

Abstract
A comprehensive description of starch biosynthesis and granule assembly remains undefined despite the central nature of starch as an energy storage molecule in plants and as a fundamental calorie source for many animals. Multiple theories regarding the starch synthase (SS)-catalyzed assembly of (α1–4)-linked d-glucose molecules into maltodextrins generally agree that elongation occurs at the non-reducing terminus based on the degradation of radiolabeled maltodextrins, although recent reports challenge this hypothesis. Surprisingly, a direct analysis of the SS catalytic product has not been reported, to our knowledge. We expressed and characterized recombinant Zea mays SSIIa and prepared pure ADP-[13CU]glucose in a one-pot enzymatic synthesis to address the polarity of maltodextrin chain elongation. We synthesized maltoheptaose (degree of polymerization 7) using ADP-[13CU]glucose, maltohexaose (degree of polymerization 6), and SSIIa. Product analysis by ESI-MS revealed that the [13CU]glucose unit was added to the non-reducing end of the growing chain, and SSIIa demonstrated a >7,850-fold preference for addition to the non-reducing end versus the reducing end. Independent analysis of [13CU]glucose added to maltohexaose by SSIIa using solution NMR spectroscopy confirmed the polarity of maltodextrin chain elongation.
Keywords: adenosine, enzyme mechanism, mass spectrometry (MS), nuclear magnetic resonance (NMR), oligosaccharide, SS2a, SSIIa, maltohexaose
Introduction
The development of starch biosynthesis by the Archaeplastida progenitor provided an efficient and dense energy storage molecule that dramatically altered terrestrial life on planet earth (1). More recently, starch formed an integral component of human evolution and societal development (2). By virtue of its importance, starch biosynthesis and molecular structure is studied intensely because fundamental questions remain: in particular, how glucose chains are synthesized and how different enzymes contribute to creating repeating three-dimensional structure in the starch granule.
Starch is composed of two types of polymers with repeating d-glucose units termed amylose and amylopectin, which constitutes ∼25 and ∼75% of the granule mass, respectively. Amylose contains ∼1,000–3,000 glucose units joined in almost exclusively α1–4-glycosidic bonds, divided into ∼3–10 “linear” chains attached to each other by α1–6 “branch” linkages (3). Amylopectin is >95% α1–4 linked with ∼5% branching α1–6 linkages and contains ∼5,000–10,000 glucose units (3, 4). Amylopectin and amylose are initially synthesized as (α1–4)-linked maltodextrins from ADP-α-d-glucose donor molecules by starch synthase enzymes, of which five classes are conserved in land plants, as shown in Fig. 1 (5). Amylose is predominately synthesized by granule-bound starch synthase (GBSS)2 (6). SSI, SSIIa, and SSIII are all involved in producing the linear chains of amylopectin, and SSIV along with SSIII appear to be involved in the initiation of granule formation (7, 8). Short to medium chain maltodextrins are modified by other enzymes including starch branching enzyme, phosphorylase, and starch-debranching enzyme to give rise to amylopectin.
FIGURE 1.

A simplified model depicting starch granule synthesis. Glucose is converted to ADP-glucose by hexokinase (HK), phosphoglucomutase (PGM), and ADP-glucose pyrophosphorylase (ADPGP). SS isoforms utilize ADP-glucose to synthesize growing maltodextrin chains that are branched by starch branching enzyme (BE) and starch-debranching enzyme (DBE). On the lower left is the “cluster model” of amylopectin organization that forms the structural component of a starch granule, cutaway in the image on the lower right to show layers of amylopectin.
Amylose and amylopectin synthesis is tightly controlled to properly assemble the higher order structure in the starch granule that is required to expel water, although surprisingly little is known about this process. According to widely accepted models, the nascent structure is radially arranged around the initiation site with the non-reducing ends facing away from the initiation site. This is thought to be favorable because the starch granules are degraded from the non-reducing termini, meaning more surface area for efficient metabolism (1). Amylopectin forms dehydrated regions of semicrystalline and amorphous lamellae with a periodicity of 9 nm (1). The overall shape and size of starch granules can vary between species and is heavily influenced by environment; however, the periodicity of the lamellae remains constant (5).
The chemical structure of amylose and amylopectin implies that the fundamental process in the construction of starch granules is elongation of α1–4-linked glucan chains by SS. The polarity of d-glucose addition to a growing maltodextrin chain remains a primary question in starch biosynthesis and would affect enzymatic mechanisms put forward by several groups (9–11). Leloir and co-workers first addressed this question and demonstrated that the [14C]glucose moiety from UDP-[14C]glucose or ADP-[14C]glucose was incorporated into both amylose and amylopectin within starch granules (12–14). Digestion of these granules with the exo-acting enzyme β-amylase released [14C]maltobiose, indicating d-glucose is added to the non-reducing end of the growing maltodextrin polymers. Further evidence for a non-reducing end addition mechanism was found upon analysis of reaction products with chemical degradation methods that distinguish polarity. In this case, [14C]glucose addition (in the form of UDP-[14C]glucose) to unlabeled maltotriose or maltotetraose using SS from starch granules occurred at the non-reducing pole (13). Robyt and co-workers (9, 15, 16) recently challenged these findings and concluded that maltodextrin elongation occurs at the reducing end using similar pulse and chase experiments that also lacked direct evaluation of the products. Both of these laboratories used enzyme mixtures from natural sources and degradation methods that are subject to errors.
The advent of recombinant protein expression techniques provided a new source of pure enzyme isoforms (8, 17–23). These studies detailed the effect of amino acid substitutions to SS enzymes, as well as kinetic characterization of substrate acceptors; however, none of these studies directly characterized the acceptor product to address the polarity of maltodextrin growth, and many show only single additions from the SS enzymes.
In this study we expressed and purified recombinant Zea mays SSIIa to address the polarity of d-glucose addition using sensitive, high resolution MS and NMR techniques. We chose maltohexaose (DP6) as the primary substrate because SSIIa was shown to elongate DP6 chains in a distributive manner (8).
Results
Expression, Purification, and Analysis of ZmSSIIa
SSIIa from Z. mays (ZmSSIIa) expressed in Escherichia coli as a single polypeptide with a molecular mass of 77.3 kDa as shown in Fig. 2. The majority of the protein expressed in an insoluble form, although soluble S-tagged ZmSSIIa constituted the predominant protein (>80%) following purification with S protein-agarose beads. This recombinant protein expression system eliminates potential contamination from other competing SS isoforms or other maltodextrin-modifying enzymes that would be present using starch granules as an enzyme source. ZmSSIIa bound to S-agarose beads served as the enzyme source for all subsequent analyses. Previous analyses using this expression system demonstrated that the kinetic parameters of the enzyme were essentially identical whether it was bound to beads or free in solution after release from the matrix (23).
FIGURE 2.
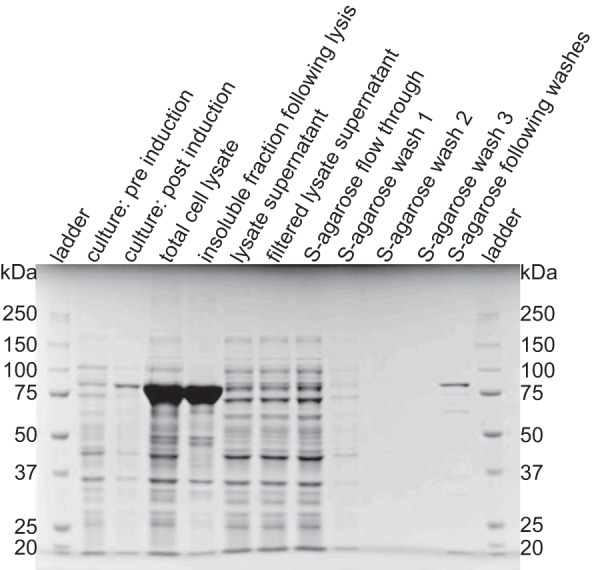
Expression and purification of ZmSSIIa analyzed by SDS-PAGE and Coomassie staining. S-tagged enzyme was expressed and purified from E. coli and captured using an S protein-agarose resin. ZmSSIIa-bound agarose beads were boiled in SDS-containing loading buffer before loading to analyze purity.
ZmSSIIa utilized ADP-glucose in the presence of maltodextrin or glycogen. The rate of ADP generation was higher with 10 mg/ml glycogen as an acceptor substrate when compared with the rate with 10 mm maltohexaose (DP6) as shown in Fig. 3A. It is difficult to directly compare the initial velocities of these two reactions because of the undefined concentration of acceptor sites in the reaction containing glycogen. We observed background intensity in reactions that contained ADP-glucose (Fig. 3B). This background increased noise in the experiments but proved smaller than the rates observed with enzyme, acceptor, and ADP-glucose present.
FIGURE 3.
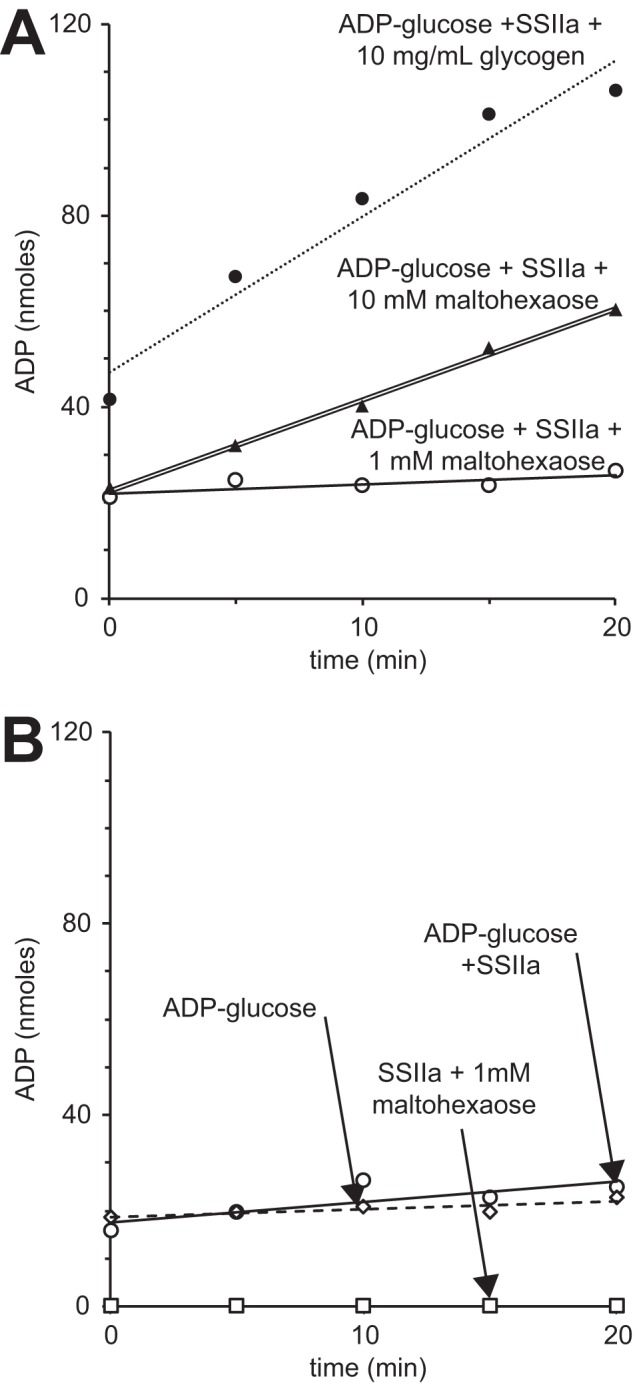
ZmSSIIa activity quantified by ADP release using the ADP-Glo reagent (Promega). A, ZmSSIIa activity using maltohexaose (DP6) or glycogen as acceptor molecules. B, the ADP-glucose preparation contributed to observed signal in the assay that was less than the ADP generated in reactions containing the combination of ADP-glucose, an acceptor substrate and SSIIa.
Analysis of the maltodextrin products revealed multiple additions to maltohexaose (DP6) during the ZmSSIIa-catalyzed reaction. TLC analysis of the substrates and products showed the near complete disappearance of ADP-glucose, the formation of primarily maltoheptaose (DP7) in the early stages of the reaction, and a distribution of larger forms (DP8–14) in the later stages from a reaction containing ZmSSIIa (Fig. 4). A similar reaction lacking the DP6 acceptor substrate showed no product formation and consistent ADP-glucose levels. MALDI-TOF MS analysis of the maltodextrin reaction products showed the same result (data not shown). The sequential addition of glucose residues to maltohexaose is consistent with reports that SSIIa acts in a distributive manner (6, 8).
FIGURE 4.
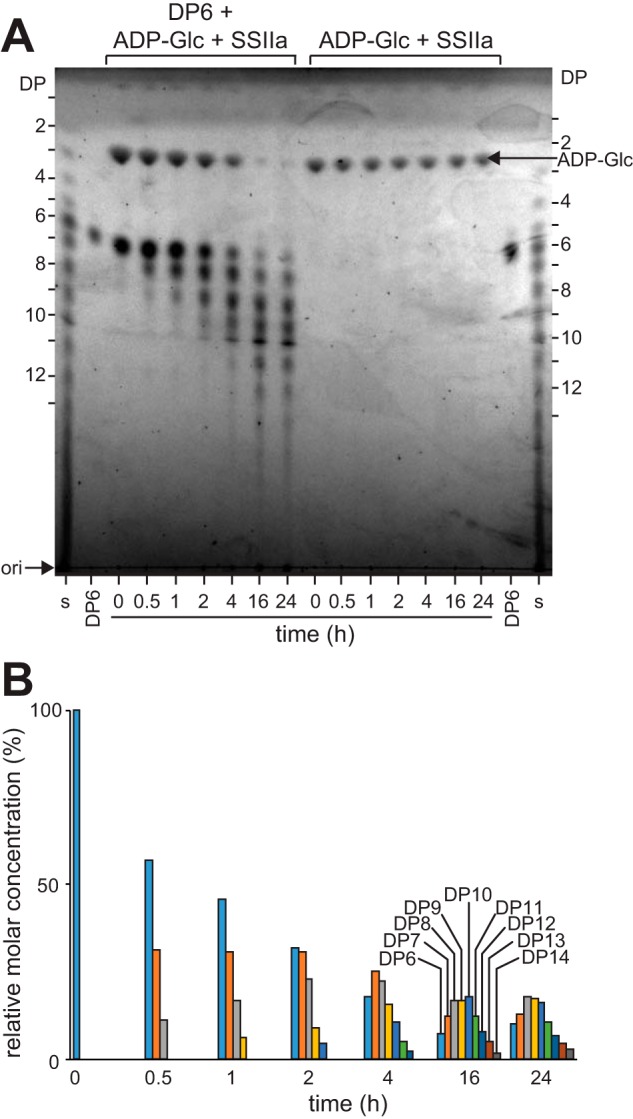
Analysis of ZmSSIIa products in a reaction containing maltohexaose (DP6) and ADP-glucose shows incorporation of multiple glucose units. A, TLC analysis showing the result of two reactions. B, quantification of band intensity from A.
These data indicate that the ZmSSIIa preparation was active in solution and catalyzed multiple additions but do not provide information regarding the polarity of maltodextrin chain growth. Once maltodextrin is extended, it is impossible to distinguish newly added glucose residues from those present in the starting material. We synthesized ADP-[13CU]glucose for use as a donor substrate in a ZmSSIIa-catalyzed reaction to eliminate isotopic symmetry of the maltodextrin product.
One-pot Synthesis of ADP-[13CU]Glucose
We pursued a one-pot, enzyme-catalyzed reaction scheme to prepare ADP-[13CU]glucose and avoid losses from purifying pathway intermediates (Fig. 5A). Hexokinase, phosphoglucomutase, a thermostable ADP-glucose pyrophosphorylase (GlgC2), and an inorganic pyrophosphorylase were active in a HEPES, pH 7.5 buffer and converted >99% of the starting [13CU]glucose to ADP-[13CU]glucose. A 1H NMR spectrum of ADP-[13CU]glucose purified by anion exchange chromatography revealed no identifiable peaks corresponding contaminating species other than a pH buffer and a chemical shift standard (4,4-dimethyl-4-silapentane-1-sulfonic acid; Fig. 5B). Seven strong peaks observed in a two-dimensional heteronuclear (1H-13C) single quantum coherence (HSQC) spectrum were consistent with a highly purified sample free of contaminating nucleotide, free glucose, or other 13C-enriched molecule (Fig. 5C). ZmSSIIa utilized the isotopically labeled donor substrate as efficiently as unlabeled ADP-glucose (data not shown).
FIGURE 5.

Synthesis and characterization ADP-[13CU]glucose. A, [13CU]glucose was completely converted using a one-pot enzymatic synthesis. B, one-dimensional NMR 1H spectrum of purified ADP-[13CU]glucose shows a high degree of purity. A chemical shift standard (4,4-dimethyl-4-silapentane-1-sulfonic acid) was added to this sample for the NMR data collection. C, two-dimensional 1H-13C HSQC of ADP-[13CU]glucose showing assigned peaks.
ESI-MS/MS Analysis of 13C-Labeled Maltodextrin
It is possible that ZmSSIIa adds glucose to the reducing end of maltodextrin, the non-reducing end, or a combination of both. We analyzed the reaction products directly to determine the polarity of ZmSSIIa-catalyzed maltodextrin chain elongation. The analytic scheme diagrammed in Fig. 6A utilizes unlabeled maltohexaose (DP6) as an acceptor substrate and ADP-[13CU]glucose as the donor. Reaction times were limited by removing enzyme-linked agarose beads to terminate catalysis. Next, reductive amination completely modified the reducing end of the product and unreacted maltodextrin substrate with N-(naphthyl)-ethylenediamine and provided a handle for reversed phase chromatography prior to ESI-MS. The MS spectrum indicated that DP7 constituted a majority of the product with a small amount converted to DP8, and differences of <0.01 Da separated the expected and observed masses for all species (Fig. 6B). We observed multiple singly charged forms corresponding to +H+, +Na+, and +K+ adducts of each reaction product and unreacted substrate using the positive instrument mode.
FIGURE 6.

Multi-dimensional MS analysis of maltoheptaose (DP7) and maltooctaose (DP8) synthesized from unlabeled maltohexaose (DP6) using ZmSSIIa and ADP-[13CU]glucose. A, the analytical scheme applied herein. Following conversion, the reducing end is modified through reductive amination. B, ESI-MS spectrum of the reaction products showing conversion to DP7 and DP8; monoisotopic masses are provided. C–E, MS-MS spectra of the parent ions for DP6 (C), DP7 (D), and DP8 (E). Ions are identified at the bottom of the figure with connections through vertical dashed gray lines and clearly indicate only modification of the DP6 non-reducing terminus.
Once ions corresponding to modified substrate and product molecules were identified in MS spectra (Fig. 6B), the polarity of [13CU]glucose addition was determined by fragmenting the parent ions in a second MS dimension. If the [13CU]glucose molecule was added to the reducing end, the [13CU]glucose would be directly attached to the N-(naphthyl)-ethylenediamine tag. However, if the [13CU]glucose molecule was added to the non-reducing end, the N-(naphthyl)-ethylenediamine tag would be attached to an unlabeled [12C]glucose unit. If both ends were modified, a mixture of 12C- and 13CU-labeled species would be identified and could be directly compared with determine the reaction preference. The isolation of specific parent ions from the first MS dimension using the Orbitrap instrument prevents cross-contamination of different parent ion species and thus selective MS/MS spectra.
Ion selection provided very high signal to noise measurements in the MS/MS spectra. The MS/MS spectrum of the DP6 substrate that corresponded to the parent ion, and a neutral loss of n*(∼162.1), which indicates loss of n [12C]glucose units where n = 1, 2, 3, 4, or 5 (Fig. 6C). As expected for the unlabeled substrate, the reducing end N-(naphthyl)-ethylenediamine tag coupled to a [12C]glucose was also found with a mass of 351.2 Da. MS/MS spectra of the labeled DP7 reaction product revealed a neutral loss of ∼168.1 Da when compared with the parent ion, indicating loss of a [13CU]glucose unit from the non-reducing end (Fig. 6D). The spectra revealed no evidence for the neutral loss of ∼162.1 Da from the parent ion. Furthermore, the second dimension spectrum contained reducing end N-(naphthyl)-ethylenediamine tag coupled to a [12C]glucose (351.2 Da) and revealed no evidence of a N-(naphthyl)-ethylenediamine tag coupled to a [13C]glucose (357.2 Da; Fig. 7). Comparison of the signal intensity of the peak at 351.2 Da to the noise level observed surrounding 357.2 Da revealed that ZmSSIIa prefers the non-reducing end of maltohexaose for >99.987% of glucose addition reactions.
FIGURE 7.

ZmSSIIa catalyzes growth at the non-reducing end of maltohexaose with at least a 7,850-fold preference over addition to the reducing end. These data represent expansions of Fig. 6C, and the molecule symbols match those in Fig. 6.
Similar degradation patterns of the DP8 reaction product in MS/MS spectra provided data complementary to that described for DP7 above (Fig. 6E). In this instance neutral loss of one labeled glucose unit (∼168.1 Da) or two labeled units (∼336.2) from the parent ion indicated addition at the non-reducing end, and [13C]glucose modified by the chemical tag and thus located at the reducing end was not detected.
NMR Analysis of 13C-Labeled Maltodextrin
Labeling maltodextrins with stable isotopes provides an opportunity to validate the ESI-MS/MS analysis of SSIIa recognition polarity with an orthogonal technique. [13CU]Glucose addition to the non-reducing end of maltodextrin can be distinguished from reducing end modification using solution NMR spectroscopy (Fig. 8). The anomeric 1H1 and 13C1 nuclei (Fig. 8A) display characteristic resonance frequencies depending upon the chemistry of the glycosidic linkage and stereochemistry of the hemiacetal moiety. A glucose residue at the reducing end of a maltodextrin will have an anomeric configuration that mutarotates between α and β forms with 1H1 resonance frequencies at 5.19 and 4.62 ppm, respectively (Fig. 8B). A glucose residue at the non-reducing end or within the interior of the polymer will be locked in the α configuration with a 1H1 frequency of 5.37 ppm. Carbon [13C] chemical shifts are likewise distinct.
FIGURE 8.

NMR analysis of maltodextrins synthesized using ZmSSIIa and ADP-[13CU]glucose. A, model of maltohexaose (DP6) showing nomenclature. B and C, the anomeric 1H regions of one-dimensional NMR 1H (B) and two-dimensional 1H-13C HSQC spectra (C) collected on a reaction of unlabeled DP6, ZmSSIIa, and ADP-[13CU]glucose creating a small amount of DP7 (comparable with the conversion in Fig. 6B). A small amount of natural abundance signal is observed, particularly for the α and β forms at the reducing end. D, HCCH-TOCSY spectra of the reaction shows clear 13C-13C transfer occurred only in a [13CU]glucose unit added to the non-reducing end of the maltodextrin as highlighted with a dashed red circle. E and F, similar experiments collected on the acceptor substrate (E) and the donor substrate (F) do not show these features.
Two-dimensional HSQC NMR spectra of a reaction similar to that described for the ESI-MS/MS analysis between maltohexaose, ADP-[13CU]glucose, and ZmSSIIa revealed 1H-13C correlations (Fig. 8C). Peaks for all three expected anomeric correlations in a mixture of maltohexaose (DP6) and maltoheptaose (DP7) were identified (non-reducing end α, reducing end α, and reducing end β). 1H-13C correlations for the free anomeric carbons are due to the ∼1% natural [13C] abundance rather than to labeling with incorporated [13CU]glucose, because they appear in spectra of unreacted maltohexaose substrate (data not shown). The peak for the bound anomeric [13C] carbons, which includes the non-reducing end, has much greater intensity than the free anomeric carbon peak. The presence of a strong peak in the HSQC spectrum corresponding to the alpha form of the non-reducing end indicated the reaction specifically modified the maltohexaose non-reducing end with [13CU]glucose.
The effect of natural [13C] abundance in NMR spectra of labeled maltodextrins may be reduced by utilizing 13C-13C pairs to probe the added [13CU]glucose unit specifically with an HCCH-TOCSY pulse sequence commonly applied to assign amino acids side chain resonances in uniformly 13C-labeled proteins (24). This experiment passes magnetization between adjacent 13C-13C pairs (1H1-13C1-13C2-1H2, for example), which by natural abundance represent only 1 in 10,000 carbon atoms, but 98% of the carbons in the [13CU, 99%]glucose. Any correlations that meet these criteria are found as off-diagonal peaks in a plot showing 1H frequency on the x and y axes. Peaks located along the diagonal (that have the same 1H frequency in both the x and y dimensions) indicate that no transfer occurred and are not informative (Fig. 8, D–F).
A two-dimensional 1H(CC)1H-TOCSY experiment on the material prepared using the reaction described above showed correlations between the 1H1 of the non-reducing end glucose (observed on the x axis) and the 1H2 nucleus (located on the vertical dashed gray line and surrounded by a dashed red circle in Fig. 8D), but no similar cross-peaks for either the α or β 1H1 glucose residue on the reducing end (marked by gray H2 labels in Fig. 8D). An artifact peak was observed (marked a in Fig. 8, D–F) but was identified as a minor contaminant from the ADP-[13CU]glucose substrate (Fig. 8F) not present in the maltohexaose substrate (Fig. 8E).
We observed a small amount of H2 signal from an HCCH-TOCSY spectrum of the unlabeled maltohexaose substrate caused by natural abundance 13C upon lowering the contour level to nearly that of noise (Fig. 8E). Although the signal from [13CU]glucose was much stronger, we explored the result of a longer magnetization transfer pathway to suppress background signals an additional 100-fold (or 1 in 1,000,000). This pathway involved three 13C transfers and can be seen in the H3 peaks in Fig. 8D (1H1-13C1-13C2-13C3-1H3). H3 signals from a reducing end glucose residue were not observed. These HCCH-TOCSY spectra agree completely with the HSQC spectra and ESI-MS/MS analysis and unambiguously identify the non-reducing end of maltohexaose as the primary acceptor site for ZmSSIIa-catalyzed maltodextrin elongation.
Discussion
This study directly characterized the chemical structure of ZmSSIIa reaction products. MS analyses that directly distinguished the position of the newly added glucosyl moiety showed a striking preference for the non-reducing end within a sensitivity limit that cannot exclude one reducing end addition for more than 7,850 glucose units incorporated. NMR spectra proved less sensitive than the ESI-MS spectra and could not independently support the >7,850:1 addition ratio, however, observation of 1H correlation signals requiring two or three adjacent 13C atoms qualitatively demonstrated that the great majority of additions are at the non-reducing end. Taken together, these two analytical techniques provide definitive evidence that ZmSSIIa catalysis is specific for the non-reducing end of a maltodextrin acceptor substrate. The techniques described here can be used to directly assess this prediction for any other plant SS or glycosyl transferases from other species.
ZmSSIIa activity was undetectable when maltohexaose was omitted from the reaction. This was evident from the lack of maltodextrin products detected either by TLC or MALDI-TOF and from the consistent ADP-glucose levels (Fig. 4A, right side). Thus, in this reconstituted system, the activity of SSIIa is strictly dependent on the presence of a maltodextrin acceptor substrate.
These results support the original conclusion of Leloir and co-workers that SS present within starch granules adds primarily to the non-reducing end (12–14). It is unclear why Robyt and Leloir arrived at different conclusions despite the use of similar techniques and comparable material sources, although both relied upon indirect analysis techniques that characterized hydrolyzed glucose units following reduction or oxidation of the maltodextrin chain (9, 15, 16, 25). The technique employed by Robyt and co-workers would have been sensitive to unanticipated starch degradation that occurred prior to chemical modification and maltodextrin hydrolysis. Such degradation could have occurred enzymatically because none of the enzyme preparations used in the previous studies were biochemically pure, or they could have resulted from unforeseen chemical reactions. The independent characterization techniques used here that employed a purified recombinant enzyme and a defined maltohexaose molecule as a potential acceptor substrate, plus the direct and targeted characterization of the reaction product, avoid any such potential artifacts.
Another explanation for differing conclusions of maltodextrin chain elongation polarity is that different SS enzymes employ distinct reaction mechanisms. It is formally possible ZmSSIIa utilizes a different mechanism than GBSS or the mixture of SS enzymes studied by the Leloir and Robyt laboratories (12, 26). This explanation appears to be unlikely because of the high degree of primary, secondary, and tertiary structural conservation across SS enzymes. SSI-IV, GBSS, and bacterial glycogen synthases are members of the orthologous GT5 family of glycosyl transferases, and although structures of all GT5 members are not available, analysis of reported structures indicates a high degree of structural similarity including rice GBSS (27), barley SSI (28), and glycogen synthase from E. coli (29, 30) or Agrobacterium tumefaciens (31). A structure of the E. coli glycogen synthase bound to maltotriose (DP3) shows the maltotriose non-reducing C4-OH proximal to the β-phosphate group of ADP, where an incoming donor glucosyl moiety would be located (29, 30). These observations suggest that GT5 family members, including SSs and glycogen synthases, employ similar mechanisms that extend maltodextrins by glucosyl addition to the non-reducing end.
Experimental Procedures
All materials were purchased from Sigma-Aldrich unless otherwise noted.
ADP-Glucose Pyrophosphorylase Purification
A pHM1 expression plasmid encoding hexahistidine-tagged wild type glgC2 from Thermotoga maritima was provided by Dr. Christopher Meyer (Cal State, Fullerton) (32, 33). E. coli BL21* cells containing the GlgC2 expression plasmid were grown in 1 liter of Luria-Bertani medium supplemented with 100 μg/ml ampicillin to an A600 of 0.5 at 37 °C in a rotary shaker. Protein expression was induced at this point with 1.5 g of l-arabinose and incubated with shaking overnight at 18 °C. The cells were resuspended in a lysis buffer containing 50 mm sodium phosphate, 300 mm sodium chloride, and 10 mm imidazole, pH 8.0, and lysed by multiple passages through an Emulsiflex C5 homogenizer (Avestin) operating at 15,000 p.s.i. The lysate was clarified by centrifugation at 16,000 × g for 1 h. The supernatant was applied to a 5-ml nickel-nitrilotriacetic acid column (Qiagen) that was washed with 5 × 5 ml of lysis buffer, 5 × 5 ml of wash buffer (50 mm sodium phosphate, 300 mm sodium chloride, 5 mm dithiothreitol, pH 8.0) with 20 mm imidazole, 3 × 5 ml of wash buffer with 100 mm imidazole, 3 × 5 ml of wash buffer with 250 mm imidazole, and 1 × 5 ml of wash buffer with 500 mm imidazole. Fractions containing GlgcC2 protein were identified using SDS-PAGE, pooled, exchanged into lysis buffer using a centrifugal filter unit (Amicon, 10-kDa molecular mass cutoff), diluted to a final concentration of 10% glycerol, and stored at −80 °C. Protein concentration was estimated using a molar extinction coefficient of 57,550 m−1 cm−1.
ADP-[13CU]Glucose Synthesis
A one-pot reaction based on the conditions presented (34) and containing 200 mm HEPES, 20 mm magnesium chloride, 10 mm [13CU]glucose, 20 mm adenosine triphosphate, 0.0016 unit/μl hexokinase (H4502; Sigma), 0.0076 unit/μl phosphoglucomutase (P3397; Sigma), 0.0008 unit/μl inorganic pyrophosphotase (l1643; Sigma), 0.14 mg/ml ADP-glucose pyrophosphorylase (GlgC2), pH 7.5 was incubated for 1 day at 25 °C. The reaction was monitored using a Bruker 700-MHz solution NMR system equipped with a 5-mm cryogenically cooled probe. The reaction was filtered through a sterile 0.22-μm filter. ADP-[13CU]glucose was purified from contaminating reaction components with a buffer containing 5% 1 m sodium hydroxide and eluted with a 0–95% gradient of 1 m ammonium acetate from a Carbo PAC PA-100 on a Dionex HPLC system (Thermo Scientific). Fractions containing ADP-[13CU]glucose were pooled and lyophilized. The resulting powder was resuspended in D2O and quantified using 1H NMR by comparison with an internal 4,4-dimethyl-4-silapentane-1-sulfonic acid standard.
SSIIa Purification
Z. mays SSIIa containing an N-terminal S-tag was expressed from a pET29:SSIIa plasmid and purified as previously described (23). The amount of protein attached to the S protein-agarose beads (EMD Millipore) was quantified by adding 10 μl of 1:1 SSIIA bead:buffer slurry to 20 μl of 8 m urea followed by centrifugation. The A280 of the resulting supernatant was measured on a NanoDrop 2000C spectrophotometer (Thermo Scientific), and SSIIa concentration was estimated using a molar extinction coefficient of 117,730 m−1 cm−1.
The SSIIa-catalyzed Reaction
Activity assays were performed after each purification. We noted that SSIIa activity reduced by ∼50% after 1 week at 4 °C, so each activity assay was performed on SSIIa that was not more than 1 week old. Reactions consisted of 20 mm glycine, 5 mm EDTA, 0.5 mg/ml bovine serum albumin, 5 mm dithiothreitol, acceptor substrate (maltohexaose or shellfish glycogen), 2–4 mm ADP-glucose or ADP-[13CU]glucose, 5 μm SSIIa, pH 8.2, and were performed with shaking at 20–37 °C with constant gentle rotation to mix the beads and the solution. Aliquots were removed to monitor the reaction progress and then centrifuged for 1 min at 0.1 × g to separate the bead-bound SSIIa from the reaction supernatant.
SSIIa Activity Assayed Using the ADP-GloTM Kinase Reagent
Reaction supernatant (5 μl) was added to 85 μl of 40 mm tris(hydroxymethyl)aminomethane, 20 mm sodium chloride, and 0.1% (w/v) bovine serum albumin, pH 7.5, in a white 96-well microtiter plate. After all time points were collected, 10 μl of the kinase detection reagent (ADP-GloTM kinase assay; Promega) was added to each well, and the plate was shaken for 5 min at 25 °C. Luminescence was measured using a Synergy 2 plate reader (Biotek) and compared with a standard solutions containing known amounts of ADP.
Thin Layer Chromatography
Reaction supernatant (2 μl) was spotted on a silica gel 60 TLC plate (EMD Millipore). The plate was developed with a running solution of 85:20:50:60 acetonitrile:ethyl acetate:isopropanol:water and dried. This process was repeated twice, and following the third drying, the plate was thoroughly dried and dipped in a solution of 95% methanol, 5% sulfuric acid, and 0.3% N-(1-naphthyl)-ethylenediamine-dihydrochloride. The plate was allowed to air dry and then incubated in a 120 °C oven for 10 min or until the appearance of uniform color development (26). The maltodextrin standard was prepared by debranching shellfish glycogen with isoamylase (10 units) (Megazyme) in pH 4.5 sodium acetate buffer at 22 °C for 24 h as previously described (35).
Analysis by MALDI-TOF MS
Reactions were analyzed using a Voyager DE Pro Star MALDI-TOF instrument (Applied Biosystems). Reaction supernatant (0.5 μl) was mixed with 0.5 μl of matrix solution (40 mg/ml 2,5-dihydroxybenzoic acid in 80:20 acetonitrile:water) and applied to the MALDI plate. The crystals were shot in reflector mode with positive polarity with the following settings: laser intensity, 2,800; accelerating voltage, 19,000; grid, 76%; guide wire, 0.2%; and delay time, 275 ns.
Analysis by ESI-MS
Reaction supernatant was further analyzed with an Agilent 1260 liquid chromatography system liquid chromatography system coupled with an ESI-MS (Q exactive hybrid quadrupole Orbitrap mass spectrometer; Thermo Scientific). First, samples were modified at the reducing end with reductive amination. Reaction supernatant (25 μl) was added to 20 μl of 0.2 m N-(naphthyl)-ethylenediamine dihydrochloride in 15% acetic acid, 5 μl of 5 m sodium cyanoborohydride and incubated at 37 °C for 15 h (35). Samples (10 μl) were injected at a flow rate of 0.03 ml/min onto a C18 reversed phase column previously equilibrated with 95% solvent A (0.1% formic acid in water) and 5% solvent B (0.1% formic acid in acetonitrile) at 20 °C. Samples were eluted using a linear gradient of 5–35% solvent B for 1–8 min followed by a linear gradient from 35–100% solvent B for 8–13 min. The ESI-MS instrument was set to positive polarity with 3,600 kV spray voltage and a mass scan range of 100–1,500 m/z. MS/MS spectra were collected on select ions by higher energy collisional dissociation using 25 eV. The data were displayed and analyzed using Thermo XCalibur Qual Browser (version 3.0.63).
Analysis of 13C-Labeled Dextrans by Nuclear Magnetic Resonance Spectroscopy
Analysis was performed on a Bruker 18.8T MHz Avance III system equipped with a 5-mm cryogenically cooled TXI probe. This system was equipped with TopSpin 3.2. Standard 1H detection, 1H-13C HSQC, and HCCH-TOCSY experiments were performed.
Author Contributions
A. W. B. and A. M. M. conceived and coordinated the study. M. E. L., D. J. F., A. M. M., and A. W. B. wrote the manuscript. M. E. L. and D. J. F. designed, performed, and analyzed the experiments. A. W. B. designed and analyzed the experiments. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Joel Nott (Iowa State University Protein Facility) for assistance with the ESI-MS data collection and analysis, Dr. D. Bruce Fulton (Iowa State University NMR Facility), and Prof. Christopher Meyer (Cal State, Fullerton) for providing the GlgC2 expression plasmid and conditions.
This work was supported by Grant R01-GM115489 from the NIGMS, National Institutes of Health and by funds from the Roy J. Carver Department of Biochemistry, Biophysics & Molecular Biology at Iowa State University. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- GBSS
- granule-bound starch synthase
- DPn
- degree of polymerization n
- HSQC
- heteronuclear single quantum coherence spectroscopy
- SS
- starch synthase
- TOCSY
- total correlation spectroscopy
- ESI
- electrospray ionization.
References
- 1. Zeeman S. C., Kossmann J., and Smith A. M. (2010) Starch: its metabolism, evolution, and biotechnological modification in plants. Annu. Rev. Plant Biol. 61, 209–234 [DOI] [PubMed] [Google Scholar]
- 2. Perry G. H., Dominy N. J., Claw K. G., Lee A. S., Fiegler H., Redon R., Werner J., Villanea F. A., Mountain J. L., Misra R., Carter N. P., Lee C., and Stone A. C. (2007) Diet and the evolution of human amylase gene copy number variation. Nat. Genet. 39, 1256–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jane J. (2009) in Starch: Chemistry and Technology (BeMiller J., and Whistler R., eds) 3rd Ed., pp. 193–236, Academic Press, Orlando, FL [Google Scholar]
- 4. Keeling P. L., and Myers A. M. (2010) Biochemistry and genetics of starch synthesis. Annu. Rev. Food Sci. Technol. 1, 271–303 [DOI] [PubMed] [Google Scholar]
- 5. Smith A. M., Denyer K., and Martin C. (1997) The synthesis of the starch granule. Annu. Rev. Plant Physiol. Plant Mol. Biol. 48, 67–87 [DOI] [PubMed] [Google Scholar]
- 6. Denyer K., Waite D., Motawia S., Møller B. L., and Smith A. M. (1999) Granule-bound starch synthase I in isolated starch granules elongates malto-oligosaccharides processively. Biochem. J. 340, 183–191 [PMC free article] [PubMed] [Google Scholar]
- 7. Szydlowski N., Ragel P., Raynaud S., Lucas M. M., Roldán I., Montero M., Muñoz F. J., Ovecka M., Bahaji A., Planchot V., Pozueta-Romero J., D'Hulst C., and Mérida A. (2009) Starch granule initiation in Arabidopsis requires the presence of either class IV or class III starch synthases. Plant Cell 21, 2443–2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cuesta-Seijo J. A., Nielsen M. M., Ruzanski C., Krucewicz K., Beeren S. R., Rydhal M. G., Yoshimura Y., Striebeck A., Motawia M. S., Willats W. G., and Palcic M. M. (2015) In vitro biochemical characterization of all barley endosperm starch synthases. Front. Plant Sci. 6, 1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mukerjea R., and Robyt J. F. (2012) De novo biosynthesis of starch chains without a primer and the mechanism for its biosynthesis by potato starch-synthase. Carbohydr. Res. 352, 137–142 [DOI] [PubMed] [Google Scholar]
- 10. Soya N., Fang Y., Palcic M. M., and Klassen J. S. (2011) Trapping and characterization of covalent intermediates of mutant retaining glycosyltransferases. Glycobiology 21, 547–552 [DOI] [PubMed] [Google Scholar]
- 11. Lee S. S., Hong S. Y., Errey J. C., Izumi A., Davies G. J., and Davis B. G. (2011) Mechanistic evidence for a front-side, S(N)i-type reaction in a retaining glycosyltransferase. Nat. Chem. Biol. 7, 631–638 [DOI] [PubMed] [Google Scholar]
- 12. Rongine de Fekete M. A., Leloir L. F., and Cardini C. E. (1960) Mechanism of starch biosynthesis. Nature 187, 918–919 [DOI] [PubMed] [Google Scholar]
- 13. Leloir L. F., Rongine de Fekete M. A., and Cardini C. E. (1961) Starch and oligosaccharide synthesis from uridine diphosphate glucose. J. Biol. Chem. 236, 636–641 [PubMed] [Google Scholar]
- 14. Recondo E., and Leloir L. F. (1961) Adenosine diphosphate glucose and starch synthesis. Biochem. Biophys. Res. Commun. 6, 85–88 [DOI] [PubMed] [Google Scholar]
- 15. Mukerjea R., Yu L., and Robyt J. F. (2002) Starch biosynthesis: mechanism for the elongation of starch chains. Carbohydr. Res. 337, 1015–1022 [DOI] [PubMed] [Google Scholar]
- 16. Mukerjea R., and Robyt J. F. (2005) Starch biosynthesis: further evidence against the primer nonreducing-end mechanism and evidence for the reducing-end two-site insertion mechanism. Carbohydr. Res. 340, 2206–2211 [DOI] [PubMed] [Google Scholar]
- 17. Imparl-Radosevich J. M., Li P., Zhang L., McKean A. L., Keeling P. L., and Guan H. (1998) Purification and characterization of maize starch synthase I and its truncated forms. Arch. Biochem. Biophys. 353, 64–72 [DOI] [PubMed] [Google Scholar]
- 18. Edwards A., Borthakur A., Bornemann S., Venail J., Denyer K., Waite D., Fulton D., Smith A., and Martin C. (1999) Specificity of starch synthase isoforms from potato. Eur. J. Biochem. 266, 724–736 [DOI] [PubMed] [Google Scholar]
- 19. Bustos R., Fahy B., Hylton C. M., Seale R., Nebane N. M., Edwards A., Martin C., and Smith A. M. (2004) Starch granule initiation is controlled by a heteromultimeric isoamylase in potato tubers. Proc. Natl. Acad. Sci. U.S.A. 101, 2215–2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Senoura T., Isono N., Yoshikawa M., Asao A., Hamada S., Watanabe K., Ito H., and Matsui H. (2004) Characterization of starch synthase I and II expressed in early developing seeds of kidney bean (Phaseolus vulgaris L.). Biosci. Biotechnol. Biochem. 68, 1949–1960 [DOI] [PubMed] [Google Scholar]
- 21. Busi M. V., Palopoli N., Valdez H. A., Fornasari M. S., Wayllace N. Z., Gomez-Casati D. F., Parisi G., and Ugalde R. A. (2008) Functional and structural characterization of the catalytic domain of the starch synthase III from Arabidopsis thaliana. Proteins 70, 31–40 [DOI] [PubMed] [Google Scholar]
- 22. Valdez H. A., Busi M. V., Wayllace N. Z., Parisi G., Ugalde R. A., and Gomez-Casati D. F. (2008) Role of the N-terminal starch-binding domains in the kinetic properties of starch synthase III from Arabidopsis thaliana. Biochemistry 47, 3026–3032 [DOI] [PubMed] [Google Scholar]
- 23. Huang B., Keeling P. L., Hennen-Bierwagen T. A., and Myers A. M. (2016) Comparative in vitro analyses of recombinant maize starch synthases SSI, SSIIa, and SSIII reveal direct regulatory interactions and thermosensitivity. Arch. Biochem. Biophys. 596, 63–72 [DOI] [PubMed] [Google Scholar]
- 24. Bax A., Clore G. M., and Gronenborn A. M. (1990) 1H1H correlation via isotropic mixing of 13C magnetization, a new three-dimensional approach for assigning 1H and 13C spectra of 13C-enriched proteins. J. Mag. Reson. 88, 425–431 [Google Scholar]
- 25. Mukerjea R., and Robyt J. F. (2005) Starch biosynthesis: the primer nonreducing-end mechanism versus the nonprimer reducing-end two-site insertion mechanism. Carbohydr. Res. 340, 245–255 [DOI] [PubMed] [Google Scholar]
- 26. Mukerjea R., Falconer D. J., Yoon S. H., and Robyt J. F. (2010) Large-scale isolation, fractionation, and purification of soluble starch-synthesizing enzymes: starch synthase and branching enzyme from potato tubers. Carbohydr. Res. 345, 1555–1563 [DOI] [PubMed] [Google Scholar]
- 27. Momma M., and Fujimoto Z. (2012) Interdomain disulfide bridge in the rice granule bound starch synthase I catalytic domain as elucidated by X-ray structure analysis. Biosci. Biotechnol. Biochem. 76, 1591–1595 [DOI] [PubMed] [Google Scholar]
- 28. Cuesta-Seijo J. A., Nielsen M. M., Marri L., Tanaka H., Beeren S. R., and Palcic M. M. (2013) Structure of starch synthase I from barley: insight into regulatory mechanisms of starch synthase activity. Acta crystallogr. D Biol. Crystallogr. 69, 1013–1025 [DOI] [PubMed] [Google Scholar]
- 29. Sheng F., Jia X., Yep A., Preiss J., and Geiger J. H. (2009) The crystal structures of the open and catalytically competent closed conformation of Escherichia coli glycogen synthase. J. Biol. Chem. 284, 17796–17807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sheng F., Yep A., Feng L., Preiss J., and Geiger J. H. (2009) Oligosaccharide binding in Escherichia coli glycogen synthase. Biochemistry 48, 10089–10097 [DOI] [PubMed] [Google Scholar]
- 31. Buschiazzo A., Ugalde J. E., Guerin M. E., Shepard W., Ugalde R. A., and Alzari P. M. (2004) Crystal structure of glycogen synthase: homologous enzymes catalyze glycogen synthesis and degradation. EMBO J. 23, 3196–3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lesley S. A., Kuhn P., Godzik A., Deacon A. M., Mathews I., Kreusch A., Spraggon G., Klock H. E., McMullan D., Shin T., Vincent J., Robb A., Brinen L. S., Miller M. D., McPhillips T. M., et al. (2002) Structural genomics of the Thermotoga maritima proteome implemented in a high-throughput structure determination pipeline. Proc. Natl. Acad. Sci. U.S.A. 99, 11664–11669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Matsui M., Guandique E., Tran M., Nisperos S., Orry A., and Meyer C. (2010) Characterization of the Thermotoga maritima ADP-glucose pyrophosphorylase: both glgC and glgD subunits are required for optimal activity and allosteric regulation. FASEB J. 24, 835.10 [Google Scholar]
- 34. Yep A., Bejar C. M., Ballicora M. A., Dubay J. R., Iglesias A. A., and Preiss J. (2004) An assay for adenosine 5′-diphosphate (ADP)-glucose pyrophosphorylase that measures the synthesis of radioactive ADP-glucose with glycogen synthase. Anal. Biochem. 324, 52–59 [DOI] [PubMed] [Google Scholar]
- 35. O'Shea M. G., and Morell M. K. (1996) High resolution slab gel electrophoresis of 8-amino-1,3, 6-pyrenetrisulfonic acid (APTS) tagged oligosaccharides using a DNA sequencer. Electrophoresis 17, 681–686 [DOI] [PubMed] [Google Scholar]