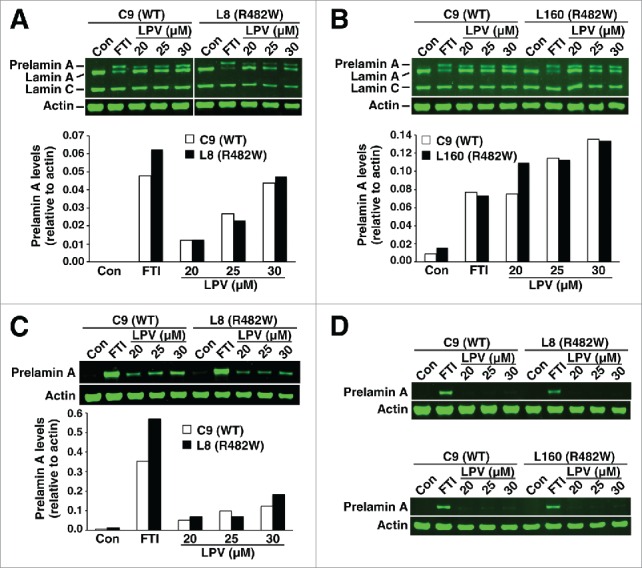
Western blots of cell extracts from wild-type (C9) and R482W fibroblasts (L8 and L160) with lamin A/C– and prelamin A–specific antibodies. Fibroblasts were grown in the absence of a drug; with a protein farnesyltransferase inhibitor (FTI); or with lopinavir (LPV), an HIV protease inhibitor that binds to ZMPSTE24 and inhibits catalytic activity. (A–B) Western blots with a polyclonal lamin A/C antibody. Bar graphs show the amount of prelamin A, relative to actin, as judged by an Odyssey infrared scanner. (C) Western blot with a prelamin A monoclonal antibody, 3C8, that recognizes both farnesylated and nonfarnesylated prelamin A. (D) Blot with a prelamin A monoclonal antibody, 7G11, that recognizes only nonfarnesylated prelamin A.
Similar results were observed in T528M and C591F fibroblasts (Fig. 3). In the absence of an FTI or lopinavir (Fig. 3), no prelamin A was found in T528M and C591F fibroblasts. Also, adding lopinavir to the medium did not lead to exaggerated accumulation of prelamin A (Fig. 3). Similar findings were observed in I299V fibroblasts (Fig. 4).
Figure 3.
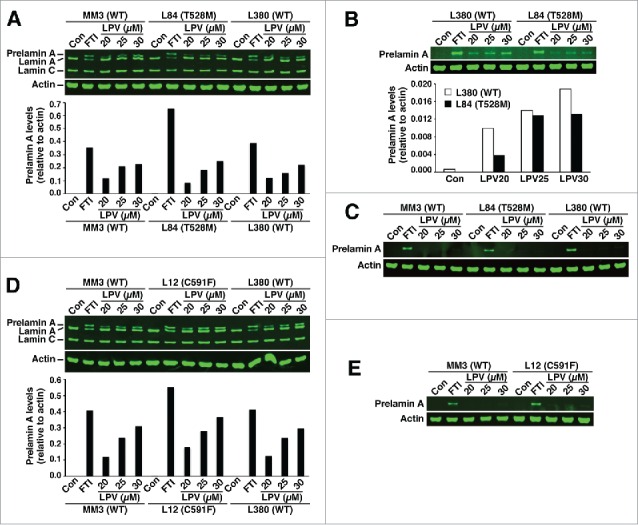
Western blots of cell extracts from wild-type fibroblasts (MM3 and L380) and FPLD fibroblasts T528M (L84) and C591F (L12). (A–C) Wild-type and T528M fibroblasts were grown without a drug or in the presence of a protein farnesyltransferase inhibitor (FTI) or lopinavir (LPV). (A) Western blot of fibroblast extracts with the lamin A/C polyclonal antibody. Bar graph shows the amount of prelamin A, relative to actin, as judged by an Odyssey infrared scanner. (B) Western blot of fibroblast extracts with the prelamin A–specific antibody 3C8. (C) Western blot with the prelamin A–specific antibody 7G11. (D–E) Wild-type and C591F fibroblasts were grown without a drug or in the presence of an FTI or LPV. Western blots of cell extracts were performed with the lamin A/C polyclonal antibody (D) or antibody 7G11(E).
Figure 4.
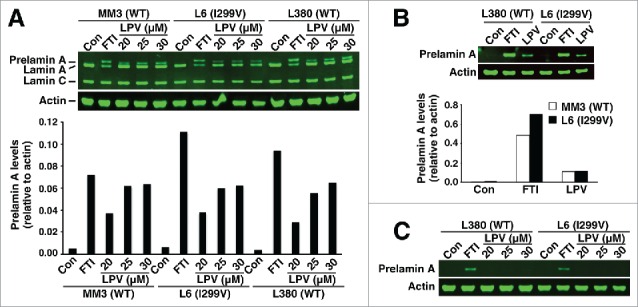
Western blots of cell extracts from wild-type fibroblasts (MM3 and L380) and I299V fibroblasts (L6). Wild-type and I299V fibroblasts were grown without a drug or in the presence of a protein farnesyltransferase inhibitor (FTI) or lopinavir (LPV). (A) Western blot of fibroblast extracts with the lamin A/C polyclonal antibody. Bar graph shows the amounts of prelamin A, relative to actin, as judged by an Odyssey infrared scanner. (B) Western blot of fibroblast extracts with antibody 3C8. The LPV concentration was 25 μM. Bar graph shows the amounts of prelamin A, relative to actin. (C) Western blot of fibroblast extracts with antibody 7G11.
A recent study pointed out that lopinavir is protein-bound and that the amount of prelamin A accumulation in peripheral blood mononuclear cells is reduced when the cells are cultured in a medium with high concentrations of protein.35 We obtained similar results in fibroblasts; increased amounts of fetal bovine serum in the medium reduced the amount of prelamin A accumulation during lopinavir treatment (Fig. S1). Adding bovine serum albumin (BSA) to the cell culture medium also reduced lopinavir-induced prelamin A accumulation (Fig. S2). In contrast, BSA had no effect on the accumulation of nonfarnesylated prelamin A in FTI-treated cells (Fig. S2).
The fact that prelamin A accumulation in response to lopinavir was similar in FPLD and wild-type fibroblasts implies that ZMPSTE24 activity is also similar in wild-type and mutant fibroblasts. If ZMPSTE24 expression had been reduced in FPLD fibroblasts, more prelamin A would have accumulated during lopinavir treatment. In control studies, we found that half-normal amounts of ZMPSTE24 expression in Zmpste24+/− fibroblasts is accompanied by twofold greater accumulation of prelamin A during lopinavir treatment (Fig. 5A–D). As expected, Zmpste24 transcripts were reduced by 50% in Zmpste24+/− fibroblasts (Fig. 5E). We also measured ZMPSTE24 transcript levels by qRT-PCR in wild-type and FPLD fibroblasts with 2 different oligonucleotide primer pairs; we observed no evidence for reduced ZMPSTE24 expression in FPLD fibroblasts (Fig. 6A–B). Similarly, there appeared to be no effect of the FPLD mutations on prelamin A transcript levels (Fig. 6C).
Figure 5.

Prelamin A accumulation in wild-type mouse fibroblasts (Zmpste24+/+) and heterozygous Zmpste24 knockout fibroblasts (Zmpste24+/−) in response to lopinavir treatment. (A–B) Western blots, using a lamin A/C polyclonal antibody, of extracts from Zmpste24+/+ fibroblasts (E12, E17) and Zmpste24+/− fibroblasts (E11, E15) after lopinavir treatment. In these studies, Zmpste24−/− fibroblasts were included as controls (KO). (C–D) Bar graphs show amounts of prelamin A, relative to lamin C, in fibroblasts E12 and E11 (C) and in fibroblasts E17 and E15 (D). (E) Half-normal levels of Zmpste24 transcripts in Zmpste24+/− fibroblasts (E11, E15), compared with Zmpste24+/+ fibroblasts (E12, E17). As controls, we included 2 different Zmpste24−/− fibroblast cell lines (E10, E14).
Figure 6.
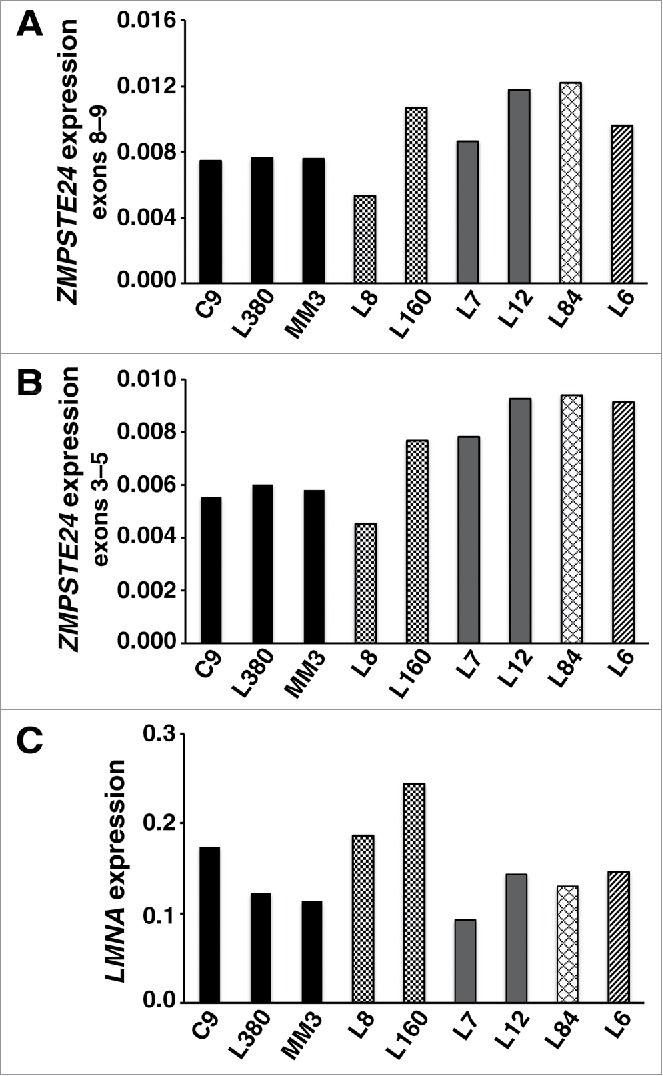
Transcript levels in wild-type fibroblasts and fibroblasts from FPLD subjects. (A–B) ZMPSTE24 transcript levels, as judged by qRT-PCR with 2 primer pairs (exons 8–9 and exons 3–5). (C) LMNA transcript levels with a primer pair that detects prelamin A transcripts. Wild-type fibroblasts: C9, L380, MM3. R482W fibroblasts: L8, L160. C591F fibroblasts: L7, L12. T528M fibroblasts: L84. I299V fibroblasts: L6.
Discussion
Rare mutations underlying progeroid syndromes, for example the LMNA mutation causing HGPS and ZMPSTE24 missense mutations that impair ZMPSTE24 activity, interfere directly with the conversion of farnesyl–prelamin A to lamin A.7,10-12 In those cases, the mechanisms for prelamin A accumulation are straightforward and make perfect sense. Several publications have suggested that the LMNA mutations causing FPLD, including the classic R482W mutation, also cause an accumulation of prelamin A—and that this accumulation is evident in cultured fibroblasts.6,22-24 Prelamin A accumulation in the setting of FPLD mutations is more difficult to understand, given that most of those mutations are located in sequences distant from those important for prelamin A processing. In the current study, we revisited whether FPLD mutations lead to prelamin A accumulation in fibroblasts. We examined fibroblasts from subjects with 4 LMNA mutations associated with lipodystrophy, including the classic R482W mutation, and found no evidence for prelamin A accumulation. Our conclusions were based on protein gel blots with a polyclonal lamin A/C antibody and a pair of prelamin A–specific monoclonal antibodies. The western blots were conducted with methods that allowed us to discern differences in the mobilities of nonfarnesylated and farnesylated prelamin A.14,16
We do not understand why earlier studies reached the conclusion that FPLD fibroblasts, including R482W fibroblasts, had an accumulation of prelamin A. One possibility is nonspecific binding of antibodies from commercial vendors. Additional formal possibilities include differences in the site of the skin biopsy, differences in cell passage, and differences in the components of the medium, although one would have to propose that those differences selectively affected FPLD fibroblasts. In the current studies, we utilized both lamin A/C polyclonal antibodies and 2 different prelamin A–specific monoclonal antibodies.
An earlier study suggested that FPLD cells have reduced ZMPSTE24 expression.23 That finding was intriguing, but it was unclear why lamin A/C missense mutations would influence ZMPSTE24 expression. In the current study, we found no evidence for reduced ZMPSTE24 expression in FPLD fibroblasts. The levels of ZMPSTE24 transcripts were not reduced in FPLD fibroblasts, nor was prelamin A accumulation exaggerated during lopinavir treatment. Had ZMPSTE24 activity been reduced in FPLD fibroblasts, we would have observed an exaggerated response to lopinavir. When Zmpste24+/− fibroblasts were treated with lopinavir, they exhibited twofold more prelamin A accumulation.
In earlier studies finding prelamin A accumulation in FPLD cells, the assumption was that the prelamin A was farnesylated.22,23 The prelamin A that accumulates in the setting of HGPS or restrictive dermopathy (ZMPSTE24 deficiency) is definitely farnesylated, as judged by metabolic labeling.13-15 Of note, progeroid disorders similar to HGPS can be caused by LMNA missense mutations that do not disrupt prelamin A processing.34,36 For example, R644C and E578V mutations in LMNA cause progeroid syndromes without causing prelamin A accumulation.36,37 In the case of R644C fibroblasts, lopinavir treatment did not lead to an exaggerated accumulation of prelamin A (not shown). Also, it is interesting that a FPLD syndrome occurs in the setting of a prelamin A truncation mutation that eliminates any possibility of prelamin A farnesylation.38
One limitation of our study is that we examined cultured fibroblasts and not adipose tissue from human subjects. Another is that the FPLD cell lines described in the literature were not available to us for study. In any case, we are confident in our findings with 4 different LMNA mutations linked to lipodystrophy, and we hope that our observations prompt additional studies. If future studies were to uncover a FPLD mutation causing substantial prelamin A accumulation, it would be very interesting to define the underlying mechanisms.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was funded by the National Heart, Lung, and Blood Institute (HL089781 to L.G.F. and HL126551 to S.G.Y.) and by the Instituto de Salud Carlos III (PI081449) and the European Regional Development Fund FEDER (D.A.-V and S.S.-I).
References
- [1].Cao H, Hegele RA. Nuclear lamin A/C R482Q mutation in canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum Mol Genet 2000; 9:109-12; PMID:10587585; http://dx.doi.org/ 10.1093/hmg/9.1.109 [DOI] [PubMed] [Google Scholar]
- [2].Shackleton S, Lloyd DJ, Jackson SN, Evans R, Niermeijer MF, Singh BM, Schmidt H, Brabant G, Kumar S, Durrington PN, et al.. LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat Genet 2000; 24:153-6; PMID:10655060; http://dx.doi.org/ 10.1038/72807 [DOI] [PubMed] [Google Scholar]
- [3].Speckman RA, Garg A, Du F, Bennett L, Veile R, Arioglu E, Taylor SI, Lovett M, Bowcock AM. Mutational and haplotype analyses of families with familial partial lipodystrophy (Dunnigan variety) reveal recurrent missense mutations in the globular C-terminal domain of lamin A/C. Am J Hum Genet 2000; 66:1192-8; PMID:10739751; http://dx.doi.org/ 10.1086/302836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lanktree M, Cao H, Rabkin SW, Hanna A, Hegele RA. Novel LMNA mutations seen in patients with familial partial lipodystrophy subtype 2 (FPLD2; MIM 151660). Clinical Genetics 2007; 71:183-6; PMID:17250669; http://dx.doi.org/ 10.1111/j.1399-0004.2007.00740.x [DOI] [PubMed] [Google Scholar]
- [5].Araujo-Vilar D, Victoria B, Gonzalez-Mendez B, Barreiro F, Fernandez-Rodriguez B, Cereijo R, Gallego-Escuredo JM, Villarroya F, Paneda-Menendez A. Histological and molecular features of lipomatous and nonlipomatous adipose tissue in familial partial lipodystrophy caused by LMNA mutations. Clinical Endocrinology 2012; 76:816-24; PMID:21883346; http://dx.doi.org/ 10.1111/j.1365-2265.2011.04208.x [DOI] [PubMed] [Google Scholar]
- [6].Caron M, Auclair M, Donadille B, Bereziat V, Guerci B, Laville M, Narbonne H, Bodemer C, Lascols O, Capeau J, et al.. Human lipodystrophies linked to mutations in A-type lamins and to HIV protease inhibitor therapy are both associated with prelamin A accumulation, oxidative stress and premature cellular senescence. Cell Death Differ 2007; 14:1759-67; PMID:17612587; http://dx.doi.org/ 10.1038/sj.cdd.4402197 [DOI] [PubMed] [Google Scholar]
- [7].Merideth MA, Gordon LB, Clauss S, Sachdev V, Smith AC, Perry MB, Brewer CC, Zalewski C, Kim HJ, Solomon B, et al.. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med 2008; 358:592-604; PMID:18256394; http://dx.doi.org/ 10.1056/NEJMoa0706898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, et al.. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003; 423:293-8; PMID:12714972; http://dx.doi.org/ 10.1038/nature01629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Young SG, Fong LG, Michaelis S. Prelamin A, Zmpste24, misshapen cell nuclei, and progeria—New evidence suggesting that protein farnesylation could be important for disease pathogenesis. J Lipid Res 2005; 46:2531-58; PMID:16207929; http://dx.doi.org/ 10.1194/jlr.R500011-JLR200 [DOI] [PubMed] [Google Scholar]
- [10].Wang Y, Lichter-Konecki U, Anyane-Yeboa K, Shaw JE, Lu JT, Ostlund C, Shin JY, Clark LN, Gundersen GG, Nagy PL, et al.. A mutation abolishing the ZMPSTE24 cleavage site in prelamin A causes a progeroid disorder. J Cell Sci 2016; 129:1975-80; PMID:27034136; http://dx.doi.org/ 10.1242/jcs.187302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Shackleton S, Smallwood DT, Clayton P, Wilson LC, Agarwal AK, Garg A, Trembath RC. Compound heterozygous ZMPSTE24 mutations reduce prelamin A processing and result in a severe progeroid phenotype. J Med Genet 2005; 42:e36; PMID:15937076; http://dx.doi.org/ 10.1136/jmg.2004.029751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Barrowman J, Wiley PA, Hudon-Miller SE, Hrycyna CA, Michaelis S. Human ZMPSTE24 disease mutations: residual proteolytic activity correlates with disease severity. Hum Mol Genet 2012; 21:4084-9403; PMID:22718200; http://dx.doi.org/ 10.1093/hmg/dds233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yang SH, Chang SY, Ren S, Wang Y, Andres DA, Spielmann HP, Fong LG, Young SG. Absence of progeria-like disease phenotypes in knock-in mice expressing a non-farnesylated version of progerin. Hum Mol Genet 2011; 20:436-44; PMID:21088111; http://dx.doi.org/ 10.1093/hmg/ddq490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Coffinier C, Hudon SE, Lee R, Farber EA, Nobumori C, Miner JH, Andres DA, Spielmann HP, Hrycyna CA, Fong LG, et al.. A potent HIV protease inhibitor, darunavir, does not inhibit ZMPSTE24 or lead to an accumulation of farnesyl-prelamin A in cells. J Biol Chem 2008; 283:9797-804; PMID:18230615; http://dx.doi.org/ 10.1074/jbc.M709629200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Dechat T, Shimi T, Adam SA, Rusinol AE, Andres DA, Spielmann HP, Sinensky MS, Goldman RD. Alterations in mitosis and cell cycle progression caused by a mutant lamin A known to accelerate human aging. Proc Natl Acad Sci USA 2007; 104:4955-60; PMID:17360326; http://dx.doi.org/ 10.1073/pnas.0700854104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Coffinier C, Hudon SE, Farber EA, Chang SY, Hrycyna CA, Young SG, Fong LG. HIV protease inhibitors block the zinc metalloproteinase ZMPSTE24 and lead to an accumulation of prelamin A in cells. Proc Natl Acad Sci U S A 2007; 104:13432-7; PMID:17652517; http://dx.doi.org/ 10.1073/pnas.0704212104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gutierrez F, Padilla S, Navarro A, Masia M, Hernandez I. Lipid abnormalities in HIV-Infected patients and lopinavir plasma concentrations. J Acquir Immune Defic Syndr 2004; 36:1107-9; PMID:15247567; http://dx.doi.org/ 10.1097/00126334-200408150-00017 [DOI] [PubMed] [Google Scholar]
- [18].Martinez E, Domingo P, Galindo MJ, Milinkovic A, Arroyo JA, Baldovi F, Larrousse M, Leon A, de Lazzari E, Gatell JM. Risk of metabolic abnormalities in patients infected with HIV receiving antiretroviral therapy that contains lopinavir-ritonavir. Clin Infect Dis 2004; 38:1017-23; PMID:15034836; http://dx.doi.org/ 10.1086/382531 [DOI] [PubMed] [Google Scholar]
- [19].Noor MA, Parker RA, O'Mara E, Grasela DM, Currie A, Hodder SL, Fiedorek FT, Haas DW. The effects of HIV protease inhibitors atazanavir and lopinavir/ritonavir on insulin sensitivity in HIV-seronegative healthy adults. AIDS 2004; 18:2137-44; PMID:15577646; http://dx.doi.org/ 10.1097/00002030-200411050-00005 [DOI] [PubMed] [Google Scholar]
- [20].Beck LA, Hosick TJ, Sinensky M. Isoprenylation is required for the processing of the lamin A precursor. J Cell Biol 1990; 110:1489-99; PMID:2335559; http://dx.doi.org/ 10.1083/jcb.110.5.1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dalton MB, Fantle KS, Bechtold HA, DeMaio L, Evans RM, Krystosek A, Sinensky M. The farnesyl protein transferase inhibitor BZA-5B blocks farnesylation of nuclear lamins and p21ras but does not affect their function or localization. Cancer Res 1995; 55:3295-304; PMID:7614464 [PubMed] [Google Scholar]
- [22].Bidault G, Garcia M, Vantyghem MC, Ducluzeau PH, Morichon R, Thiyagarajah K, Moritz S, Capeau J, Vigouroux C, Bereziat V. Lipodystrophy-linked LMNA p.R482W mutation induces clinical early atherosclerosis and in vitro endothelial dysfunction. Arterioscler Thromb Vasc Biol 2013; 33:2162-71; PMID:23846499; http://dx.doi.org/ 10.1161/ATVBAHA.113.301933 [DOI] [PubMed] [Google Scholar]
- [23].Afonso P, Auclair M, Boccara F, Vantyghem MC, Katlama C, Capeau J, Vigouroux C, Caron-Debarle M. LMNA mutations resulting in lipodystrophy and HIV protease inhibitors trigger vascular smooth muscle cell senescence and calcification: Role of ZMPSTE24 downregulation. Atherosclerosis 2016; 245:200-211; PMID:26724531; http://dx.doi.org/ 10.1016/j.atherosclerosis.2015.12.012 [DOI] [PubMed] [Google Scholar]
- [24].Capanni C, Mattioli E, Columbaro M, Lucarelli E, Parnaik VK, Novelli G, Wehnert M, Cenni V, Maraldi NM, Squarzoni S, et al.. Altered pre-lamin A processing is a common mechanism leading to lipodystrophy. Hum Mol Genet 2005; 14:1489-502; PMID:15843404; http://dx.doi.org/ 10.1093/hmg/ddi158 [DOI] [PubMed] [Google Scholar]
- [25].Coffinier C, Jung HJ, Li Z, Nobumori C, Yun UJ, Farber EA, Davies BS, Weinstein MM, Yang SH, Lammerding J, et al.. Direct synthesis of lamin A, bypassing prelamin a processing, causes misshapen nuclei in fibroblasts but no detectable pathology in mice. J Biol Chem 2010; 285:20818-26; PMID:20439468; http://dx.doi.org/ 10.1074/jbc.M110.128835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Davies BS, Barnes RH 2nd, Tu Y, Ren S, Andres DA, Spielmann HP, Lammerding J, Wang Y, Young SG, Fong LG. An accumulation of non-farnesylated prelamin A causes cardiomyopathy but not progeria. Hum Mol Genet 2010; 19:2682-94; PMID:20421363; http://dx.doi.org/ 10.1093/hmg/ddq158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chang SY, Hudon-Miller SE, Yang SH, Jung HJ, Lee JM, Farber E, Subramanian T, Andres DA, Spielmann HP, Hrycyna CA, et al.. Inhibitors of protein geranylgeranyltransferase-I lead to prelamin A accumulation in cells by inhibiting ZMPSTE24. J Lipid Res 2012; 53:1176-82; PMID:22448028; http://dx.doi.org/ 10.1194/jlr.M026161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jung HJ, Coffinier C, Choe Y, Beigneux AP, Davies BS, Yang SH, Barnes RH 2nd, Hong J, Sun T, Pleasure SJ, et al.. Regulation of prelamin A but not lamin C by miR-9, a brain-specific microRNA. Proc Natl Acad Sci U S A 2012; 109:E423-431; PMID:22308344; http://dx.doi.org/ 10.1073/pnas.1111780109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Fong LG, Ng JK, Meta M, Cote N, Yang SH, Stewart CL, Sullivan T, Burghardt A, Majumdar S, Reue K, et al.. Heterozygosity for Lmna deficiency eliminates the progeria-like phenotypes in Zmpste24-deficient mice. Proc Natl Acad Sci USA 2004; 101:18111-6; PMID:15608054; http://dx.doi.org/ 10.1073/pnas.0408558102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 2008; 3:1101-8; PMID:18546601; http://dx.doi.org/ 10.1038/nprot.2008.73 [DOI] [PubMed] [Google Scholar]
- [31].Araujo-Vilar D, Loidi L, Dominguez F, Cabezas-Cerrato J. Phenotypic gender differences in subjects with familial partial lipodystrophy (Dunnigan variety) due to a nuclear lamin A/C R482W mutation. Hormone and Metabolic Research 2003; 35:29-35; PMID:12669268; http://dx.doi.org/ 10.1055/s-2003-38388 [DOI] [PubMed] [Google Scholar]
- [32].Araujo-Vilar D, Lado-Abeal J, Palos-Paz F, Lattanzi G, Bandin MA, Bellido D, Dominguez-Gerpe L, Calvo C, Perez O, Ramazanova A, et al.. A novel phenotypic expression associated with a new mutation in LMNA gene, characterized by partial lipodystrophy, insulin resistance, aortic stenosis and hypertrophic cardiomyopathy. Clin Endocrinol 2008; 69:61-8; PMID:18031308; http://dx.doi.org/ 10.1111/j.1365-2265.2007.03146.x [DOI] [PubMed] [Google Scholar]
- [33].Savage DB, Soos MA, Powlson A, O'Rahilly S, McFarlane I, Halsall DJ, Barroso I, Thomas EL, Bell JD, Scobie I, et al.. Familial partial lipodystrophy associated with compound heterozygosity for novel mutations in the LMNA gene. Diabetologia 2004; 47:753-6; PMID:15298354; http://dx.doi.org/ 10.1007/s00125-004-1360-4 [DOI] [PubMed] [Google Scholar]
- [34].Verstraeten VL, Broers JL, van Steensel MA, Zinn-Justin S, Ramaekers FC, Steijlen PM, Kamps M, Kuijpers HJ, Merckx D, Smeets HJ, et al.. Compound heterozygosity for mutations in LMNA causes a progeria syndrome without prelamin A accumulation. Hum Mol Genet 2006; 15:2509-22; PMID:16825282; http://dx.doi.org/ 10.1093/hmg/ddl172 [DOI] [PubMed] [Google Scholar]
- [35].Perrin S, Cremer J, Faucher O, Reynes J, Dellamonica P, Micallef J, Solas C, Lacarelle B, Stretti C, Kaspi E, et al.. HIV protease inhibitors do not cause the accumulation of prelamin A in PBMCs from patients receiving first line therapy: the ANRS EP45 “aging” study. PLoS One 2012; 7:e53035; PMID:23285253; http://dx.doi.org/ 10.1371/journal.pone.0053035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Csoka AB, Cao H, Sammak PJ, Constantinescu D, Schatten GP, Hegele RA. Novel lamin A/C gene (LMNA) mutations in atypical progeroid syndromes. J Med Genet 2004; 41:304-8; PMID:15060110; http://dx.doi.org/ 10.1136/jmg.2003.015651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Toth JI, Yang SH, Qiao X, Beigneux AP, Gelb MH, Moulson CL, Miner JH, Young SG, Fong LG. Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc Natl Acad Sci USA 2005; 102:12873-8; PMID:16129834; http://dx.doi.org/ 10.1073/pnas.0505767102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Le Dour C, Schneebeli S, Bakiri F, Darcel F, Jacquemont ML, Maubert MA, Auclair M, Jeziorowska D, Reznik Y, Bereziat V, et al.. A homozygous mutation of prelamin-A preventing its farnesylation and maturation leads to a severe lipodystrophic phenotype: new insights into the pathogenicity of nonfarnesylated prelamin-A. J Clin Endocrinol Metab 2011; 96:E856-862; PMID:21346069; http://dx.doi.org/ 10.1210/jc.2010-2234 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.