
Fig. 4.
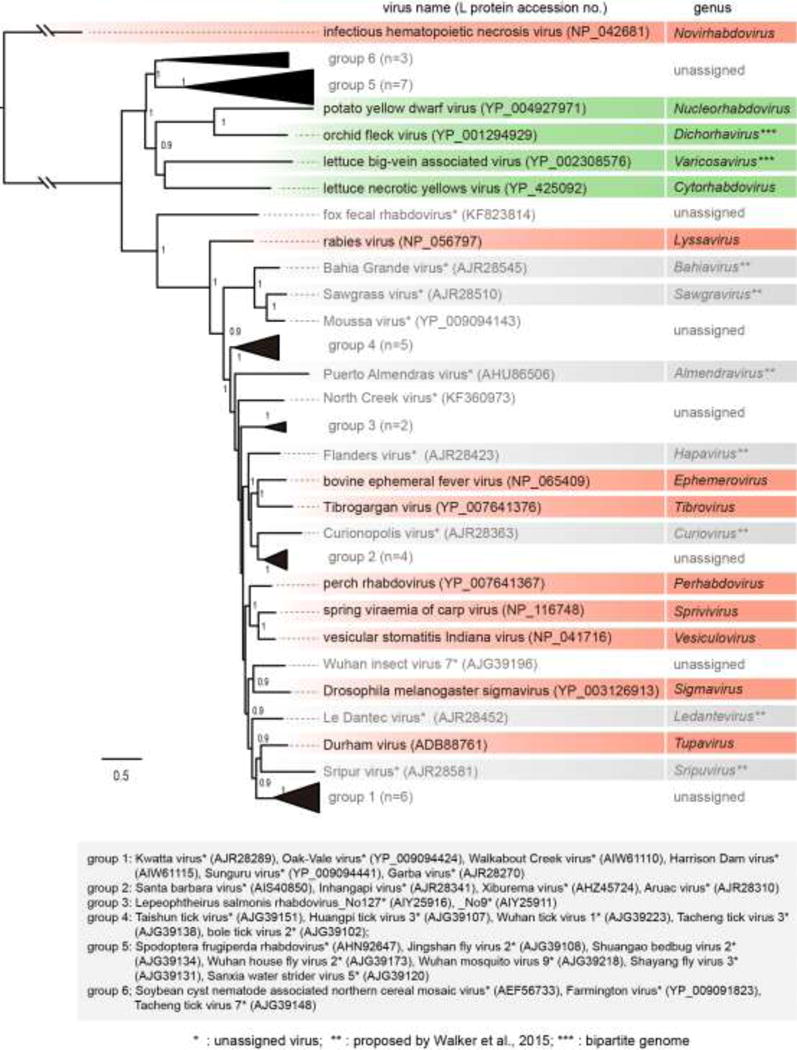
Phylogenetic relationships of members of the family Rhabdoviridae. The tree was constructed by the maximum likelihood (ML) method as described previously (Kondo et al., 2015). The entire L polymerase sequences were aligned using MAFFT 7.0 (Katoh and Toh, 2008) under default settings and ambiguously aligned regions were removed using Gblocks 0.91b (Talavera and Castresana, 2007) with all the options of less stringent selection. A model LG with + I + G + F was selected as the best fit model using PhyML 3.0 (Guindon et al., 2010) with automatic model selection by Smart Model Selection (SMS). The resulting tree was visualized with the FigTree 1.3.1 (http://tree.bio.ed.ac.uk/software/). Numbers at the nodes indicate aLRT values determined using an SH-like calculation (values less than 0.9 are not displayed). Virus names (the member of type species of genera and other selected unclassified rhabdoviruses) and GenBank/Refseq accession numbers (within parentheses) of L proteins are shown. Distantly related rhabdoviruses (group 1 to 6; n, number of sequences) that formed monophyletic clades, probably establishing additional genera in the family, were collapsed into a black triangle. The names and accession numbers of these viruses are shown below the tree.