

Abstract
Mitochondrial encephalomyopathy with lactic acid and stroke-like episodes (MELAS) is a multisystem mitochondrial disorder that typically presents in childhood. We describe the follow-up of a patient who was diagnosed with late-onset MELAS at the age of 49. Her clinical course includes sensorineural hearing loss, seizures, and multiple episodes of stroke-like metabolic crises. Molecular genetic testing on whole blood revealed 31% heteroplasmy of a m.3243A > G variant in the mtDNA, the causative variant in approximately 80% of MELAS cases. The original diagnostic criteria for MELAS required the onset of stroke-like episodes prior to 40 years of age but this case and others demonstrate that onset may be delayed in certain individuals. Therefore, MELAS should be included in the differential diagnosis of stroke-like episodes in patients of any age.
Abbreviations: MELAS, Mitochondrial encephalomyopathy with lactic acid and stroke-like episodes; mtDNA, Mitochondrial DNA
1. Introduction
MELAS is one of the most common matrilineal mitochondrial disorders [1]. Though clinically heterogeneous, MELAS classically manifests as stroke-like episodes in the parietal, temporal, or occipital lobes that can result in hemianopia, hemiparesis, and progressive neurodegeneration [1]. Other common clinical features of this multisystem disorder include short stature, sensorineural hearing loss, seizures, external ophthalmoplegia, headaches, dementia, encephalopathy, cardiomyopathy, nausea, vomiting, exercise intolerance, lactic acidosis, limb muscle weakness, and ataxia [2], [3].
The typical course of MELAS is characterized by normal development interrupted by episodes of neurologic dysfunction beginning in childhood, most commonly before 20 years of age. Thus, MELAS is an important differential diagnosis to stroke in young patients [3], [4]. The disorder progresses in a relapsing-remitting pattern, with acute neurometabolic crises overlaying a progressive neurological decline, often with dementia.
Currently, over 30 pathogenic variants in mitochondrial DNA (mtDNA) are reported to cause MELAS [5], [6], [7]. The most common variant, accounting for over 80% of MELAS cases [8], is the mitochondrial m.3243A > G variant. This point mutation in the MT-TL1 gene results in a defective mitochondrial transfer RNA for leucine. The consequent disruption of mitochondrial protein translation leads to impaired respiratory chain function which causes reduced ATP production via aerobic metabolism. As is typical of mitochondrial disorders, MELAS severely affects organ systems with the highest energy demands, including the brain and skeletal muscle [9]. The pathologic mechanism of stroke-like episodes in MELAS has not been fully elucidated but recent investigations have pointed to a small vessel angiopathy resulting from endothelial cell mitochondrial dysfunction.
The traditional criteria for a clinical diagnosis of MELAS are as follows: (1) a stroke-like episode prior to age 40 years; (2) seizures and/or dementia, constituting encephalopathy, and (3) lactic acidosis and/or ragged-red fibers on muscle biopsy [10]. However, these criteria do not offer a complete picture of the broad spectrum of presentations that have been identified, most notably late onset after age 40 [2], [3], [5], [6]. Clinical presentations suggestive of a MELAS warrant pursuit of molecular testing [6], [7]. In patients with a history of stroke-like episodes, characteristic lesions may be visible on brain imaging and may help distinguish MELAS from other causes of cerebral ischemia. If molecular studies in blood do not reveal any causal pathogenic variants there is the possibility of false negative results. If clinical suspicion remains high additional testing on other tissues such as skin fibroblasts or muscle can be considered [7]. Current guidelines do not recommend invasive testing such as skeletal muscle biopsy unless genetic testing is inconclusive [6], [7].
Once diagnosed, MELAS requires multi-specialist management. Treatments center on support of mitochondrial function with vitamin and antioxidant supplementation, prevention of acute neurological deterioration (rapid reduction in consciousness level that is unexplained, such as stroke-like episodes), progressive neurodegeneration, anticonvulsive prophylaxis, and symptom management [6], [7]. In some centers, acute neurologic decompensation is managed with intravenous glucose and l-arginine, although data is conflicting whether this is efficacious [6], [7], [11], [12], [13]. Long-term supplementation with l-arginine and l-citrulline may increase nitric oxide production, improve endothelial function, maintain cerebral blood flow, and reduce the incidence and severity of stroke-like episodes [7]. However, the evidence base for such treatment regimens remains insufficient [6], [7].
The majority of patients with MELAS experience symptom onset before the age of 20 years, and presentation after age 40 is rare [2], [5], [7]. A review of the literature reveals limited published cases of late-onset MELAS. Patients carrying novel mitochondrial DNA point mutations m.3256C > T, m.4332G > A, and m.13635C > A have presented with a stroke-like episode at the ages of 36, 47, and 55 years, respectively [4], [5], [14]. More pertinent to the present case are the few reports of patients with the classic m.3243A > G variant presenting after the age of 40 years. Such reports include a 50-year-old man with diabetes and deafness who presented with headache, aphasia, psychiatric symptoms, and seizures [3]; a 53-year-old man who presented with a stroke-like episode characterized by hallucination and delusion, muscle weakness, and loss of consciousness [1]; A 55-year-old woman who developed aphasia and delirium in conjunction with herpes encephalitis [15]; and a 60-year-old man who presented with stroke-like episodes and encephalopathy [4]. Onset of disease can be precipitated by febrile illness or injury in cases of late onset disease [6]. The level of heteroplasmy in blood (and other tissues including skin fibroblasts and muscle) for the m.3243A > G variant has been correlated with the severity and presentation of disease [16]. When there is 50–90% mutant mtDNA heteroplasmy the most typical presentation is classic MELAS syndrome; however, lower heteroplasmy levels are associated with autism, hearing loss, and diabetes [17]. At very high levels of mutant mtDNA heteroplasmy in blood, patients can present with Leigh syndrome and early lethality [6], [17].
Here we describe a case of a late-onset MELAS with stroke-like episodes and seizures associated with the m.3243A > G mutation. We also review the pertinent clinical characteristics, current recommendations for management of these patients, and summarize current understanding of the pathobiology of disease.
2. Case report
Our patient was diagnosed with MELAS at 49 years of age. Her first stroke-like episode was a transient confusional state with two convulsions during which she lost consciousness, occurring at 49 years of age. Additional episodes were characterized by transient aphasia, disorientation, and combativeness. Approximately three months later, she presented with a generalized tonic-clonic seizure and multiple confusional spells with preserved consciousness. Subsequent workup raised suspicion for MELAS, and she was referred to the genetics clinic. Her clinical course is notable for sensorineural hearing loss, generalized tonic-clonic seizures, and four stroke-like episodes between the ages of 49 and 50 years. These acute events resulted in mild neurologic deficits, predominantly involving language fluency.
The patient reported normal prenatal development, a full-term delivery, and normal birth weight and length, with no concerns for her development in childhood or young adulthood. She first noted hearing loss in her mid-30s that progressed symmetrically. Currently, she wears bilateral hearing aids and uses a combination of verbal and sign language to communicate. Her hearing loss is progressive and thought due to be purely sensorineural with no cortical involvement.
Genetic testing of 16 of the most common mutations in mitochondrial DNA (ARMS qPCR analysis by GeneDx) revealed a m.3243A > G transition in the MT-TL1 gene. Heteroplasmy of the blood sample was approximated at 31%, confirming a clinical diagnosis of MELAS.
Family history for hearing loss was significant in the proband's mother, older brother, and maternal aunt, but none of these relatives have experienced seizures or stroke-like episodes. Both of the patient's grandmothers died of strokes in their 70s–80s.
Shortly after her diagnosis, the patient experienced another acute neurologic decompensation (stroke-like episode). She was hospitalized and treated with intravenous glucose and l-arginine. A head computed topography (CT) scan revealed areas of hypodensity in the cortex and subcortex of the left occipital lobe consistent with ischemic damage (see Fig. 1). Magnetic resonance imaging (MRI) of the brain during the subsequent asymptomatic phase demonstrated left occipital lobe cystic encephalomalacia and white matter gliosis, bilateral temporal lobe cortical and subcortical abnormalities, and small vessel ischemic damage in the corona radiata. There were no areas of restricted diffusion or evidence of acute infarct. These findings are all consistent with MELAS. Antiepileptic prophylaxis with levetiracetam, phenytoin, and lamotrigine was initiated and effectively reduced seizure activity but caused excessive sedation and confusion. Tapering the doses of these medications reduced side effects while maintaining remission from seizures. Long-term neurological sequelae were limited to difficulties with language, especially word-finding.
Fig. 1.
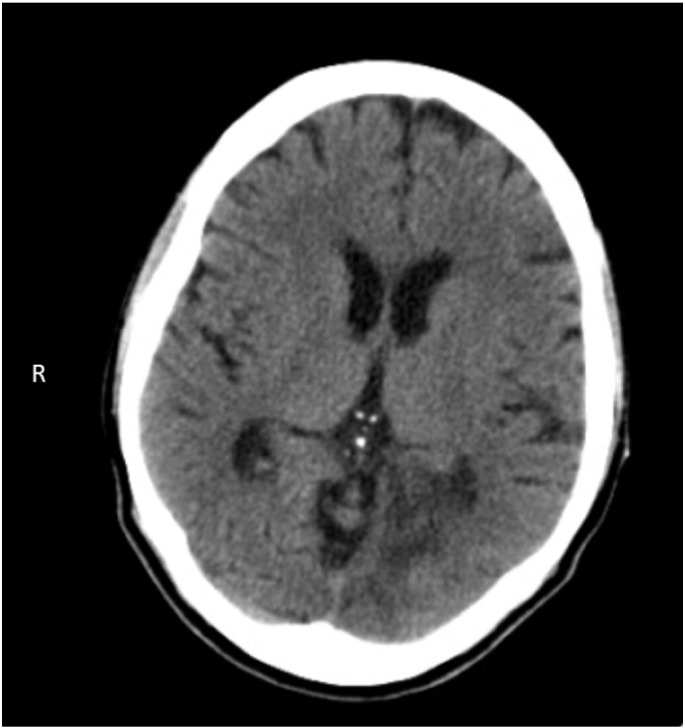
CT head after initial symptoms at age 49 demonstrating left occipital cortical and subcortical hypointensity consistent with a possible stroke-like event.
Additional pertinent findings in the patient's medical history include vision loss with a history of ocular histoplasmosis, hypothyroidism (diagnosed at age 30 years), nausea, and migraines. She has no history of diabetes. Electrocardiogram and echocardiogram revealed no evidence of cardiomyopathy secondary to mitochondrial dysfunction.
The patient's condition is well managed on multiple pharmaceutical therapies. Treatments aimed at supporting mitochondrial energy production include supplementation with l-arginine (5000 mg bid), ubiquinone (coenzyme Q10) (800 mg bid), vitamin B12 (1000 mg qd), vitamin C (1000 mg bid), vitamin E (400 mg bid), calcium (1200 mg qd) with vitamin D. She also takes levothyroxine (50 mg qd) for hypothyroidism. Antiepileptic drug therapy includes lamotrigine (250 mg bid), levetiracetam (2000 mg bid), phenytoin (tid: 50 mg; 100 mg; 200 mg), and clonazapan (1 mg qd). Current doses are well tolerated and the patient has experienced no breakthrough seizures or strokes on this regimen. Previous treatment of hyperlipidemia with statin therapy was discontinued due to the documented effect of statins decreasing endogenous synthesis of coenzyme Q10 [18]. Cerebral vasculature imaging has been reassuring, mitigating concerns for the risk of atherosclerotic stroke. The patient is advised to avoid valproate and to exercise caution if anesthesia or antibiotics are indicated, as these drug classes may interfere with mitochondrial function.
At 54 years of age, the patient is in good overall physical health and remains highly active, including an hour-long daily yoga practice. She has not had any further major events. She has no impairment in her basic or instrumental activities of daily living, though she reports difficulty with word finding, name recall, imbalance with quick turns, generalized fatigue, irritability, and depression, which she feels is related to her hearing loss. She scores one point on the NIH Stroke Scale for language (reduced verbal fluency) but is otherwise cognitively stable. Latest physical exam demonstrates a well-appearing female who is short-statured (154 cm), slender (weight, 46.5 kg and BMI, 19.6 kg/m2), and normally proportioned. She has with mild diffusely decreased muscle bulk (4/5) and weakness in the deltoid and iliopsoas muscle bilaterally. Heel-toe and tandem walking are normal, Romberg sign is negative. She could do a single unsupported squat and rise but appeared to require modification with excessive flexing of the hip to accomplish without support. Ophthalmology exam reveals normal confrontation visual field, motility and ductions however also showed pigmentary mottling in the left and a central scar in the right eye. Most recent labs demonstrate normal levels of AST/ALT, random glucose and creatinine. She receives routine follow-up with neurology, audiology, and ophthalmology along with regular blood work to monitor carnitine and lactate levels. She consults with genetics as needed for updates on new therapies and clinical trials. She wears a MedicAlert bracelet and carries a letter stating she has MELAS.
The patient has two adult children, both of whom have been counseled on the risk of MELAS that they have presumably inherited from their mother. Neither has sought genetic testing to determine the rate of heteroplasmy in their mtDNA. One has reported hearing loss and fatigue, while the other remains healthy. They have been advised to carry an emergency note explaining their family history in the event of an acute decompensation.
3. Discussion
Onset of stroke-like episodes in MELAS varies greatly but typically occurs in childhood. About 70% of individuals present before age 20 years and over 90% present before age 40 [6], [7]. We have described the case of a woman with genetically confirmed MELAS who presented with her first stroke-like episode at the age of 49. Aside from the late age of onset, her clinical course is classic for MELAS, including hearing loss, stroke-like episodes, and seizures.
Of particular interest in current discussions of MELAS is the patho-mechanism of the stroke-like episodes. Continuing elucidation of this mechanism has provided for development of effective new treatments. Two complementary theories predominate in the literature are metabolic and ischemic. First, mitochondrial dysfunction, particularly impaired neuronal oxidative energy production, results in a high degree of nonoxidative glycolysis, as indicated by elevated lactate in the serum and cerebrospinal fluid of MELAS patients [9], [18]. Second, cerebral hypoperfusion results in hypoxemic damage to neurons, which are particularly susceptible to ischemic insults.
The impaired cerebral perfusion, in turn, has two putative causes. The first is a small vessel angiopathy due to compensatory mitochondrial proliferation in the endothelium and vascular smooth muscle. The second is reduced production of the vasodilator nitric oxide. This may result from reduced activity of nitric oxide synthase (NOS), an enzyme that catalyzes conversion of l-arginine to nitric oxide [6], [7], [11]. Alternatively, deficiency of the nitric oxide precursor's l-citrulline and l-arginine has been observed in MELAS. Supplementation with l-citrulline and l-arginine is hypothesized to increase nitric oxide production, improve endothelial function and cerebral blood flow, and reduce the incidence and/or severity of stroke-like episodes [11], [13].
Like other mitochondrial disorders, MELAS poses a diagnostic challenge. It is characterized by multisystem involvement with a broad range of possible clinical manifestations, many of which are non-specific. Indeed, acute presentations of MELAS can be mistaken for vascular strokes, epilepsy, encephalopathy, infection, psychosis, and other neurodegenerative disorders [19]. Rarely does the patient's first presentation exhibit the complete phenotype of their disease burden; it may take decades to fully develop [9]. Moreover, MELAS demonstrates significant clinical heterogeneity even among individuals carrying the same mutation or within the same family. Expansion of the phenotype to include onset after 40 years makes it important to have an index of suspicion even in older individuals.
A diagnosis of MELAS should be considered in the setting of atypical strokes, even in older patients [6], [7], [16]. Particularly suspicious are regions of ischemic damage traversing multiple cerebrovascular territories, as revealed by brain CT or MRI scans. Such a pattern suggests an underlying cause related to disrupted energy production or microvasculature, rather than the more common pattern of large-vessel involvement associated with cardiovascular causes. Additional indicators of mitochondrial dysfunction include hearing loss, myopathy, or diabetes [7]. Thus, the appropriate clinical scenario should prompt inclusion of MELAS in the differential diagnosis regardless of age and should warrant molecular workup for underlying mitochondrial derangements.
The reason for late onset of neurological symptoms in certain patients with MELAS is unclear. Some investigations suggest a correlation between mtDNA variant load and disease burden [7]. Given this correlation, we posit that the relatively low rate of heteroplasmy in our patient (31% in blood) contributed to her late symptom onset and overall mild clinical course. The heteroplasmy rates of our patient's muscle and brain tissue, which may be more predictive of course and severity, are unknown.
Given the maternal inheritance of mtDNA, late-onset MELAS has unique implications for female patients in terms of reproductive choices. All children of an affected female will inherit the mutation in mtDNA, but unpredictable levels of heteroplasmy and variable expressivity inhibit accurate prognosis. Diagnosis of MELAS after childbearing years precludes use of this knowledge to inform decisions regarding family planning. Our patient has described a sense of guilt and anxiety over the uncertain health risks that her two adult children have inherited. As more cases of females with late-onset MELAS are identified, there will be a need for skilled genetic and psychosocial counseling to assist patients and their families as they navigate such challenges.
Most importantly, this case of late-onset MELAS underscores the importance of prompt diagnosis on initial presentation, as early intervention may improve outcomes. In particular, recent evidence indicates that supplementation with the nitric oxide precursors l-arginine and l-citrulline may offer greater protection if started earlier in life [11], [12], [13]. Our patient did not come to medical attention with neurologic symptoms of MELAS until age 49, but she has nonetheless done well with long-term antiepileptic therapy and metabolic supplementation.
In summary, medical knowledge of MELAS is still expanding, and many questions regarding its pathogenesis and natural history remain unanswered. With greater awareness and improved diagnosis of patients with atypical presentations, the recognized clinical spectrum of MELAS will continue to expand.
Disclosures
None.
Contribution statement
Kiri Sunde - organization, manuscript preparation of first draft. Patrick Blackburn - review and critique. Anvir Cheema - review and critique. Jennifer Gass - review and critique. Kimberly J. Guthrie - genetic counselor, review and critique. Sarah Macklin - genetic counselor, review and critique. Paldeep Atwal - medical geneticist, project execution, design, review and critique.
Acknowledgements
The authors would like to thank the patient for her permission to publish this manuscript.
References
- 1.Narita H. A case with late-onset MELAS with hallucination and delusion. No To Shinkei. 2004;56(4):345–349. [PubMed] [Google Scholar]
- 2.Kimata K.G. A case of late-onset MELAS. Arch. Neurol. 1998;55(725):722–725. doi: 10.1001/archneur.55.5.722. [DOI] [PubMed] [Google Scholar]
- 3.Kisanuki Y.Y. Late-onset mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes with bitemporal lesions. Arch. Neurol. 2006;63(8):1200–1201. doi: 10.1001/archneur.63.8.1200. [DOI] [PubMed] [Google Scholar]
- 4.Bataillard M. Atypical MELAS syndrome associated with a new mitochondrial tRNA glutamine point mutation. Neurology. 2001;56(3):405–407. doi: 10.1212/wnl.56.3.405. [DOI] [PubMed] [Google Scholar]
- 5.Jeppesen T.D. Late onset of stroke-like episode associated with a 3256C–>T point mutation of mitochondrial DNA. J. Neurol. Sci. 2003;214(1–2):17–20. doi: 10.1016/s0022-510x(03)00168-0. [DOI] [PubMed] [Google Scholar]
- 6.DiMauro S, Hirano, M., MELAS. In: Pagon, RA, Adam, MP, Ardinger, HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; (1993–2016. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1233/, 2001 Feb 27 [Updated 2013 Nov 21]).
- 7.Parikh S. Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genitourin. Med. 2015;17(9):689–701. doi: 10.1038/gim.2014.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goto Y. Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS): a correlative study of the clinical features and mitochondrial DNA mutation. Neurology. 1992;42(3 Pt 1):545–550. doi: 10.1212/wnl.42.3.545. [DOI] [PubMed] [Google Scholar]
- 9.Macmillan C., Lach B., Shoubridge E.A. Variable distribution of mutant mitochondrial DNAs (tRNA(Leu[3243])) in tissues of symptomatic relatives with MELAS: the role of mitotic segregation. Neurology. 1993;43(8):1586–1590. doi: 10.1212/wnl.43.8.1586. [DOI] [PubMed] [Google Scholar]
- 10.Hirano M. MELAS: an original case and clinical criteria for diagnosis. Neuromuscul. Disord. 1992;2(2):125–135. doi: 10.1016/0960-8966(92)90045-8. [DOI] [PubMed] [Google Scholar]
- 11.El-Hattab A.W. Impaired nitric oxide production in children with MELAS syndrome and the effect of arginine and citrulline supplementation. Mol. Genet. Metab. 2016;117(4):407–412. doi: 10.1016/j.ymgme.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koga Y. MELAS and l-arginine therapy. Mitochondrion. 2007;7(1–2):133–139. doi: 10.1016/j.mito.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 13.Koga Y. l-arginine improves the symptoms of strokelike episodes in MELAS. Neurology. 2005;64(4):710–712. doi: 10.1212/01.WNL.0000151976.60624.01. [DOI] [PubMed] [Google Scholar]
- 14.Vanniarajan A. Clinical and genetic uniqueness in an individual with MELAS. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2006;141B(5):440–444. doi: 10.1002/ajmg.b.30302. [DOI] [PubMed] [Google Scholar]
- 15.Sharfstein S.R. Adult-onset MELAS presenting as herpes encephalitis. Arch. Neurol. 1999;56(2):241–243. doi: 10.1001/archneur.56.2.241. [DOI] [PubMed] [Google Scholar]
- 16.Olsson C. Level of heteroplasmy for the mitochondrial mutation A3243G correlates with age at onset of diabetes and deafness. Hum. Mutat. 1998;12(1):52–58. doi: 10.1002/(SICI)1098-1004(1998)12:1<52::AID-HUMU8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 17.Picard M. Progressive increase in mtDNA 3243A > G heteroplasmy causes abrupt transcriptional reprogramming. Proc. Natl. Acad. Sci. U. S. A. 2014;111(38):E4033–E4042. doi: 10.1073/pnas.1414028111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilichowski E. Quantitative proton magnetic resonance spectroscopy of cerebral metabolic disturbances in patients with MELAS. Neuropediatrics. 1999;30(5):256–263. doi: 10.1055/s-2007-973500. [DOI] [PubMed] [Google Scholar]
- 19.Leff A.P. Complex partial status epilepticus in late-onset MELAS. Epilepsia. 1998;39(4):438–441. doi: 10.1111/j.1528-1157.1998.tb01397.x. [DOI] [PubMed] [Google Scholar]