

Abstract
Purpose
ABT-751 is an orally bioavailable sulfonamide that binds the colchicine site of beta-tubulin, thus inhibiting microtubule polymerizaton. A prior phase I study established the recommended dose in children with solid tumors as 200 mg/m2 PO daily × 7 days every 21 days and subjects with neuroblastoma experienced prolonged stable disease. We conducted a phase 2 study in children with progressive neuroblastoma to determine if ABT-751 prolonged the time to progression (TTP) compared to a hypothesized standard based on a historical control population.
Experimental Design
Children and adolescents (n=91) with a median (range) age 7.7 (2.3-21.5) years and progressive neuroblastoma were enrolled and stratified by disease status into disease measureable by CT/MRI (n=47) or disease assessable by 123I-metaiodobenzylguanine scintigraphy (MIBG, n=44). Response was evaluated using RECIST for measureable disease and the Curie scale for MIBG-avid disease.
Results
ABT-751 was well tolerated. Two complete responses and four partial responses were achieved. The median TTP was 42 days (95% CI: 36, 56) in the measureable disease stratum and 45 days (95% CI: 42, 85) in the MIBG-avid disease stratum. These values are similar to TTP in the historical control group (n=136, median TTP 42 days). One-year progression free (PFS) and overall survival (OS) for the combined strata (n=91) were 13%±4%, 48%±5%, respectively.
Conclusions
Although ABT-751 has many characteristics of an ideal maintenance agent for neuroblastoma, the low objective response rate and failure to prolong TTP indicate that ABT-751 is not sufficiently active to warrant further development for neuroblastoma.
Keywords: neuroblastoma, time to progression, clinical trial, microtubule inhibitor, childhood cancer
Introduction
For children and adolescents with high risk neuroblastoma who receive intensive multimodality induction and consolidation with autologous stem cell transplant, survival has been improved by the addition of the differentiating agent isotretinoin(1) and immunotherapy including the chimeric anti-disialoganglioside (GD2) monoclonal antibody ch 14.18.(2, 3) However, up to 50% of children with high-risk neuroblastoma experience recurrent disease that is almost uniformly fatal, necessitating improvements in current therapy.(3, 4) The goal of post chemotherapy maintenance treatment would be to eradicate minimal residual disease and improve survival. Ideally, maintenance therapy should include agents that have activity against neuroblastoma, can be easily administered, and have a toxicity profile that is tolerable after intensive induction and consolidation therapy.
ABT-751 (Abbott Laboratories, Abbott Park, IL), an orally bioavailable sulfonamide, binds the colchicine site on beta-tubulin and inhibits polymerization of microtubules.(5) The ABT-751 IC50 was less than 3 μM in neuroblastoma cell lines and less than 6 μM in other pediatric solid tumor cell lines.(6) In xenograft models, ABT-751 resulted in regression of rhabdomyosarcoma and Wilms tumors and prolonged time to tumor progression in neuroblastoma xenografts.(7) In children with relapsed solid tumors, the recommended dose of ABT-751 was 200 mg/m2 administered orally once daily for 7 days every 21 days. Dose-limiting toxicities were neuropathy, hypertension, and fatigue. Non-dose-limiting toxicities included anemia, abdominal pain, nausea, constipation, anorexia, fever, and weight loss. Myelosuppression was minimal and no significant cumulative toxicities were observed.(8) In children, the time to peak ABT-751 plasma concentration was 2 h and half-life was 5.1 h. The exposure (AUC0‐∞) in children receiving 200 mg/m2 was 91 μg·h/ml.(9) In the expanded phase 1 trial in children, the median event-free survival (EFS) was 9.3 weeks for 45 children with neuroblastoma and 3.3 weeks for 26 children with other solid tumors (p<0.0001). No complete or partial responses were documented using RECIST.(6)
Based on the ease of administration, acceptable toxicity profile, prolonged disease stabilization and EFS observed in heavily pre-treated population with neuroblastoma enrolled on the phase I studies of ABT-751, this phase 2 study was designed to evaluate PFS and assess the objective response rate of ABT-751 in children and adolescents with progressive neuroblastoma. This was the first clinical trial to incorporate PFS as a primary endpoint for a phase 2 study in children and adolescents with relapsed neuroblastoma. Eligibility criteria and disease assessments were tailored to determine disease progression and comparison to a standard derived from a historical control. Measures of clinical benefit including patient reported health related quality of life were evaluated throughout the trial as secondary objectives to support a PFS endpoint. In addition, an optional pharmacokinetic study of a suspension formulation of ABT-751was performed.
Materials and Methods
Eligibility
Patients (<22 years old) with refractory or relapsed neuroblastoma were eligible if they had radiographic evidence of disease progression. For patients with stable disease or previously irradiated sites of disease, a biopsy was required to demonstrate viable neuroblastoma in order to be considered evaluable for response. A performance status of at least 50% assessed using Karnofsky score (patients > 16 years old) or Lansky scale (patients ≤ 16 years old) was required.
There was no eligibility limitation to the number of prior relapses or prior therapeutic modalities. However, patients were required to have recovered from acute toxicity of therapy and be at least 2 weeks from the last dose of myelosuppressive therapy, 1 week from the last dose of retinoid therapy or growth factor support, 4 weeks from completion of external beam radiation therapy, 6 weeks from therapeutic 131I-MIBG, 2 months from autologous or 4 months from allogeneic stem cell transplant, and 30 days from the last dose of investigational agent or immunotherapy.
Adequate organ function was defined as normal serum creatinine, bilirubin less than or equal to 1.5 times the upper limit of normal (× ULN), alanine aminotransferase less than 5 × ULN, and left ventricular shortening fraction greater than 27% by echocardiogram. Residual neurological toxicity from disease or prior therapy must have been stable and less than or equal to grade 2 in severity. Adequate bone marrow function included hemoglobin greater than 7.5 g/dL (transfusion permitted), absolute neutrophil count greater than 250/μL, and platelet count greater than 25,000/μL without platelet transfusion for at least 7 days.
Patients were excluded if they were pregnant or breastfeeding, had allergy to sulfa medication, previously received ABT-751, or had a serious medical illness that would preclude evaluation. Patients were also excluded if they had disease restricted to bone marrow or had isolated elevated urinary catecholamines
Patients were enrolled in one of two strata: Stratum 1 - Patients with RECIST (Response Evaluation in Solid Tumors) measurable disease on CT or MRI scan, with or without 123I-MIBG positive disease, with or without bone marrow metastases. Stratum 2 – Patients with evaluable disease by 123I-MIBG scintigraphy (positive at a minimum of one site) with or without bone marrow metastases but without RECIST measureable disease.
Informed consent and assent were obtained according to local IRB guidelines. Participating hospitals met all institutional, FDA, ICH/GCP, COG and NCI requirements for human subject research.
Drug Administration
ABT-751 was supplied by Abbott Laboratories (Abbott Park, IL) as 25 mg and 100 mg hard gel capsules or 25 mg/mL suspension. A dosing nomogram was used to prescribe 200 mg/m2/dose, orally, once daily, for 7 days every 21 days and for subsequent dose reductions for toxicity, if necessary. The maximum daily dose was 300 mg, a maximum of 52 cycles was permitted. Docusate sodium was recommended as prophylaxis for constipation.
Trial Design and Primary Endpoint
The primary objective was to determine if the time to progression (TTP) in patients with relapsed or refractory, progressive neuroblastoma treated with ABT-751 was prolonged compared to a hypothesized model of the standard, which was derived from a historical control cohort (Figure 2D). For patients in stratum 1, progression was defined using RECIST. In stratum 2, progression was based on the appearance of a new lesion on MIBG scan. Disease evaluations were performed at baseline and after cycles, 2, 4, 6, 8, 10, 14 and then after every 4th cycle. Clinical progression declared by the treating physician without tumor imaging was considered progressive disease.
Figure 2.
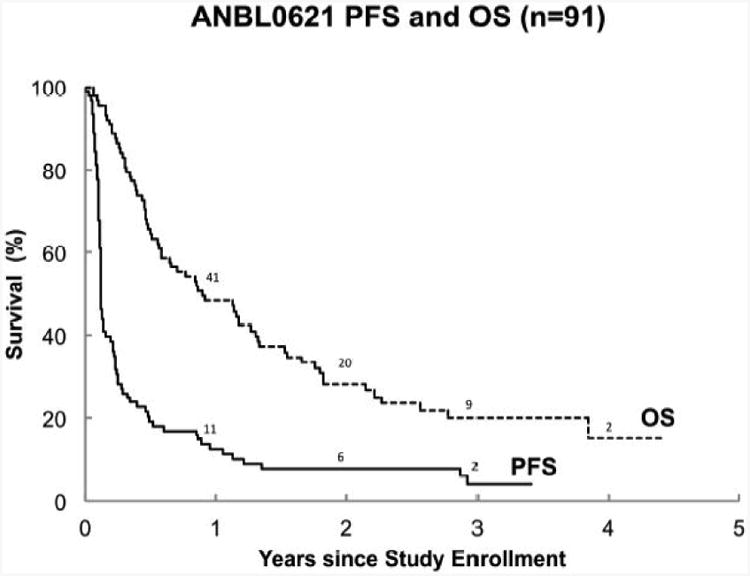
(A) Progression-free survival (PFS) and overall survival (OS) for all patients (n=91). The 1-year PFS and OS were 13%±4% and 48%±5%, respectively. The median TTP was 44 days (95% CI: 42, 56).
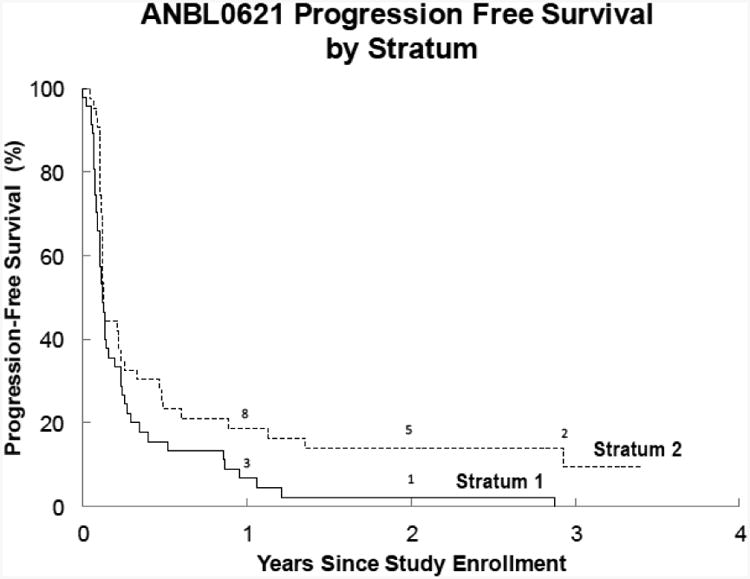
(B) Progression-free survival (PFS) for Stratum 1 (measureable disease by RECIST) and Stratum 2 (MIBG evaluable disease). The 1-year PFS was 7%±4% for stratum 1 and 19%±6% for stratum 2 (p=0.03). The median TTP was 42 days (95% CI: 36,56) in stratum 1 and 45 days (95%CI: 42, 85) in stratum 2.
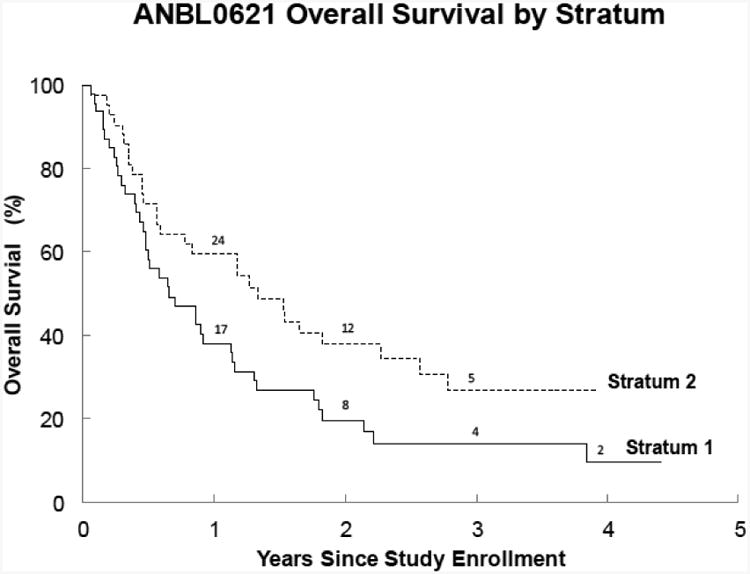
(C) Overall survival (OS) for Stratum 1 (Measurable disease) and Stratum 2 (MIBG evaluable). The 1-year OS was 30%±7% for stratum 1 and 60%±8% for stratum 2 (p=0.04).
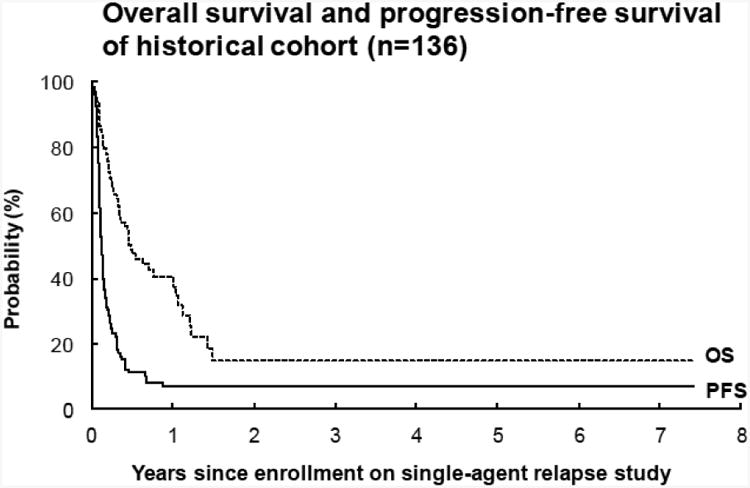
(D) Overall survival and progression-free survival of the historical cohort (n=136), comprised of Phase 1 and 2 studies CCG-8607, ANBL00321, NANT-0103, CCG-0926, and CCG-0961. The median TTP was 42 days.
The historical control group included patients who were enrolled on Phase 1 and 2 studies with similar eligibility criteria to this study including CCG-8607 (Phase 2 13-cis-retinoic acid), ANBL0321 (Phase 2 Fenretinide), NANT2001-03 (Phase 1 CEP-701), CCG-0926 (Phase 1 All Trans Retinoic Acid/Interferon alpha 2A), and CCG-0961(Phase 2 All Trans Retinoic Acid/Interferon alpha 2A). In the historical control patients (n=136), the 42-day PFS was 50%± 4% which is analogous to a median TTP of 42 days.
Secondary Endpoints
Objective Response
In stratum 1, RECIST (Version 1.0) was used to determine the objective (Complete Response + Partial Response) response rate. In stratum 2, the Curie Scale(10) was used to determine response. Clinical decisions were based on institutional assessment of overall response. All imaging studies were centrally reviewed (GK or HAJ). Central review of images obtained at least 4 weeks after initial response was required to establish a confirmed radiographic response.
Toxicity and Dose Limiting Toxicity (DLT)
Toxicity was graded according to Common Terminology Criteria for Adverse Events (CTCAE v 3.0, http://ctep/info.nih.gov) except for motor or sensory neuropathy which was graded as previously described.(11) Hematological and non-hematological DLT were defined separately. Any grade 3 or 4 non-hematological toxicity related to ABT-751 or failure to recover to grade 1 or baseline by cycle day 28 was considered to be a DLT. Since patients with compromised bone marrow function (baseline ANC less than 1000/μL or platelet count less than 100,00/μL) were eligible to enroll on this study, the definition of hematological DLT was based on hematological function at enrollment and was defined as failure to recover to levels previously required by the eligibility criteria or to 80% of enrollment ANC and platelet count (for those with compromised bone marrow) by day 28 of a treatment cycle.
Dose limiting toxicity that occurred during drug administration (day 1-7 of any cycle) resulted in discontinuation of ABT-751 for the duration of the cycle. Patients who experienced a DLT (days 1-7 or after day 7) that resolved to baseline or grade 1 prior to day 42 of the cycle could proceed to the next cycle at a reduced dose. Two dose reductions for toxicity were permitted. If toxicity did not resolve by day 42, protocol therapy was discontinued.
Heath Related Quality of Life (HRQL) and Measures of Clinical Benefit
HRQL was assessed using PedsQL™ version 4.0 Generic Core Scale.(12, 13) The 23 item self-administered or parent-proxy-administered scale measures domains in physical, emotional, social and school functioning. The PedsQL™ was administered to patients and proxy at baseline prior to each cycle, and at the off-protocol-therapy visit. Items on the PedsQL are reversed scored and linearly transformed to a 0-100 scale with higher scores indicating better health-related quality of life.(14) Measures of clinical benefit included physician's assessment of performance status using either Lansky or Karnofsky scores, number of hospitalizations and number of blood product transfusions per cycle.
Pharmacokinetics of ABT-751 Suspension
Optional pharmacokinetic samples were obtained after the first dose of ABT-751 in children receiving the suspension formulation. Limited plasma sampling, HPLC tandem mass spectrometer assay, and pharmacokinetic analysis were performed as previously described.(9)
Statistical Analysis Plan
The primary objective of the study compares the median time to progression (TTP) observed on ABT-751 to a hypothesized gold standard derived from an exponential model of the failure rate corresponding to a median PFS of 42 days.(15) TTP was defined as the shortest of the following times:
date of enrollment to date of documentation of progressive disease by any imaging modality or clinical progression; or,
date of enrollment to date of death from any cause.
For the PFS analysis, the survival times for patients who did not experience progression or death were handled as censored observations.
All patients who received at least one dose of ABT-751 were included in the analysis of the primary endpoint. Within each stratum separately, if the lower bound of a 95% confidence interval on the observed TTP excluded (was greater than) 42 days, then ABT-751 would be considered to have superior PFS within a given stratum compared to the standard. A sample size of 44 patients per stratum provided ≥85% power to detect an increase in median TTP from 42 days (standard) to 89 days, where the latter corresponds to a hazard ratio (relative failure rate) of 0.474 (assuming exponential failure distribution). Overall survival was calculated from the time of study enrollment to the time of death from any cause. Kaplan-Meier curves of PFS and OS, including the 1–year point estimates ± standard error, are presented.
Responders were defined as patients who achieved a best overall response of complete or partial response at any time on the study including patients who achieved ≥PR and later have progressive disease or relapse. Within each stratum, the objective response rate was calculated as the number of responders divided by the number of patients who got a least one dose of ABT-751.
All patients who received at least 1 dose of ABT-751 were evaluable for toxicity. A patient was counted only once for a given toxicity for the worst grade of that toxicity reported in a given cycle. The change from baseline (best score at any time after enrollment minus baseline score) of performance status, and HRQL per PedsQL™ Generic Core Scale version 4.0 were tabulated. A Wilcoxon signed-rank test was used to determine if the change from baseline differed from zero for the PedsQL™. Pharmacokinetic parameters including maximum concentration (Cmax), time to Cmax (Tmax), half-life, apparent clearance, and area under concentration time curve (AUC) were descriptively summarized.
Results
Patient Population
Between March 2007 and August 2009, 92 patients were enrolled at 28 participating Children's Oncology Group institutions. Ninety-one patients met all eligibility criteria and received at least one dose of ABT-751 (Figure 1). Patient characteristics are presented in Table 1.
Figure 1. ANBL0621 Consort Diagram.
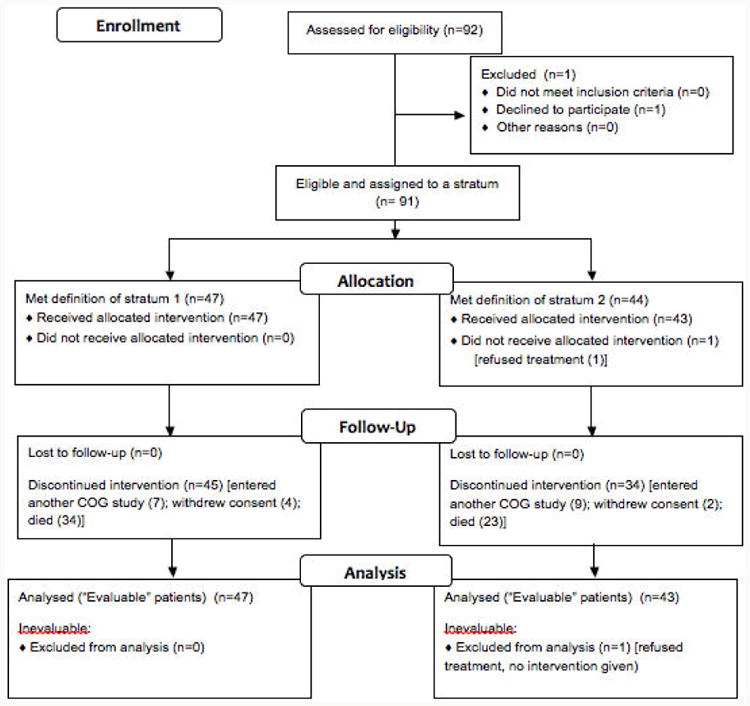
Table 1. Patient Characteristics.
| All | Stratum 1 | Stratum 2 | |
|---|---|---|---|
|
| |||
| N | 91 | 47 | 44 |
|
| |||
| Males (%) | 57 (63%) | 26 (55%) | 31 (70%) |
|
| |||
| Age, median (range) years | |||
|
| |||
| At enrollment (n=91) | 7.7 (2.3-21.5) | 7.5 (2.3-21.5) | 8.5 (3.7-19.6) |
|
| |||
| At diagnosis (n=91) | 4.5 (0.6-18.8) | 4.3 (0.6-18.8) | 4.6 (0.6-16.5) |
|
| |||
| Median (range) time from diagnosis to first relapse (years)* | 2.1 (0.4-4.8) n=39 | 1.9 (0.4-4.2) n=22 | 2.1 (0.4-4.8) n=17 |
|
| |||
| Race | |||
| White | 60 (76%) | 30 (73%) | 30 (79%) |
| Black | 17 (22%) | 9 (22%) | 8 (21%) |
| Other | 2 (2%) | 2 (5%) | 0 (0%) |
| Unknown | 12 | 6 | 6 |
|
| |||
| Tumor Biology | |||
| MYCN Amplified (38 known) | 6 (16%) | 5 (21%) | 1 (7%) |
| High MKI (11 known) | 0 (0%) | 0 (0%) | 0 (0%) |
| Diploid (36 known) | 21 (58%) | 13 (64%) | 8 (62%) |
|
| |||
| Extent of Disease | |||
| Measurable Disease | |||
| Soft Tissue Mass | 44 | 44 | - |
| Bone Lesion (MRI) | 2 | 2 | - |
| Unknown | 1 | 1 | - |
|
| |||
| Bone (MIBG or Bone Scan) | |||
| Positive | 75 | 31 | 44 |
| Negative | 1 | 1 | - |
| Unknown | 15 | 15 | - |
|
| |||
| Bone Marrow (Percent tumor by histopathology) | |||
| Negative | 24 | 12 | 12 |
| 1-24% | 27 | 12 | 15 |
| 25-49% | 3 | 0 | 3 |
| 50-90% | 5 | 3 | 2 |
| >90% | 5 | 4 | 1 |
| Unknown | 27 | 16 | 11 |
Outcome data were available prior to ANBL0621 study enrollment for 39 patients
Primary Endpoint: Time to Progression
The median TTP (Figure 2B) was 42 days (95% CI: 36, 56) for patients with measureable disease (Stratum 1) and 45 days (95% CI: 42, 85) for patients with disease assessed by MIBG (Stratum 2). In each stratum, the 95% CI included 42 days; the median TTP was not higher than the hypothesized model of the standard.
Objective Response
Central review of all imaging studies occurred for every subject enrolled. Summary of central review of overall best response is presented in Table 2. In stratum 1, there was one confirmed partial response (PR). In stratum 2, there were two complete responses (one confirmed) and three confirmed PRs.
Table 2. Central Review Assessment of Best Response to ABT-751 therapy.
| Number (%) of patients | |||
|---|---|---|---|
| Overall (n=90)* | Stratum 1 (n=47) | Stratum 2 (n=43)* | |
| Complete Response | 2 | 0 | 2 |
| Partial Response | 4 | 1 | 3 |
| Stable Disease | 32 | 19 | 13 |
| Progressive Disease | 52 | 27 | 25 |
| Responders (≥PR) | 6 (7%) 95% CI: (2%, 12%) | 1 (2%) 95% CI: (0%, 6%) | 5 (12%) 95% CI: (2%, 21%) |
Of 91 eligible patients, one was not evaluable for response (refused therapy).
CI = 95% confidence Interval
Toxicity
ABT-751 was well tolerated. The median (range) number of cycles of therapy delivered was 2 (<1-50). Toxicities were similar to those previously reported with ABT-751 in children including fatigue, anorexia, nausea and vomiting, constipation, ileus, decreased LVEF, peripheral motor and sensory neuropathy including pain, hepatic transaminase elevations, and mood disturbances. Nine patients discontinued protocol therapy due to toxicity (n=3), withdrawal of consent after non-dose limiting toxicity (n=4), or because the treating physician recommended an alternative treatment not related to toxicity (n=2). Toxicities resulting in discontinuation of ABT-751 included delayed neutrophil or platelet recovery for more than 42 days, neuropathy, transient grade 2 decrease in LVEF, infection and weight loss. No cumulative toxicity was observed in patients receiving up to 50 cycles of therapy.
Survival, HRQL and Measures of Clinical Benefit
The one-year progression-free (PFS) and overall survival (OS) for the combined strata were 13%±4%, and 48%±5%, respectively (Figure 2A). PFS was higher for patients in the MIBG stratum (stratum 2) compared to patients with measurable disease (stratum 1) (p=0.03). Measures of clinical benefit are summarized in Table 3. Transfusion of red cells or platelets occurred in 27% of patients (24/90) and 6% of cycles (31/518). Sixty-three percent of patients did not require any hospitalization. There was no change in performance status during ABT-751 administration. In addition, there was no change in patient or proxy reported outcome of physical, emotional, social and school functioning using the PedsQL v 4.0 total score.
Table 3. Measures of Clinical Benefit.
| All | Stratum 1 | Stratum 2 | ||||
|---|---|---|---|---|---|---|
| n | median (range) | n | median (range) | n | median (range) | |
| Performance Status | ||||||
| Baseline | 89 | 100 (50 to 100) | 46 | 100 (50 to 100) | 43 | 100 (90 to 100) |
| Off Therapy Score | 89 | 90 (40 to 100) | 47 | 90 (40 to 100) | 42 | 100 (60 to 100) |
| Change: (Best score minus baseline) | 88 | 0 (-50 to 20) | 46 | 0 (-50 to 20) | 42 | 0 (-40 to 10) |
| Quality of Life: PedsQL v 4.0 | ||||||
| Baseline* Composite Score | 71 | 76 (29-100) | 34 | 68 (29-100) | 37 | 80 (44-99) |
| Off Therapy Composite Score | 62 | 77 (0-100) | 30 | 70 (0-100) | 32 | 82 (40-100) |
| Change: (off therapy minus baseline) p-value | 62 | +1 (-38 to +24) (p=0.80) | 30 | +1 (-38 to +24) (p=0.98) | 32 | +1 (-37 to +24) (p=0.83) |
Subject-Proxy Correlation: R2=0.82 overall, R2=0.86 stratum 1, and R2=0.74 stratum 2.
Pharmacokinetics of ABT-751 Suspension
Forty-three children or adolescents received ABT-751 suspension, 37% (16/43) participated in pharmacokinetics. The median (range) age of patients participating in pharmacokinetics was 4.8 (1.7-15.7) years. Plasma pharmacokinetic parameters of ABT-751 and metabolites are presented in Table 4.
Table 4. Median (Range) Plasma Pharmacokinetic Parameters of ABT-751 Suspension.
| Subjects n=16 | ABT-751 | ABT-751 Glucuronide | ABT-751 Sulfate |
|---|---|---|---|
| Cmax (μg/mL) | 15.3 (7.3-22.4) | 8.4 (3.7-13.4) | 11.8 (4.7-22.4) |
| Tmax (h) | 1 (1-3) | 3 (3-10) | 3 (1-6) |
| AUC0-24 (μg·h/mL) | 73.6 (45.3-164.3) | 85.1 (18.5-171.2) | 131.7 (63.8-255.7) |
| AUC0-∞ (μg·h/mL) | 77.5 (46.8-169.3) | 92.8 (28.1-270.6) | 150.1 (94.7-295.6) |
| CL/F (mL/min/m2) | 40.8 (18.6-60.8) | - | - |
| T1/2 (h) | 5.8 (4.4-11.1) | 6.6 (4.5-19.2) | 7.4 (5.5-17.4) |
Discussion
This is the first phase 2 clinical trial in children and adolescents with relapsed neuroblastoma to use TTP as the primary endpoint. An accurate assessment of TTP in this population was assured through enrollment stratification (measureable or 123I-MIBG evaluable disease), exclusion of patients with isolated bone marrow relapse of neuroblastoma, an eligibility requirement for progressive disease at enrollment, and adherence to timing of tumor restaging evaluations. This design is especially valuable for relatively rare cancers of childhood in which placebo-controlled randomized phase 2 designs may not be feasible.
ABT-751 did not extend the TTP in children and adolescents with relapsed or refractory neuroblastoma compared to the hypothesized standard derived from the historical control. Although comparison to historical control has significant limitations, this was mitigated by using clinical trials with similar eligibility criteria and by performing the comparison to a fixed standard rather than directly to the historical data. Based on five clinical trials of inactive agents in relapsed or refractory neuroblastoma patients we established a TTP for an inactive agent as 42 days.
ABT-751 did not improve TTP in either stratum 1 or stratum 2; the 95% CI in each case included 42 days. Although it was not an objective of this study to demonstrate a difference in PFS by stratum, the fact that PFS was longer for patients in the MIBG stratum (stratum 2) compared to patients with measurable disease (stratum 1) indicates that stratification based on extent of disease and primary imaging modality may be advisable in future clinical trials for patients with relapsed neuroblastoma
The toxicity profile of ABT-751 was similar to prior experience in children except for the observed incidence of myelosuppression. Myelosuppression is likely multi-factorial; patients with relapsed or refractory neuroblastoma have limited bone marrow reserve due to tumor involvement and prior intensive therapy. The eligibility criteria for enrollment in our study permitted compromised bone marrow function. Using a definition of dose limiting hematological toxicity relative to enrollment hematological function, three children discontinued therapy for myelosuppression or delay in blood count recovery (>42 days). Despite marginal bone marrow reserve, transfusion requirements were minimal and the majority of patients did not require hospitalization. No cumulative toxicity was observed.
Development of palatable oral liquid formulations for new agents is essential for development of oral agents in children. In this trial, the median age at enrollment was 7.7 years and 48% of enrolled children received ABT-751 suspension. Pharmacokinetics of the suspension appear similar to capsule formulation previously studied.(9) Patient participation in the optional pharmacokinetic study demonstrates that using a limited sampling strategy, a pharmacokinetic study to evaluate new formulations can be performed in the phase 2 setting.
In summary, ABT-751 is an agent with a pediatric oral formulation and schedule of administration and toxicity profile that are amenable to use in the maintenance phase of therapy for children and adolescents with neuroblastoma. The low objective response rate and failure to prolong TTP indicate that ABT-751 is not sufficiently active to warrant further development in children or adolescents with neuroblastoma. However, the detailed reporting of enrollment characteristics, stratification based on disease status, and sufficient sample size of this study may serve as a robust historical control group for future clinical trials using TTP as an endpoint in children and adolescents with relapsed neuroblastoma.
Statement of Translational Relevance.
The pediatric phase 1 trials of the orally bioavailable tubulin binding agent ABT-751 demonstrated a favorable toxicity profile, including minimal myelosuppression, and prolonged disease stabilization and event free survival in children and adolescents with heavily pre-treated neuroblastoma. Based on these observations, this phase 2 clinical trial of ABT-751 for progressive neuroblastoma utilized time to progression (TTP) as a primary endpoint to assess the drug's activity and potential utility as a maintenance agent in front line treatment regimens for neuroblastoma. Pharmacokinetics, health related quality of life and measures of clinical benefit were secondary objectives. Detailed reporting of enrollment characteristics, stratification based on disease status, and sample size of this study serve as a robust historical control group for future clinical trials in this population. This trial design may be valuable for childhood cancers in which placebo-controlled randomized phase 2 designs may not be feasible.
Acknowledgments
Financial Support: This trial was sponsored by Abbott Laboratories (Abbott Park, IL). The Children's Oncology Group is funded by the National Cancer Institute, CA 098453.
Grant Support: The Children's Oncology Group is funded by the National Cancer Institute of the NIH: Chair's grant U10 CA 098453 and the Statistical and Data Center grant U10 CA98413.
Footnotes
Conflict of Interest: During the conduct of this study, AK and GG were employed by Abbott Laboratories. No other authors have financial conflict of interest to disclose.
References
- 1.Matthay K, Villablanca JG, Seeger RC, et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis retinoic acid. N Engl J of Med. 1999;341:1164–73. doi: 10.1056/NEJM199910143411601. [DOI] [PubMed] [Google Scholar]
- 2.Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010;363:1324–34. doi: 10.1056/NEJMoa0911123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369:2106–20. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- 4.Park JR, Eggert A, Caron H. Neuroblastoma: biology, prognosis, and treatment. Hematol Oncol Clin North Am. 2010;24:65–86. doi: 10.1016/j.hoc.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 5.Galmarini CM. ABT-751 (Abbott) Curr Opin Investig Drugs. 2005;6:623–30. [PubMed] [Google Scholar]
- 6.Meany HJ, Sackett DL, Maris JM, Ward Y, Krivoshik A, Cohn SL, et al. Clinical outcome in children with recurrent neuroblastoma treated with ABT-751 and effect of ABT-751 on proliferation of neuroblastoma cell lines and on tubulin polymerization in vitro. Pediatr Blood Cancer. 2010;54:47–54. doi: 10.1002/pbc.22267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morton CL, Favours EG, Mercer KS, Boltz CR, Crumpton JC, Tucker C, et al. Evaluation of ABT-751 against childhood cancer models in vivo. Invest New Drugs. 2007;25:285–95. doi: 10.1007/s10637-007-9042-y. [DOI] [PubMed] [Google Scholar]
- 8.Fox E, Maris JM, Widemann BC, Meek K, Goodwin A, Goodspeed W, et al. A phase 1 study of ABT-751, an orally bioavailable tubulin inhibitor, administered daily for 7 days every 21 days in pediatric patients with solid tumors. Clin Cancer Res. 2006;12:4882–7. doi: 10.1158/1078-0432.CCR-06-0534. [DOI] [PubMed] [Google Scholar]
- 9.Fox E, Maris JM, Cohn SL, Goodspeed W, Goodwin A, Kromplewski M, et al. Pharmacokinetics of orally administered ABT-751 in children with neuroblastoma and other solid tumors. Cancer Chemother Pharmacol. 2010;66:737–43. doi: 10.1007/s00280-009-1218-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ady N, Zucker JM, Asselain B, Edeline V, Bonnin F, Michon J, et al. A new 123I-MIBG whole body scan scoring method--application to the prediction of the response of metastases to induction chemotherapy in stage IV neuroblastoma. Eur J Cancer. 1995;31A:256–61. doi: 10.1016/0959-8049(94)00509-4. [DOI] [PubMed] [Google Scholar]
- 11.Fox E, Maris JM, Widemann BC, Goodspeed W, Goodwin A, Kromplewski M, et al. A phase I study of ABT-751, an orally bioavailable tubulin inhibitor, administered daily for 21 days every 28 days in pediatric patients with solid tumors. Clin Cancer Res. 2008;14:1111–5. doi: 10.1158/1078-0432.CCR-07-4097. [DOI] [PubMed] [Google Scholar]
- 12.Varni JW, Burwinkle TM, Seid M, Skarr D. The PedsQL 4.0 as a pediatric population health measure: feasibility, reliability, and validity. Ambul Pediatr. 2003;3:329–41. doi: 10.1367/1539-4409(2003)003<0329:tpaapp>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 13.Varni JW, Burwinkle TM, Katz ER, Meeske K, Dickinson P. The PedsQL in pediatric cancer: reliability and validity of the Pediatric Quality of Life Inventory Generic Core Scales, Multidimensional Fatigue Scale, and Cancer Module. Cancer. 2002;94:2090–106. doi: 10.1002/cncr.10428. [DOI] [PubMed] [Google Scholar]
- 14.Varni JW, et al. The Peds QL™ as a pediatric population health measure:feasibility, reliability, and validity. Ambulatory Pediatrics. 2003;3:329–41. doi: 10.1367/1539-4409(2003)003<0329:tpaapp>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 15.Woolson R. Rank tests and a one sample logrank test for comparing observed survival data to a standard population. Biometrics. 1981;37:687–96. [Google Scholar]