

Abstract
Metabolism of the essential amino acid L-tryptophan (TRP) is implicated in a number of neurological conditions including depression, neurodegenerative diseases, and cancer. The TRP catabolite kynurenine (KYN) has recently emerged as an important neuroactive factor in brain tumor pathogenesis, with additional studies implicating KYN in other types of cancer. Often highlighted as a modulator of the immune response and a contributor to immune escape for malignant tumors, it is well known that KYN has effects on the production of the co-enzyme nicotinamide adenine dinucleotide (NAD+), which can have a direct impact on DNA repair, replication, cell division, redox signaling, and mitochondrial function. Additional effects of KYN signaling are imparted through its role as an endogenous agonist for the aryl hydrocarbon receptor (AhR), and it is largely through activation of the AhR that KYN appears to mediate malignant progression in gliomas. We have recently reported on the ability of KYN signaling to modulate expression of human DNA polymerase kappa (hpol κ), a translesion enzyme involved in bypass of bulky DNA lesions and activation of the replication stress response. Given the impact of KYN on NAD+ production, AhR signaling, and translesion DNA synthesis, it follows that dysregulation of KYN signaling in cancer may promote malignancy through alterations in the level of endogenous DNA damage and replication stress. In the following perspectives article, we discuss the connections between KYN signaling, DNA damage tolerance, and genomic instability, as they relate to cancer.
Keywords: Aryl hydrocarbon receptor, cancer treatment, DNA replication, glioma/glioblastoma, kynurenine, polymerase, translesion DNA synthesis
Graphical Abstract
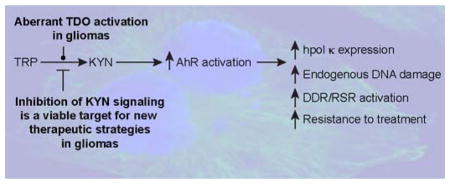
Aberrant kynurenine (KYN) signaling in gliomas promotes malignancy
L-tryptophan (TRP) is an essential amino acid in humans that has important neuroactive properties. Most TRP (~95%) in the body is metabolized to KYN by one of three enzymes: tryptophan 2,3-dioxygenase (TDO; also called TDO2), indoleamine 2,3-dioxygenase 1 (IDO1) and 2 (IDO2) (Figure 1).1 KYN is a precursor to both neuroprotective and neurotoxic molecules, such as 3-hydroxy-L-kynurenine (3-HK, neurotoxic), 3-hydroxyanthranilic acid (3-HAA, neurotoxic), kynurenic acid (KYNA, neuroprotective), picolinic acid (PIC, neuroprotective), and quinolinic acid (QUIN, neurotoxic).2 Metabolites of the KYN pathway (KP) act as N-methyl-D-aspartate (NMDA) receptor agonists/antagonists, glutamate receptor antagonists, and can contribute to the production of free radicals in the central nervous system (CNS).1 As discussed below, KYN is an endogenous agonist for the aryl hydrocarbon receptor (AhR), a transcription factor best known for its role in activation of genes following exposure to xenobiotics. Moreover, production of the co-enzyme nicotinamide adenine dinucleotide (NAD+), which is used in many biological pathways including ATP production, p53-mediated stress responses and DNA repair, depends on the KYN metabolite QUIN. QUIN has been shown to increase the production of free radicals, leading to oxidative stress, DNA damage, and increased Poly(ADP-ribose) polymerase 1 (PARP-1) activity.3–5 NAD+ influences DNA repair and gene expression through its role as a substrate for PARP-1.6 Poly(ADP-ribosylation) of acceptor proteins near sites of DNA damage influences the recruitment of DNA repair factors. PARP-1 activity also influences gene expression (through interactions with transcriptional co-activators) and cellular energy balance (through modulation of cellular ATP levels). Thus, aberrant KYN signaling and subsequent formation of the NAD+ precursor QUIN can act through PARP-1 to modulate genomic integrity.
Figure 1.

KYN signaling can have multifaceted effects on gliomas. Major steps in the breakdown of TRP through the KYN pathway are shown and important properties are noted. The effects of certain intermediates in the KYN pathway on glioma biology are listed in the box to the right.
There are a number of neurological pathologies associated with alterations in KYN signaling including Alzheimer’s disease, amyotrophic lateral sclerosis, Huntington’s disease, multiple sclerosis, and Parkinson’s disease. The physiology and clinical implications related to KYN signaling in the CNS is complex and has been reviewed in detail elsewhere.1 Needless to say, there are many aspects of how KYN and its metabolites affect neurodegenerative and autoimmune diseases that are under intense investigation as possible routes to new therapies. Likewise, the role of KYN signaling in cancer is multi-faceted and new molecular features continue to be reported. A landmark study in 2011 revealed a previously unrecognized connection between TDO-mediated elevation of KYN levels, subsequent activation of the AhR, and malignant progression in gliomas.7 Additionally, increases in the KYN/TRP ratio and/or increased expression of KYN pathway enzymes have been observed in adult T-cell leukemia/lymphoma,8 breast cancer,9, 10 cervical cancer,11 colorectal cancer,12 meningiomas,13 and non-small cell lung cancer.14 The effect of increased KYN signaling on tumor biology is multifaceted, but several metabolites of TRP catabolism, including KYN, can promote malignant properties in cancer (Figure 1). TRP catabolism has been linked to an immunosuppressive effect in cancer both by depleting Trp, which induces T-cell hyporesponsiveness and apoptosis, and by accumulation of immunosuppressive trp catabolites, such as KYN.15 Elevated KYN levels can suppress allogeneic T-cell proliferation, promote tumor cell survival, as well as motility, in glioma cells.7 While many studies have focused on the role of KYN in immune escape, there is now evidence supporting the notion that aberrant KYN signaling can alter genome stability in gliomas through changes in DNA repair and replication factors.
Glioma pathobiology, molecular features, and clinical impact
Gliomas, which arise from glial cells, are the most common type of primary brain tumor.16 Glial cells provide support and protection for neurons. They are the most common cells in the brain and consist of three subtypes: astrocytes, oligodendrocytes, and ependymal cells. More than three quarters of gliomas are astrocytomas, which are generally thought to arise from astrocytes, although it is unclear if they arise from differentiated astrocytes, astroglial progenitors, or neural stem cells.16 Other types are oligodendrogliomas and ependymomas, and tumors consisting of mixed types also occur.16 The most malignant form of glioma, glioblastoma multiforme (GBM or simply glioblastoma), is the most common of all malignant brain and CNS tumors, accounting for 46.1% of these patients.16 It is predicted that > 12,000 people will be diagnosed with GBM in 2016.16 Symptoms vary widely depending on where the tumor is located and the rate of growth. Recurrence rate for GBM is approximately 90%.17 In addition to being the most prevalent type of malignant tumor in the central nervous system, gliomas are also one of the deadliest forms of cancer. Glioblastoma is defined by the World Health Organization (WHO) as a grade IV astrocytoma. The current one-year relative survival rate for GBM is 37.2% and for five years is 5.1%.16 The median survival time of GBM patients has hovered near 12 months for the past decade18 despite the intense pursuit of new therapeutic strategies.
Morphologically, GBMs are characterized by widespread infiltration throughout the brain, which often makes surgical removal difficult. Additional complications arise from the extreme resistance to chemo- and radio- therapies, destruction of normal brain tissue, uncontrolled cellular proliferation, high vascularization, and rampant genomic instability observed in GBMs.18 These characteristics are common to most cancers,19 however, the degree of genomic instability in particular is much higher than in other epithelial carcinomas.20 As the name “multiforme” suggests, GBMs have significant intratumoral heterogeneity, which is also related to a high level of genomic instability. Understanding glioma-specific mechanisms promoting increased levels of replication stress and endogenous DNA damage is a vital area of research.
Aberrant DNA damage signaling and replication stress in gliomas
The poor prognosis associated with higher stage gliomas is due in part to the fact that these tumors, especially GBMs, generally exhibit little response to therapies, such as ionizing radiation and chemotherapy.18 This inherent resistance may be due to the fact that these cancers exhibit constitutive, widespread, and robust activation of the replication stress response and DNA damage signaling.20 Indeed, it has been shown that glioma cells with higher DNA damage response and cell cycle checkpoint activation are more resistant to irradiation than those with less activation.21 Since the prolonged presence of DNA breaks induces apoptosis or senescence, the ability to tolerate stress at the fork and repair DNA damage is essential for tumor cells to survive treatment with DNA damaging agents.22–24
Elevated levels of replication stress and DNA damage markers in gliomas have been observed before the onset of chemotherapy, suggesting that these events are intrinsic to the biology of the disease.20 Both low- and high-grade human gliomas exhibit elevated γH2AX levels, activation of the ATM-Chk2-p53 pathway, and increased 53BP1 foci formation.20 Chk1 activation, Rad17 phosphorylation, replication protein A (RPA) foci, and ssDNA gaps are all indicative of ongoing elevated replication stress in GBMs and are thought to be a major source of genomic instability in these tumors.20 There is evidence of elevated oxidative damage to DNA (i.e. increased 8-oxoguanine) in some GBM cell lines and tumors.20 However, the presence of high γH2AX staining in tumor sections with low 8-oxoguanine staining indicates that oxidative damage is not the main source of DNA damage inducing the replication stress response.20 Interestingly, the highest level of DNA damage signaling has been observed in grade II astrocytomas (based on γH2AX staining).20 Typically, DNA damage response activation is thought to act as a barrier to tumor progression, and this does seem to be the case in early stages of glioma. However, the fact that mutant p53 was most prevalent in GBM tissue sections with high constitutive DNA damage response activation supports a model where persistent replication stress selects for mutations that facilitate escape from cell cycle checkpoints.20 Selection through mutation is dependent upon mechanisms that promote tolerance of DNA damage and the resolution of replication stress.
Translesion DNA synthesis: a central mediator of DNA damage tolerance
DNA damage and replication stress responses play an essential role in helping maintain genomic stability through the activation of pathways that cause cell cycle arrest when DNA damage is detected, allowing the damage to be repaired.24 However, if there is unrepaired damage or damage that occurs during S-phase, lesions may be bypassed in lieu of actual repair to preserve fork stability and ensure completion of replication and avoid prolonged stalling, which can lead to fork collapse, double strand break formation, and increased genomic instability.25 The direct bypass of lesions occurs through a process known as translesion DNA synthesis (TLS). Evolution has retained multiple different types of DNA polymerases (pols) that are classified into different families, including A, B, C, D, X, Y, and reverse transcriptases.26 TLS is carried out by a group of specialized DNA pols, of which the Y-family members are the most versatile at bypassing a wide-variety of lesions (Figure 2A). In humans, the Y-family consists of pol eta (pol η), pol iota (pol ι), pol kappa (pol κ), and Rev1.27 The B-family member pol zeta (pol ζ) is also important for many TLS events.28–30
Figure 2.

Translesion DNA synthesis is a central mediator of DNA damage tolerance. A. Prototypical finger, palm, thumb, and little finger domains for the human Y-family members are shown in blue, red, green, and purple, respectively. The N-clasp of hpol κ and N-digit of hRev1 are shown in orange. Distinctive structural features allow the enzymes to accommodate unique nt base pairs (bp) and facilitate bypass of blocks to replication (PDB codes: eta – 4DL2; kappa – 2W7P; iota – 3EPG; hRev1 – 2AQ4). B. Replisomes encounter a barrier to replication, stalling B-family replicative pols (B-pol) (upper left panel). Immediate “on-the-fly” bypass of lesions occurs via binding of TLS pols, such as Y-family pols and pol ζ, to Rev1, and through interactions with PCNA. If the replication fork slows and stalls, resulting in binding of ssDNA by RPA and activation of RSR mechanisms, ATR-mediated USP1 degradation allows levels of Ub-PCNA to increase. Levels of ubiquitinated PCNA (Ub-PCNA) are increased by Rad6/Rad18 activity and decreased by USP1 activity. Y-family pol expression and post-translational modifications are also altered to promote increased cellular concentrations of the TLS pols, further amplifying the strength of Y-family pol association with Ub-PCNA and promoting TLS past the block to replication (lower right panel).
TLS pols are recruited to sites of DNA damage/replication stress to allow direct bypass of lesions (Figure 2B). Recruitment of the Y-family pols to the replication fork is a complicated process and some aspects (e.g. order of recruitment, extent of protein-protein interactions, cell-cycle dependence) are not completely understood.25, 31 Like most other DNA pols, Y-family members possess a short PCNA interacting peptide (or PIP box), which facilitates binding to a hydrophobic patch on the sliding clamp.32, 33 The interaction with PCNA through the PIP box has been shown to stimulate Y-family pol activity (at least for some members), but the stimulation is likely related to a decreased rate of dissocation from the DNA substrate and not due to altered conformational dynamics.32, 34–36
Ubiquitination of PCNA is important for the recruitment of Y-family pols in certain instances. Y-family pols contain ubiquitin-binding domains that are important for interactions with ubiquitinated PCNA that stabilize the TLS pols at the fork.37–41 Replication fork stalling can lead to uncoupling of pol and helicase action, which can generate regions of ssDNA that are recognized as signs of replication stress. Formation of ssDNA gaps leads to the activation of Rad18 and cleavage of USP1, a de-ubiquitinating isopeptidase that removes the ubiquitin from mono-ubiquitinated PCNA.42 Rad18 binds Rad6 and this complex catalyzes the mono-ubiquitination PCNA25, 31, increasing the affinity of PCNA for TLS pols.37, 38, 43–45 There is a constant cycle of PCNA ubiquitination/deubiquitination, and it is through the degradation of the deubiquitinase USP1 in response to ATR activation and subsequent phosphorylation of the Chk1 kinase that the balance is shifted towards stable ubiquitinated-PCNA.46 In addition to post-translational regulation of TLS, there is evidence that Y-family pols track with replication forks in the absence of damage signaling (i.e. in the absence of ATR activation and subsequent stabilization of Ub-PCNA).47, 48 This so-called “on the fly” TLS likely serves to help bypass endogenous barriers to replication, such as G-quadruplexes, and the basal level of DNA damage that results from normal cellular metabolism.48, 49
In addition to interactions with PCNA, the Y-family pols can interact with each other to help recruit and stabilize the TLS pols near sites of damage. For example, Rev1 is capable of binding pol κ, pol η and pol ι, as well as pol ζ.50–53 Rev1 also interacts with PCNA and is thought to serve as a scaffold between PCNA and the other TLS pols.40, 54–56 Both pol η and ι are localized in replication foci in response to DNA damage.31, 57 Ubiquitination of PCNA increases the number of contacts between these enzymes and PCNA through their ubiquitin-binding domains.25, 32 Pol κ, which has two ubiquitin binding domains25, is thought to be recruited to the replication fork when PCNA is mono- or poly-ubiquitinated in response to specific types of damage or during prolonged periods of replication stress.25, 60–62 Polyubiquitination of PCNA occurs through the actions of Rad18, which is able to bind not only to Rad6 but also to the Rad5 E3 ligase63, and the Rad5/MMS2/UBC13 complex.25, 31
Due to their unusually spacious catalytic sites, TLS pols can accommodate many different lesions that cause high-fidelity replicative pols to stall.27 The bypass of lesions by Y-family pols can occur in either an “error-free” manner, where the correct nucleotide is inserted opposite the damage, or in an “error-prone” manner, with the wrong nucleotide inserted. TLS pols lack exonuclease activity and, therefore, cannot readily correct any errors they may make.48, 62, 64, 65 The fidelity of the bypass depends upon several factors, such as the type of DNA lesion and the catalytic attributes of the specific pol carrying out bypass.48 For example, Rev1 promotes proficient and error-free synthesis through N2-adducts on guanines because this enzyme is specialized for the incorporation of dCTP through pairing with arginine in its active site rather than the template base.66 Similarly, hpol κ has been shown to play an important role in the accurate bypass of bulky-N2-dG adducts and cell survival following exposure to benzo[a]pyrene.67–69 The recruitment of pol κ to sites of benzo[a]pyrene dihydrodiol epoxide-induced replication stress is mediated by the action of the Rad18 E3 ligase70
In addition to the role for pol κ in catalyzing bypass of DNA lesions, there is also evidence that pol κ DNA synthesis is important for Chk1 activation.71 Phosphorylation of Chk1 following treatment with hydroxyurea was decreased in pol κ-deficient cells relative to cells expressing pol κ.71 The activity of pol κ has been shown to be involved in the formation of short replication intermediates in regions undergoing replication stress.71 These intermediates possess free 5′ termini that are crucial for the binding of the 9-1-1 complex to DNA through Rad9.72 It is, therefore, not surprising that pol κ depletion also affects recruitment of the Rad9 subunit to chromatin.71 The 9-1-1 complex consists of three proteins-Rad9, Hus1, and Rad1-that interact in a heterotrimeric complex.73 This complex binds to DNA in response to replication stress and facilitates the activation of Chk1 through phosphorylation by the ATR kinase. Chk1 regulates S-phase progression, G2/M arrest, activation of DNA repair, TLS, and mediates apoptotic signaling.73, 74 Thus, one would predict that one consequence of hpol κ over-expression would be constitutive activation of the RSR.
Mis-regulation of TLS occurs in cancer, is related to disease progression, and can affect response to treatment
Y-family pols have been linked both to suppressing mutations in some instances and to promoting mutagenesis in others.64, 75–78 This balance between protective and deleterious effects is likely a major reason evolution has retained multiple regulatory mechanisms that either allow or disallow TLS pols access to the fork. Perhaps not surprisingly, DNA damage tolerance components have been reported to be mis-regulated in many different cancers76, 79, 80 and are involved in resistance to many chemotherapeutics75, 81, 82 (Table 1).
Table 1.
Notable bypass properties of the human Y-family pols and roles in cancer
| Notable bypass properties | Role in cancer | |
|---|---|---|
| Rev1 | ||
| Pol κ | ||
| Pol η | ||
| Pol ι |
Of particular interest, the mis-regulated expression of hpol κ has been observed in multiple types of cancer.79, 80, 83–85 For example, higher stage gliomas have been found to over-express DNA damage tolerance components.86 Over-expression of hpol κ was observed in 23 out of 40 glioma patients (57.5%) and hpol ι in 11 of 40 (27.5%), while there was no observable change in hpol η expression between glioma patients and normal tissue.84 Additionally, 72 out of 104 (69.2%) formalin-fixed, paraffin embedded human glioma specimens had positive hpol κ staining and 33 (31.7%) were positive for hpol ι. Importantly, hpol κ expression is correlated with advanced stages and hpol κ expression has been identified as an independent prognostic indicator of a poor outcome for glioma patients.86 It is important to note that over-expression of hpol κ, either in animals or cell culture, has been shown to induce DNA breaks, stimulate homologous recombination, and promote aneuploidy.76 Also, aberrant recruitment of hpol κ to the replication fork leads to a decrease in replication fork speed and an increase in genomic instability.87 The manifestation of these properties related to aberrant expression of hpol κ in cancer patients would certainly be detrimental.
Mis-regulation of Y-family pols has been linked to increased resistance to chemotherapies. An increase in hpol η expression, for example, causes an increased resistance to the chemotherapeutic cis-diamminedichloroplatinum(II) (cisplatin or CDDP).75, 88, 89 Replicative pols alpha (pol α), delta (pol δ), and epsilon (pol ε) cannot copy past intrastrand cross-linking CDDP adducts.90 Nucleotide excision repair (NER) is known to play a major part in the removal of CDDP-induced intra- and inter-strand crosslinks;91–94 however, pol η appears to play a significant role in bypass of unrepaired intrastrand platinum adducts.75, 88, 89, 95 Knockdown of hpol η with siRNA hypersensitized cells to cisplatin;89 and SV40-transformed fibroblasts from an XP-V patient were found to be significantly more sensitive to CDDP than clones that expressed recombinant wild-type pol η.75 XP-V cells had more S phase arrest induced by CDDP treatment than cells expressing pol η.75, 96 CDDP treatment of SV40-transformed normal fibroblast (MRC5) cells also led to an increase in the number of cells with nuclear pol η foci.75 Additionally, XP-V cells treated with CDDP had a reduction in the length of nascent DNA strands and increased DNA damage signaling compared to CDDP treated cells expressing pol η.96
The expression of pol η has also been found to correlate with poor response to oxaliplatin.97 Oxaliplatin has a very similar mechanism to CDDP, causing intra- and inter-strand platinum crosslinks. In tumor tissue from metastatic gastric adenocarcinoma patients, positive pol η expression was only found in 28.75% (23 out of 80), and this expression was moderate.97 However, expression of pol η was linked to an increase in resistance to oxaliplatin and a decrease in patient survival (8 months median survival for pol η positive versus 14 months for pol η negative; P < 0.001).97
Although often associated with bypass of bulky DNA adducts, there are several studies implicating pol κ in the bypass of adducts formed following exposure to methylating agents. For example, mouse pol κ−/− cells are hypersensitive to methyl methanesulfonate (MMS).98 A separate study found that expression of hpol κ could rescue a repair-deficient yeast strain exposed to MMS.99 Additional work led to the conclusion that hpol κ aids in the bypass of adducts following treatment with the methylating agent methyl nitrosourea (MNU).100 Treatment of HeLa cells with MNU leads to co-localization of hpol κ and PCNA, indicating that hpol κ is recruited to the replication fork to help bypass MNU-induced DNA lesions.100 In that study, knock down of hpol κ increased the cytotoxicity of MNU, but not MMS, in MMR-deficient HeLa cells.100 These results conflict with the work done with MMS and mouse pol κ-deficient cells, as well as other studies implicating pol κ in the tolerance of MMS-induced DNA damage. While there is no direct evidence to suggest that hpol κ modulates the efficacy of the commonly used chemotherapeutic temozolomide (TMZ), it is noteworthy that pol κ has been implicated in providing resistance to the same types of DNA lesions imparted by TMZ.
KYN activation of AhR promotes up-regulation of hpol κ
While numerous reports have shown that Y-family pols exhibit altered levels in many different cancers, there are very few studies investigating the mechanisms promoting mis-regulation of TLS. Several studies have examined the effect of post-translational modifications on PCNA, as well as ubiquitination of the Y-family pols themselves in regulating these enzymes.37, 38, 101–103 Other studies have examined transcriptional regulation of Y-family pols. For example, the promoter region of the gene encoding for pol κ contains stimulating protein-1 (Sp1) and cyclic AMP-responsive element (Cre)-binding sites.80 Mutations at either one of the Cre-binding sites or the Sp1-binding site leads to a decrease in pol κ transcription.80 Loss of these regulatory factors was implicated in the down-regulation of hpol κ observed in colorectal cancer. Additionally, a series of important studies established that expression of the POLK gene in mammals is stimulated following exposure to molecules, such as 3-MC and B[a]P, that activate the AhR pathway.104–106
The AhR is a basic helix-loop-helix transcription factor that is best known for its role in the response to xenobiotics via induction of cytochrome P450 expression.107 AhR activation is also known to play a role in developmental processes, as well as inflammation and tumorigenesis.7, 108 It is activated by polycyclic aromatic hydrocarbons, such as 2,3,7,8-tetrachlorodibenzodioxin (TCDD), benzo[a]pyrene (B[a]P), 7,2-dimethylbenz[a]anthracene (DMBA), and 3-methylcholanthrene (3MC).107–111 Importantly, KYN is an endogenous AhR agonist. As noted above, the most widely known AhR target genes are the cytochrome P450s CYP1A1 and CYP1B1,111 which are involved in phase I metabolism of xenobiotics and the bio-activation of certain carcinogens. The bio-activation of some AhR ligands can generate reactive intermediates capable of forming covalent DNA adducts, which require Y-family pol activity to bypass. For example, hpol κ has been shown to accurately bypass B[a]P-N2-dG adducts.68, 69, 76, 105 Mouse cells lacking pol κ were three times more sensitive to B[a]P.69 Additionally, the mutation rate in pol κ-deficient cells was 10-fold higher than in wild-type cells following exposure to B[a]P, with pol κ-deficient cells producing more G-to-T mutations in response to B[a]P treatment.69 A separate study of mouse embryo fibroblasts suggested that at least two-thirds of B[a]P-N2-dG adducts in these cells were bypassed by pol κ and again found that cells lacking pol κ were much more prone to mutagenesis.112 These results are consistent with a role for pol κ in cellular responses to bulky DNA adducts that can result from bio-activation of AhR agonists.
Given the relationship between AhR signaling and pol κ, we were intrigued by the study building a case for aberrant stimulation of AhR activity by KYN in the promotion of malignancy in glioblastomas.7 We hypothesized that the AhR pathway might be the major pathway leading to the over-expression of hpol κ reported in higher stage gliomas (Figure 3). Indeed, we conclude from our studies that the AhR signaling pathway does regulate hpol κ mRNA and protein levels in GBM cells, as treatment with the AhR agonist 3MC increased hpol κ expression and treatment with an AhR antagonist decreased its expression.113 Furthermore, blocking KYN signaling in GBM-derived cells with a small-molecule inhibitor of TDO (680C91) led to a dramatic decrease in hpol κ protein levels.113
Figure 3.

KYN signaling modulates genomic stability in gliomas and is a viable target for new therapeutic strategies. A. The cartoon schematic is shown to illustrate the apparent relationship between KYN signaling, AhR activation, DNA damage tolerance, and genome maintenance in gliomas. B. Targeted therapies that inhibit KYN signaling could attenuate the malignant properties of gliomas by reducing endogenous levels of DNA damage and inhibiting pathways that promote resistance to genotoxic treatments, such as radio- and chemo-therapy.
We went on to hypothesize that the mis-regulation of hpol κ through aberrant activity of TDO promotes the inherent genomic instability that is often seen with GBM. To assess DNA damage in GBM-derived cells, we measured micronuclei (MN) formation. Inhibition of TDO activity resulted in decreased MN formation in multiple GBM-derived cell lines. Similar decreases in MN formation were observed when AhR signaling was inhibited with the small-molecule CH-223191, as well as following RNAi-mediated knock-down of hpol κ expression.113 Simultaneous inhibition of TDO activity and AhR activation did not result in an additive effect on MN formation. Similarly, combining siRNA-mediated knock-down of hpol κ with inhibition of either TDO activity or AhR activation failed to reduce MN levels beyond those observed for inhibition of individual components of the triad, suggestive of these processes functioning together to influence genomic stability in glioma cells (i.e. TDO activity -> AhR signaling -> hpol κ expression).113 It is reasonable to assume that KYN signaling will be found to affect additional DNA damage and replication stress response factors. If such an assumption proves true, then it would go some way towards explaining why blocking KYN signaling has been found to act synergistically with genotoxic anti-cancer drugs in multiple models of cancer.114–118
Modulation of genomic maintenance through inhibition of KYN signaling as a potential anti-cancer treatment in gliomas
The first treatment employed for gliomas is typically surgery, but diffuse infiltration throughout the brain in advanced stages of the disease makes a surgical cure impossible. Radiation and chemotherapies are then used to kill as many of the remaining tumor cells as possible, and combining these treatments have succeeded in extending the survival time for some GBM patients.119–124 Some of the most exciting progress in the development of new treatments for advanced stage brain tumors is the use of oncolytic poliovirus (PVSRIPO) infused directly into the tumor, although this treatment is still in clinical trials.125 The majority of GBM patients continue to be treated with some combination of radiation and/or chemotherapy.
TMZ is one of the few genotoxic anti-cancer drugs able to cross the blood-brain barrier, and is one of the most common chemotherapeutics used to treat GBM.124, 126, 127 TMZ is a pH-dependent pro-drug that hydrolyzes to form a reactive methyldiazonium ion.128 The active form of TMZ primarily reacts with guanine to produce unstable N7-methyl-2′-deoxyguanine (m7dG) and stable O6-methyl-2′-deoxyguanine (O6-MeG) adducts.129 In the absence of repair by glycosylases, the spontaneous depurination of m7dG produces abasic sites that, like O6-MeG, are both cytotoxic and mutagenic.130 Numerous studies in tumor cell lines and patients have correlated high levels of O6-MeG repair with poor response to TMZ and other DNA alkylating agents.130–134 Repair of O6-MeG occurs through the action of the protein alkylguanine alkyltransferase (AGT, also referred to as methylguanine methyltransferase, MGMT), which irreversibly transfers the methyl group from the O6 atom of O6-MeG to a reactive cysteine residue.135–137 Hydroxyurea, procarbazine, cisplatin, and nitrosamines, such as carmustine, lomustine, and nimustine are among the other genotoxic agents often used to treat GBM.138 Ultimately, all of these genotoxic agents fail to eliminate GBM from the patient’s body. Therefore, it is of utmost importance that new routes to effective treatments for GBM patients are pursued and validated.
It is a well-known fact that increased DNA repair/damage tolerance can limit the effectiveness of genotoxic anti-cancer treatments. It is possible that modulation of the KYN pathway could be used to enhance existing genotoxic treatments by diminishing the ability of tumors to handle DNA damage (Figure 3). There are several clinical trials testing IDO inhibitors in cancer patients either alone or in combination with existing drugs, including trials to treat both adult and pediatric brain tumors with a combination of indoximod and TMZ (NCT02502708 and NCT02052648). TDO has, thus far, received less attention than IDO in therapies attempting to reverse immune-suppression in cancer, but it is emerging as an important target for new strategies to treat glioblastoma. A potential weakness in treatments for GBM that combine IDO inhibitors with TMZ is related to the fact that TDO seems to be the major source of KYN production in these tumors.7 Future clinical studies will likely include radiation with chemotherapy and inhibition of KYN signaling, as pre-clinical models have shown positive results when IDO-blocking drugs were combined with chemo- and radio-therapy.115–117 Finally, the rationale for combining inhibitors of the KYN pathway with existing anti-cancer drugs is solely based on the idea that TDO/IDO activity modulates immunologic responses. These studies might benefit from a greater understanding of the effect that inhibiting KYN signaling has upon genomic maintenance in tumors. Additional investigations should explore the effect of inhibiting the KYN pathway on DNA damage and replication stress responses in vivo to examine whether effects observed in cell culture are also manifest in whole organisms. Ultimately, exploring the relationship between KYN signaling and genomic maintenance in malignant gliomas may aid in the design of new and better therapeutic strategies for what continues to be a deadly disease.
Acknowledgments
FUNDING SOURCES
This work was supported by National Institutes of Health Grant R01CA183895 (R.L.E.) with additional support from the University of Arkansas for Medical Sciences Translational Research Institute (CTSA Grant Award UL1TR000039), and the UAMS College of Medicine. This research was also supported by a grant from the Arkansas Breast Cancer Research Programs.
We thank Dr. Sarah Eddy for assistance in the preparation of Figure 2.
ABBREVIATIONS
- AhR
aryl hydrocarbon receptor
- GBM
glioblastoma multiforme
- Kyn
kynurenine
- TDO
tryptophan-2,3-dioxygenase
- TLS
translesion DNA synthesis
- TMZ
temozolomide
- Trp
tryptophan
Biographies
Dr. April C.L. Bostian is a native of Springdale, AR. She obtained her B.Sc. in Biological Sciences from Arkansas Tech University in 2010. She obtained her Ph.D. from the University of Arkansas for Medical Sciences in May of 2016 under the mentorship of Dr. Robert L. Eoff. Her dissertation research focused on the mis-regulation of the Y-family polymerase κ in glioblastoma by the AhR pathway.
Dr. Robert L. Eoff is a native of Harrison, AR. He obtained his B.Sc. in General Chemistry from Henderson State University in 2000 and his Ph.D. in Biochemistry and Molecular Biology under the mentorship of Dr. Kevin Raney at the University of Arkansas for Medical Sciences in 2005. He is currently an Associate Professor in the Department of Biochemistry and Molecular Biology at the University of Arkansas for Medical Sciences where his research team studies the molecular mechanisms of translesion synthesis and the role of DNA damage tolerance in the etiology of cancer.
References
- 1.Vécsei L, Szalárdy L, Fülöp F, Toldi J. Kynurenines in the CNS: recent advances and new questions. Nat Rev Drug Discov. 2013;12:64–82. doi: 10.1038/nrd3793. [DOI] [PubMed] [Google Scholar]
- 2.Adams S, Braidy N, Bessede A, Brew BJ, Grant R, Teo C, Guillemin GJ. The kynurenine pathway in brain tumor pathogenesis. Cancer Res. 2012;72:5649–5657. doi: 10.1158/0008-5472.CAN-12-0549. [DOI] [PubMed] [Google Scholar]
- 3.Goda K, Kishimoto R, Shimizu S, Hamane Y, Ueda M. Quinolinic acid and active oxygens. Possible contribution of active Oxygens during cell death in the brain. Adv Exp Med Biol. 1996;398:247–254. [PubMed] [Google Scholar]
- 4.Perez-De La Cruz V, Carrillo-Mora P, Santamaria A. Quinolinic Acid, an endogenous molecule combining excitotoxicity, oxidative stress and other toxic mechanisms. Int J Tryptophan Res. 2012;5:1–8. doi: 10.4137/IJTR.S8158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Platenik J, Stopka P, Vejrazka M, Stipek S. Quinolinic acid-iron(ii) complexes: slow autoxidation, but enhanced hydroxyl radical production in the Fenton reaction. Free Radic Res. 2001;34:445–459. doi: 10.1080/10715760100300391. [DOI] [PubMed] [Google Scholar]
- 6.Surjana D, Halliday GM, Damian DL. Role of nicotinamide in DNA damage, mutagenesis, and DNA repair. J Nucleic Acids. 2010;2010 doi: 10.4061/2010/157591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, Schumacher T, Jestaedt L, Schrenk D, Weller M, Jugold M, Guillemin GJ, Miller CL, Lutz C, Radlwimmer B, Lehmann I, von Deimling A, Wick W, Platten M. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478:197–203. doi: 10.1038/nature10491. [DOI] [PubMed] [Google Scholar]
- 8.Masaki A, Ishida T, Maeda Y, Suzuki S, Ito A, Takino H, Ogura H, Totani H, Yoshida T, Kinoshita S, Narita T, Ri M, Kusumoto S, Inagaki A, Komatsu H, Niimi A, Ueda R, Utsunomiya A, Inagaki H, Iida S. Prognostic Significance of Tryptophan Catabolism in Adult T-cell Leukemia/Lymphoma. Clin Cancer Res. 2015;21:2830–2839. doi: 10.1158/1078-0432.CCR-14-2275. [DOI] [PubMed] [Google Scholar]
- 9.Heng B, Lim CK, Lovejoy DB, Bessede A, Gluch L, Guillemin GJ. Understanding the role of the kynurenine pathway in human breast cancer immunobiology. Oncotarget. 2016;7:6506–6520. doi: 10.18632/oncotarget.6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lyon DE, Walter JM, Starkweather AR, Schubert CM, McCain NL. Tryptophan degradation in women with breast cancer: a pilot study. BMC Res Notes. 2011;4:156. doi: 10.1186/1756-0500-4-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferns DM, Kema IP, Buist MR, Nijman HW, Kenter GG, Jordanova ES. Indoleamine-2,3-dioxygenase (IDO) metabolic activity is detrimental for cervical cancer patient survival. Oncoimmunology. 2015;4:e981457. doi: 10.4161/2162402X.2014.981457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Santhanam S, Alvarado DM, Ciorba MA. Therapeutic targeting of inflammation and tryptophan metabolism in colon and gastrointestinal cancer. Transl Res. 2016;167:67–79. doi: 10.1016/j.trsl.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bosnyak E, Kamson DO, Guastella AR, Varadarajan K, Robinette NL, Kupsky WJ, Muzik O, Michelhaugh SK, Mittal S, Juhasz C. Molecular imaging correlates of tryptophan metabolism via the kynurenine pathway in human meningiomas. Neuro-Oncology. 2015;17:1284–1292. doi: 10.1093/neuonc/nov098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suzuki Y, Suda T, Furuhashi K, Suzuki M, Fujie M, Hahimoto D, Nakamura Y, Inui N, Nakamura H, Chida K. Increased serum kynurenine/tryptophan ratio correlates with disease progression in lung cancer. Lung Cancer. 2010;67:361–365. doi: 10.1016/j.lungcan.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 15.Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res. 2012;72:5435–5440. doi: 10.1158/0008-5472.CAN-12-0569. [DOI] [PubMed] [Google Scholar]
- 16.Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro-Oncology. 2015;17(Suppl 4):iv1–iv62. doi: 10.1093/neuonc/nov189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weller M, Cloughesy T, Perry JR, Wick W. Standards of care for treatment of recurrent glioblastoma--are we there yet? Neuro-Oncology. 2013;15:4–27. doi: 10.1093/neuonc/nos273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, Chin L, DePinho RA, Cavenee WK. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 19.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 20.Bartkova J, Hamerlik P, Stockhausen MT, Ehrmann J, Hlobilkova A, Laursen H, Kalita O, Kolar Z, Poulsen HS, Broholm H, Lukas J, Bartek J. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene. 2010;29:5095–5102. doi: 10.1038/onc.2010.249. [DOI] [PubMed] [Google Scholar]
- 21.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 22.Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- 23.Carr AM. DNA structure dependent checkpoints as regulators of DNA repair. DNA Repair (Amst) 2002;1:983–994. doi: 10.1016/s1568-7864(02)00165-9. [DOI] [PubMed] [Google Scholar]
- 24.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 25.Chang DJ, Cimprich KA. DNA damage tolerance: when it’s OK to make mistakes. Nat Chem Biol. 2009;5:82–90. doi: 10.1038/nchembio.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Filee J, Forterre P, Sen-Lin T, Laurent J. Evolution of DNA polymerase families: evidences for multiple gene exchange between cellular and viral proteins. J Mol Evol. 2002;54:763–773. doi: 10.1007/s00239-001-0078-x. [DOI] [PubMed] [Google Scholar]
- 27.Prakash S, Johnson RE, Prakash L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem. 2005;74:317–353. doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- 28.Nelson JR, Lawrence CW, Hinkle DC. Thymine-thymine dimer bypass by yeast DNA polymerase zeta. Science. 1996;272:1646–1649. doi: 10.1126/science.272.5268.1646. [DOI] [PubMed] [Google Scholar]
- 29.Makarova AV, Burgers PM. Eukaryotic DNA polymerase zeta. DNA Repair (Amst) 2015;29:47–55. doi: 10.1016/j.dnarep.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Livneh Z, Ziv O, Shachar S. Multiple two-polymerase mechanisms in mammalian translesion DNA synthesis. Cell Cycle. 2010;9:729–735. doi: 10.4161/cc.9.4.10727. [DOI] [PubMed] [Google Scholar]
- 31.Lehmann AR, Niimi A, Ogi T, Brown S, Sabbioneda S, Wing JF, Kannouche PL, Green CM. Translesion synthesis: Y-family polymerases and the polymerase switch. DNA Repair (Amst) 2007;6:891–899. doi: 10.1016/j.dnarep.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 32.Acharya N, Yoon JH, Gali H, Unk I, Haracska L, Johnson RE, Hurwitz J, Prakash L, Prakash S. Roles of PCNA-binding and ubiquitin-binding domains in human DNA polymerase eta in translesion DNA synthesis. Proc Natl Acad Sci (U S A) 2008;105:17724–17729. doi: 10.1073/pnas.0809844105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vidal AE, Kannouche P, Podust VN, Yang W, Lehmann AR, Woodgate R. Proliferating cell nuclear antigen-dependent coordination of the biological functions of human DNA polymerase iota. J Biol Chem. 2004;279:48360–48368. doi: 10.1074/jbc.M406511200. [DOI] [PubMed] [Google Scholar]
- 34.Haracska L, Johnson RE, Unk I, Phillips B, Hurwitz J, Prakash L, Prakash S. Physical and functional interactions of human DNA polymerase eta with PCNA. Mol Cell Biol. 2001;21:7199–7206. doi: 10.1128/MCB.21.21.7199-7206.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haracska L, Johnson RE, Unk I, Phillips BB, Hurwitz J, Prakash L, Prakash S. Targeting of human DNA polymerase iota to the replication machinery via interaction with PCNA. Proc Natl Acad Sci (U S A) 2001;98:14256–14261. doi: 10.1073/pnas.261560798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haracska L, Unk I, Johnson RE, Phillips BB, Hurwitz J, Prakash L, Prakash S. Stimulation of DNA synthesis activity of human DNA polymerase kappa by PCNA. Mol Cell Biol. 2002;22:784–791. doi: 10.1128/MCB.22.3.784-791.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bienko M, Green CM, Crosetto N, Rudolf F, Zapart G, Coull B, Kannouche P, Wider G, Peter M, Lehmann AR, Hofmann K, Dikic I. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science. 2005;310:1821–1824. doi: 10.1126/science.1120615. [DOI] [PubMed] [Google Scholar]
- 38.Guo C, Tang TS, Bienko M, Parker JL, Bielen AB, Sonoda E, Takeda S, Ulrich HD, Dikic I, Friedberg EC. Ubiquitin-binding motifs in REV1 protein are required for its role in the tolerance of DNA damage. Mol Cell Biol. 2006;26:8892–8900. doi: 10.1128/MCB.01118-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Plosky BS, Vidal AE, Fernandez de Henestrosa AR, McLenigan MP, McDonald JP, Mead S, Woodgate R. Controlling the subcellular localization of DNA polymerases iota and eta via interactions with ubiquitin. EMBO J. 2006;25:2847–2855. doi: 10.1038/sj.emboj.7601178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wood A, Garg P, Burgers PM. A ubiquitin-binding motif in the translesion DNA polymerase Rev1 mediates its essential functional interaction with ubiquitinated proliferating cell nuclear antigen in response to DNA damage. J Biol Chem. 2007;282:20256–20263. doi: 10.1074/jbc.M702366200. [DOI] [PubMed] [Google Scholar]
- 41.Garg P, Burgers PM. Ubiquitinated proliferating cell nuclear antigen activates translesion DNA polymerases eta and REV1. Proc Natl Acad Sci (U S A) 2005;102:18361–18366. doi: 10.1073/pnas.0505949102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang TT, Nijman SM, Mirchandani KD, Galardy PJ, Cohn MA, Haas W, Gygi SP, Ploegh HL, Bernards R, D’Andrea AD. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat Cell Biol. 2006;8:339–347. doi: 10.1038/ncb1378. [DOI] [PubMed] [Google Scholar]
- 43.Kannouche PL, Wing J, Lehmann AR. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell. 2004;14:491–500. doi: 10.1016/s1097-2765(04)00259-x. [DOI] [PubMed] [Google Scholar]
- 44.Bi X, Barkley LR, Slater DM, Tateishi S, Yamaizumi M, Ohmori H, Vaziri C. Rad18 regulates DNA polymerase kappa and is required for recovery from S-phase checkpoint-mediated arrest. Mol Cell Biol. 2006;26:3527–3540. doi: 10.1128/MCB.26.9.3527-3540.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Watanabe K, Tateishi S, Kawasuji M, Tsurimoto T, Inoue H, Yamaizumi M. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004;23:3886–3896. doi: 10.1038/sj.emboj.7600383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fox JT, Lee KY, Myung K. Dynamic regulation of PCNA ubiquitylation/deubiquitylation. FEBS Letters. 2011;585:2780–2785. doi: 10.1016/j.febslet.2011.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Edmunds CE, Simpson LJ, Sale JE. PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol Cell. 2008;30:519–529. doi: 10.1016/j.molcel.2008.03.024. [DOI] [PubMed] [Google Scholar]
- 48.Friedberg EC, Lehmann AR, Fuchs RP. Trading places: how do DNA polymerases switch during translesion DNA synthesis? Mol Cell. 2005;18:499–505. doi: 10.1016/j.molcel.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 49.Sale JE, Batters C, Edmunds CE, Phillips LG, Simpson LJ, Szuts D. Timing matters: error-prone gap filling and translesion synthesis in immunoglobulin gene hypermutation. Philos Trans R Soc Lond B Biol Sci. 2009;364:595–603. doi: 10.1098/rstb.2008.0197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guo C, Fischhaber PL, Luk-Paszyc MJ, Masuda Y, Zhou J, Kamiya K, Kisker C, Friedberg EC. Mouse Rev1 protein interacts with multiple DNA polymerases involved in translesion DNA synthesis. EMBO J. 2003;22:6621–6630. doi: 10.1093/emboj/cdg626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ohashi E, Murakumo Y, Kanjo N, Akagi J, Masutani C, Hanaoka F, Ohmori H. Interaction of hREV1 with three human Y-family DNA polymerases. Genes Cells. 2004;9:523–531. doi: 10.1111/j.1356-9597.2004.00747.x. [DOI] [PubMed] [Google Scholar]
- 52.Tissier A, Kannouche P, Reck MP, Lehmann AR, Fuchs RP, Cordonnier A. Co-localization in replication foci and interaction of human Y-family members, DNA polymerase pol eta and REVl protein. DNA Repair (Amst) 2004;3:1503–1514. doi: 10.1016/j.dnarep.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 53.Murakumo Y, Ogura Y, Ishii H, Numata S, Ichihara M, Croce CM, Fishel R, Takahashi M. Interactions in the error-prone postreplication repair proteins hREV1, hREV3, and hREV7. J Biol Chem. 2001;276:35644–35651. doi: 10.1074/jbc.M102051200. [DOI] [PubMed] [Google Scholar]
- 54.Ross AL, Simpson LJ, Sale JE. Vertebrate DNA damage tolerance requires the C-terminus but not BRCT or transferase domains of REV1. Nucleic Acids Res. 2005;33:1280–1289. doi: 10.1093/nar/gki279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guo C, Sonoda E, Tang TS, Parker JL, Bielen AB, Takeda S, Ulrich HD, Friedberg EC. REV1 protein interacts with PCNA: significance of the REV1 BRCT domain in vitro and in vivo. Mol Cell. 2006;23:265–271. doi: 10.1016/j.molcel.2006.05.038. [DOI] [PubMed] [Google Scholar]
- 56.de Groote FH, Jansen JG, Masuda Y, Shah DM, Kamiya K, de Wind N, Siegal G. The Rev1 translesion synthesis polymerase has multiple distinct DNA binding modes. DNA Repair (Amst) 2011;10:915–925. doi: 10.1016/j.dnarep.2011.04.033. [DOI] [PubMed] [Google Scholar]
- 57.Kannouche P, Fernandez de Henestrosa AR, Coull B, Vidal AE, Gray C, Zicha D, Woodgate R, Lehmann AR. Localization of DNA polymerases eta and iota to the replication machinery is tightly co-ordinated in human cells. EMBO J. 2003;22:1223–1233. doi: 10.1093/emboj/cdf618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ogi T, Kannouche P, Lehmann AR. Localisation of human Y-family DNA polymerase kappa: relationship to PCNA foci. J Cell Sci. 2005;118:129–136. doi: 10.1242/jcs.01603. [DOI] [PubMed] [Google Scholar]
- 59.Bergoglio V, Bavoux C, Verbiest V, Hoffmann JS, Cazaux C. Localisation of human DNA polymerase kappa to replication foci. J Cell Sci. 2002;115:4413–4418. doi: 10.1242/jcs.00162. [DOI] [PubMed] [Google Scholar]
- 60.Lin JR, Zeman MK, Chen JY, Yee MC, Cimprich KA. SHPRH and HLTF act in a damage-specific manner to coordinate different forms of postreplication repair and prevent mutagenesis. Mol Cell. 2011;42:237–249. doi: 10.1016/j.molcel.2011.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Motegi A, Liaw HJ, Lee KY, Roest HP, Maas A, Wu X, Moinova H, Markowitz SD, Ding H, Hoeijmakers JH, Myung K. Polyubiquitination of proliferating cell nuclear antigen by HLTF and SHPRH prevents genomic instability from stalled replication forks. Proc Natl Acad Sci (U S A) 2008;105:12411–12416. doi: 10.1073/pnas.0805685105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chang DJ, Lupardus PJ, Cimprich KA. Monoubiquitination of proliferating cell nuclear antigen induced by stalled replication requires uncoupling of DNA polymerase and mini-chromosome maintenance helicase activities. J Biol Chem. 2006;281:32081–32088. doi: 10.1074/jbc.M606799200. [DOI] [PubMed] [Google Scholar]
- 63.Ulrich HD. Protein-protein interactions within an E2-RING finger complex. Implications for ubiquitin-dependent DNA damage repair. J Biol Chem. 2003;278:7051–7058. doi: 10.1074/jbc.M212195200. [DOI] [PubMed] [Google Scholar]
- 64.Edmunds CE, Simpson LJ, Sale JE. PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol Cell. 2008;30:519–529. doi: 10.1016/j.molcel.2008.03.024. [DOI] [PubMed] [Google Scholar]
- 65.Freudenthal BD, Gakhar L, Ramaswamy S, Washington MT. Structure of monoubiquitinated PCNA and implications for translesion synthesis and DNA polymerase exchange. Nat Struct Mol Biol. 2010;17:479–484. doi: 10.1038/nsmb.1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nair DT, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Rev1 employs a novel mechanism of DNA synthesis using a protein template. Science. 2005;309:2219–2222. doi: 10.1126/science.1116336. [DOI] [PubMed] [Google Scholar]
- 67.Bi X, Slater DM, Ohmori H, Vaziri C. DNA polymerase kappa is specifically required for recovery from the benzo[a]pyrene-dihydrodiol epoxide (BPDE)-induced S-phase checkpoint. J Biol Chem. 2005;280:22343–22355. doi: 10.1074/jbc.M501562200. [DOI] [PubMed] [Google Scholar]
- 68.Choi JY, Angel KC, Guengerich FP. Translesion synthesis across bulky N2-alkyl guanine DNA adducts by human DNA polymerase kappa. J Biol Chem. 2006;281:21062–21072. doi: 10.1074/jbc.M602246200. [DOI] [PubMed] [Google Scholar]
- 69.Ogi T, Shinkai Y, Tanaka K, Ohmori H. Pol κ protects mammalian cells against the lethal and mutagenic effects of benzo[a]pyrene. Proc Natl Acad Sci (U S A) 2002;99:15548–15553. doi: 10.1073/pnas.222377899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bi X, Barkley LR, Slater DM, Tateishi S, Yamaizumi M, Ohmori H, Vaziri C. Rad18 regulates DNA polymerase kappa and is required for recovery from S-phase checkpoint-mediated arrest. Mol Cell Biol. 2006;26:3527–3540. doi: 10.1128/MCB.26.9.3527-3540.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Betous R, Pillaire MJ, Pierini L, van der Laan S, Recolin B, Ohl-Seguy E, Guo C, Niimi N, Gruz P, Nohmi T, Friedberg E, Cazaux C, Maiorano D, Hoffmann JS. DNA polymerase kappa-dependent DNA synthesis at stalled replication forks is important for CHK1 activation. EMBO J. 2013;32:2172–2185. doi: 10.1038/emboj.2013.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ellison V, Stillman B. Biochemical characterization of DNA damage checkpoint complexes: clamp loader and clamp complexes with specificity for 5′ recessed DNA. PLoS Biol. 2003;1:E33. doi: 10.1371/journal.pbio.0000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Parrilla-Castellar ER, Arlander SJ, Karnitz L. Dial 9-1-1 for DNA damage: the Rad9-Hus1-Rad1 (9-1-1) clamp complex. DNA Repair (Amst) 2004;3:1009–1014. doi: 10.1016/j.dnarep.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 74.Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–429. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 75.Albertella MR, Green CM, Lehmann AR, O’Connor MJ. A role for polymerase eta in the cellular tolerance to cisplatin-induced damage. Cancer Res. 2005;65:9799–9806. doi: 10.1158/0008-5472.CAN-05-1095. [DOI] [PubMed] [Google Scholar]
- 76.Bavoux C, Leopoldino AM, Bergoglio V, JOW, Ogi T, Bieth A, Judde JG, Pena SD, Poupon MF, Helleday T, Tagawa M, Machado C, Hoffmann JS, Cazaux C. Up-regulation of the error-prone DNA polymerase κ promotes pleiotropic genetic alterations and tumorigenesis. Cancer Res. 2005;65:325–330. [PubMed] [Google Scholar]
- 77.Bétous R, Rey L, Wang G, Pillaire MJ, Puget N, Selves J, Biard DS, Shin-ya K, Vasquez KM, Cazaux C, Hoffmann JS. Role of TLS DNA polymerases eta and kappa in processing naturally occurring structured DNA in human cells. Mol Carcinog. 2009;48:369–378. doi: 10.1002/mc.20509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Loeb LA, Bielas JH, Beckman RA. Cancers exhibit a mutator phenotype: clinical implications. Cancer Res. 2008;68:3551–3557. doi: 10.1158/0008-5472.CAN-07-5835. [DOI] [PubMed] [Google Scholar]
- 79.Albertella MR, Lau A, O’Connor MJ. The overexpression of specialized DNA polymerases in cancer. DNA Repair (Amst) 2005;4:583–593. doi: 10.1016/j.dnarep.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 80.Lemée F, Bavoux C, Pillaire MJ, Bieth A, Machado CR, Pena SD, Guimbaud R, Selves J, Hoffmann JS, Cazaux C. Characterization of promoter regulatory elements involved in downexpression of the DNA polymerase kappa in colorectal cancer. Oncogene. 2007;26:3387–3394. doi: 10.1038/sj.onc.1210116. [DOI] [PubMed] [Google Scholar]
- 81.Moraes MC, de Andrade AQ, Carvalho H, Guecheva T, Agnoletto MH, Henriques JA, Sarasin A, Stary A, Saffi J, Menck CF. Both XPA and DNA polymerase eta are necessary for the repair of doxorubicin-induced DNA lesions. Cancer Lett. 2012;314:108–118. doi: 10.1016/j.canlet.2011.09.019. [DOI] [PubMed] [Google Scholar]
- 82.Ceppi P, Novello S, Cambieri A, Longo M, Monica V, Lo Iacono M, Giaj-Levra M, Saviozzi S, Volante M, Papotti M, Scagliotti G. Polymerase eta mRNA expression predicts survival of non-small cell lung cancer patients treated with platinum-based chemotherapy. Clin Cancer Res. 2009;15:1039–1045. doi: 10.1158/1078-0432.CCR-08-1227. [DOI] [PubMed] [Google Scholar]
- 83.Pan Q, Fang Y, Xu Y, Zhang K, Hu X. Down-regulation of DNA polymerases kappa, eta, iota, and zeta in human lung, stomach, and colorectal cancers. Cancer Lett. 2005;217:139–147. doi: 10.1016/j.canlet.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 84.Wang H, Wu W, Wang HW, Wang S, Chen Y, Zhang X, Yang J, Zhao S, Ding HF, Lu D. Analysis of specialized DNA polymerases expression in human gliomas: association with prognostic significance. Neuro-Oncology. 2010;12:679–686. doi: 10.1093/neuonc/nop074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang Y, Seimiya M, Kawamura K, Yu L, Ogi T, Takenaga K, Shishikura T, Nakagawara A, Sakiyama S, Tagawa M, JOW Elevated expression of DNA polymerase kappa in human lung cancer is associated with p53 inactivation: Negative regulation of POLK promoter activity by p53. Int J Oncol. 2004;25:161–165. [PubMed] [Google Scholar]
- 86.Wang H, Wu W, Wang HW, Wang S, Chen Y, Zhang X, Yang J, Zhao S, Ding HF, Lu D. Analysis of specialized DNA polymerases expression in human gliomas: association with prognostic significance. Neuro-Oncology. 2010;12:679–686. doi: 10.1093/neuonc/nop074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jones MJ, Colnaghi L, Huang TT. Dysregulation of DNA polymerase kappa recruitment to replication forks results in genomic instability. EMBO J. 2012;31:908–918. doi: 10.1038/emboj.2011.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ho TV, Scharer OD. Translesion DNA synthesis polymerases in DNA interstrand crosslink repair. Environ Mol Mutagen. 2010;51:552–566. doi: 10.1002/em.20573. [DOI] [PubMed] [Google Scholar]
- 89.Hicks JK, Chute CL, Paulsen MT, Ragland RL, Howlett NG, Gueranger Q, Glover TW, Canman CE. Differential roles for DNA polymerases eta, zeta, and REV1 in lesion bypass of intrastrand versus interstrand DNA cross-links. Mol Cell Biol. 2010;30:1217–1230. doi: 10.1128/MCB.00993-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hoffmann JS, Pillaire MJ, Maga G, Podust V, Hubscher U, Villani G. DNA polymerase beta bypasses in vitro a single d(GpG)-cisplatin adduct placed on codon 13 of the HRAS gene. Proc Natl Acad Sci (U S A) 1995;92:5356–5360. doi: 10.1073/pnas.92.12.5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Reardon JT, Vaisman A, Chaney SG, Sancar A. Efficient nucleotide excision repair of cisplatin, oxaliplatin, and Bis-aceto-ammine-dichloro-cyclohexylamine-platinum(IV) (JM216) platinum intrastrand DNA diadducts. Cancer Res. 1999;59:3968–3971. [PubMed] [Google Scholar]
- 92.Furuta T, Ueda T, Aune G, Sarasin A, Kraemer KH, Pommier Y. Transcription-coupled nucleotide excision repair as a determinant of cisplatin sensitivity of human cells. Cancer Res. 2002;62:4899–4902. [PubMed] [Google Scholar]
- 93.Wu X, Fan W, Xu S, Zhou Y. Sensitization to the cytotoxicity of cisplatin by transfection with nucleotide excision repair gene xeroderma pigmentosun group A antisense RNA in human lung adenocarcinoma cells. Clin Cancer Res. 2003;9:5874–5879. [PubMed] [Google Scholar]
- 94.Hansson J, Wood RD. Repair synthesis by human cell extracts in DNA damaged by cis- and trans-diamminedichloroplatinum(II) Nucleic Acids Res. 1989;17:8073–8091. doi: 10.1093/nar/17.20.8073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vaisman A, Masutani C, Hanaoka F, Chaney SG. Efficient translesion replication past oxaliplatin and cisplatin GpG adducts by human DNA polymerase eta. Biochemistry. 2000;39:4575–4580. doi: 10.1021/bi000130k. [DOI] [PubMed] [Google Scholar]
- 96.Sokol AM, Cruet-Hennequart S, Pasero P, Carty MP. DNA polymerase eta modulates replication fork progression and DNA damage responses in platinum-treated human cells. Sci Rep. 2013;3:3277. doi: 10.1038/srep03277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Teng KY, Qiu MZ, Li ZH, Luo HY, Zeng ZL, Luo RZ, Zhang HZ, Wang ZQ, Li YH, Xu RH. DNA polymerase eta protein expression predicts treatment response and survival of metastatic gastric adenocarcinoma patients treated with oxaliplatin-based chemotherapy. J Trans Med. 2010;8:126. doi: 10.1186/1479-5876-8-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Takenaka K, Ogi T, Okada T, Sonoda E, Guo C, Friedberg EC, Takeda S. Involvement of vertebrate Pol kappa in translesion DNA synthesis across DNA monoalkylation damage. J Biol Chem. 2006;281:2000–2004. doi: 10.1074/jbc.M506153200. [DOI] [PubMed] [Google Scholar]
- 99.Plosky BS, Frank EG, Berry DA, Vennall GP, McDonald JP, Woodgate R. Eukaryotic Y-family polymerases bypass a 3-methyl-2′-deoxyadenosine analog in vitro and methyl methanesulfonate-induced DNA damage in vivo. Nucleic Acids Res. 2008;36:2152–2162. doi: 10.1093/nar/gkn058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lupari E, Ventura I, Marcon F, Aquilina G, Dogliotti E, Fortini P. Pol kappa partially rescues MMR-dependent cytotoxicity of O6-methylguanine. DNA Repair (Amst) 2012;11:579–586. doi: 10.1016/j.dnarep.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 101.Bienko M, Green CM, Sabbioneda S, Crosetto N, Matic I, Hibbert RG, Begovic T, Niimi A, Mann M, Lehmann AR, Dikic I. Regulation of translesion synthesis DNA polymerase eta by monoubiquitination. Mol Cell. 2010;37:396–407. doi: 10.1016/j.molcel.2009.12.039. [DOI] [PubMed] [Google Scholar]
- 102.Bomar MG, D’Souza S, Bienko M, Dikic I, Walker GC, Zhou P. Unconventional ubiquitin recognition by the ubiquitin-binding motif within the Y family DNA polymerases iota and Rev1. Mol Cell. 2010;37:408–417. doi: 10.1016/j.molcel.2009.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Guo C, Tang TS, Bienko M, Dikic I, Friedberg EC. Requirements for the interaction of mouse Pol kappa with ubiquitin and its biological significance. J Biol Chem. 2008;283:4658–4664. doi: 10.1074/jbc.M709275200. [DOI] [PubMed] [Google Scholar]
- 104.Brauze DRAA. The effect of aryl hydrocarbon receptor ligands on the expression of polymerase (DNA directed) kappa (Pol k), polymerase RNA II (DNA directed) polypeptide A2 (PolR2a), CYP1B1 and CYP1A1 genes in rat liver. Environ Toxicol Pharmacol. 2012;34:819–825. doi: 10.1016/j.etap.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 105.Ogi T, Mimura J, Hikida M, Fujimoto H, Fujii-Kuriyama Y, Ohmori H. Expression of human and mouse genes encoding polkappa: testis-specific developmental regulation and AhR-dependent inducible transcription. Genes Cells. 2001;6:943–953. doi: 10.1046/j.1365-2443.2001.00478.x. [DOI] [PubMed] [Google Scholar]
- 106.Ogi T, Shinkai Y, Tanaka K, Ohmori H. Pol kappa protects mammalian cells against the lethal and mutagenic effects of benzo[a]pyrene. Proc Natl Acad Sci (U S A) 2002;99:15548–15553. doi: 10.1073/pnas.222377899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol. 2003;43:309–334. doi: 10.1146/annurev.pharmtox.43.100901.135828. [DOI] [PubMed] [Google Scholar]
- 108.Denison MS, Heath-Pagliuso S. The Ah receptor: a regulator of the biochemical and toxicological actions of structurally diverse chemicals. Bull Environ Contam Toxicol. 1998;61:557–568. doi: 10.1007/pl00002973. [DOI] [PubMed] [Google Scholar]
- 109.Denison MS, Pandini A, Nagy SR, Baldwin EP, Bonati L. Ligand binding and activation of the Ah receptor. Chem Biol Interact. 2002;141:3–24. doi: 10.1016/s0009-2797(02)00063-7. [DOI] [PubMed] [Google Scholar]
- 110.Hankinson O. The aryl hydrocarbon receptor complex. Annu Rev Pharmacol Toxicol. 1995;35:307–340. doi: 10.1146/annurev.pa.35.040195.001515. [DOI] [PubMed] [Google Scholar]
- 111.Bock KW. Aryl hydrocarbon or dioxin receptor: biologic and toxic responses. Rev Physiol Biochem Pharmacol. 1994;125:1–42. doi: 10.1007/BFb0030908. [DOI] [PubMed] [Google Scholar]
- 112.Avkin S, Goldsmith M, Velasco-Miguel S, Geacintov N, Friedberg EC, Livneh Z. Quantitative analysis of translesion DNA synthesis across a benzo[a]pyrene-guanine adduct in mammalian cells: the role of DNA polymerase kappa. J Biol Chem. 2004;279:53298–53305. doi: 10.1074/jbc.M409155200. [DOI] [PubMed] [Google Scholar]
- 113.Bostian AC, Maddukuri L, Reed MR, Savenka T, Hartman JH, Davis L, Pouncey DL, Miller GP, Eoff RL. Kynurenine signaling increases DNA polymerase kappa expression and promotes genomic instability in glioblastoma cells. Chem Res Toxicol. 2016;29:101–108. doi: 10.1021/acs.chemrestox.5b00452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hanihara M, Kawataki T, Oh-Oka K, Mitsuka K, Nakao A, Kinouchi H. Synergistic antitumor effect with indoleamine 2,3-dioxygenase inhibition and temozolomide in a murine glioma model. J Neurosurg. 2015:1–8. doi: 10.3171/2015.5.JNS141901. [DOI] [PubMed] [Google Scholar]
- 115.Hou DY, Muller AJ, Sharma MD, DuHadaway J, Banerjee T, Johnson M, Mellor AL, Prendergast GC, Munn DH. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res. 2007;67:792–801. doi: 10.1158/0008-5472.CAN-06-2925. [DOI] [PubMed] [Google Scholar]
- 116.Li M, Bolduc AR, Hoda MN, Gamble DN, Dolisca SB, Bolduc AK, Hoang K, Ashley C, McCall D, Rojiani AM, Maria BL, Rixe O, MacDonald TJ, Heeger PS, Mellor AL, Munn DH, Johnson TS. The indoleamine 2,3-dioxygenase pathway controls complement-dependent enhancement of chemo-radiation therapy against murine glioblastoma. J Immunother Cancer. 2014;2:21. doi: 10.1186/2051-1426-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med. 2005;11:312–319. doi: 10.1038/nm1196. [DOI] [PubMed] [Google Scholar]
- 118.Munn DH. Blocking IDO activity to enhance anti-tumor immunity. Front Biosci (Elite Ed) 2012;4:734–745. doi: 10.2741/e414. [DOI] [PubMed] [Google Scholar]
- 119.Friedman HS, Kerby T, Calvert H. Temozolomide and treatment of malignant glioma. Clin Cancer Res. 2000;6:2585–2597. [PubMed] [Google Scholar]
- 120.Hegi ME, Diserens AC, Godard S, Dietrich PY, Regli L, Ostermann S, Otten P, Van Melle G, de Tribolet N, Stupp R. Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin Cancer Res. 2004;10:1871–1874. doi: 10.1158/1078-0432.ccr-03-0384. [DOI] [PubMed] [Google Scholar]
- 121.Quinn JA, Desjardins A, Weingart J, Brem H, Dolan ME, Delaney SM, Vredenburgh J, Rich J, Friedman AH, Reardon DA, Sampson JH, Pegg AE, Moschel RC, Birch R, McLendon RE, Provenzale JM, Gururangan S, Dancey JE, Maxwell J, Tourt-Uhlig S, Herndon JE, 2nd, Bigner DD, Friedman HS. Phase I trial of temozolomide plus O6-benzylguanine for patients with recurrent or progressive malignant glioma. J Clin Oncol. 2005;23:7178–7187. doi: 10.1200/JCO.2005.06.502. [DOI] [PubMed] [Google Scholar]
- 122.Quinn JA, Jiang SX, Reardon DA, Desjardins A, Vredenburgh JJ, Rich JN, Gururangan S, Friedman AH, Bigner DD, Sampson JH, McLendon RE, Herndon JE, 2nd, Walker A, Friedman HS. Phase II trial of temozolomide plus O6-benzylguanine in adults with recurrent, temozolomide-resistant malignant glioma. J Clin Oncol. 2009;27:1262–1267. doi: 10.1200/JCO.2008.18.8417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Quinn JA, Pluda J, Dolan ME, Delaney S, Kaplan R, Rich JN, Friedman AH, Reardon DA, Sampson JH, Colvin OM, Haglund MM, Pegg AE, Moschel RC, McLendon RE, Provenzale JM, Gururangan S, Tourt-Uhlig S, Herndon JE, 2nd, Bigner DD, Friedman HS. Phase II trial of carmustine plus O6-benzylguanine for patients with nitrosourea-resistant recurrent or progressive malignant glioma. J Clin Oncol. 2002;20:2277–2283. doi: 10.1200/JCO.2002.09.084. [DOI] [PubMed] [Google Scholar]
- 124.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 125.Gromeier M, Lachmann S, Rosenfeld MR, Gutin PH, Wimmer E. Intergeneric poliovirus recombinants for the treatment of malignant glioma. Proc Natl Acad Sci (U S A) 2000;97:6803–6808. doi: 10.1073/pnas.97.12.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sathornsumetee S, Reardon DA, Desjardins A, Quinn JA, Vredenburgh JJ, Rich JN. Molecularly targeted therapy for malignant glioma. Cancer. 2007;110:13–24. doi: 10.1002/cncr.22741. [DOI] [PubMed] [Google Scholar]
- 127.Johnson DR, O’Neill BP. Glioblastoma survival in the United States before and during the temozolomide era. J Neuro-Oncol. 2012;107:359–364. doi: 10.1007/s11060-011-0749-4. [DOI] [PubMed] [Google Scholar]
- 128.Stevens MF, Newlands ES. From triazines and triazenes to temozolomide. Eur J Cancer. 1993;29A:1045–1047. doi: 10.1016/s0959-8049(05)80221-7. [DOI] [PubMed] [Google Scholar]
- 129.Friedman HS, Kerby T, Calvert H. Temozolomide and treatment of malignant glioma. Clin Cancer Res. 2000;6:2585–2597. [PubMed] [Google Scholar]
- 130.Fu D, Calvo JA, Samson LD. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer. 2012;12:104–120. doi: 10.1038/nrc3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JE, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R. MGMT gene silencing and benefit from temozolomide in glioblastoma. New Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 132.Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB, Herman JG. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. New Engl J Med. 2000;343:1350–1354. doi: 10.1056/NEJM200011093431901. [DOI] [PubMed] [Google Scholar]
- 133.Hegi ME, Diserens AC, Godard S, Dietrich PY, Regli L, Ostermann S, Otten P, Van Melle G, de Tribolet N, Stupp R. Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin Cancer Res. 2004;10:1871–1874. doi: 10.1158/1078-0432.ccr-03-0384. [DOI] [PubMed] [Google Scholar]
- 134.Uno M, Oba-Shinjo SM, Camargo AA, Moura RP, Aguiar PH, Cabrera HN, Begnami M, Rosemberg S, Teixeira MJ, Marie SK. Correlation of MGMT promoter methylation status with gene and protein expression levels in glioblastoma. Clinics. 2011;66:1747–1755. doi: 10.1590/S1807-59322011001000013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Baer JC, Freeman AA, Newlands ES, Watson AJ, Rafferty JA, Margison GP. Depletion of O6-alkylguanine-DNA alkyltransferase correlates with potentiation of temozolomide and CCNU toxicity in human tumour cells. Brit J Cancer. 1993;67:1299–1302. doi: 10.1038/bjc.1993.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Pegg AE. Multifaceted roles of alkyltransferase and related proteins in DNA repair, DNA damage, resistance to chemotherapy, and research tools. Chem Res Toxicol. 2011;24:618–639. doi: 10.1021/tx200031q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Olsson M, Lindahl T. Repair of alkylated DNA in Escherichia coli. Methyl group transfer from O6-methylguanine to a protein cysteine residue. J Biol Chem. 1980;255:10569–10571. [PubMed] [Google Scholar]
- 138.Torisu M, Katano M, Higashi D, Kinugasa T, Soma GI. Proceedings of the 12th Annual Meeting of the Society of Biotherapeutic Approaches 6 December, 2008 Fukuoka, Japan Preface. Anticancer Res. 2009:29. [Google Scholar]
- 139.He X, Ye F, Zhang J, Cheng Q, Shen J, Chen H. REV1 genetic variants associated with the risk of cervical carcinoma. Eur J Epidem. 2008;23:403–409. doi: 10.1007/s10654-008-9251-5. [DOI] [PubMed] [Google Scholar]
- 140.Sharma S, Canman CE. REV1 and DNA polymerase zeta in DNA interstrand crosslink repair. Environ Mol Mutagen. 2012;53:725–740. doi: 10.1002/em.21736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ogi T, Lehmann AR. The Y-family DNA polymerase kappa (pol kappa) functions in mammalian nucleotide-excision repair. Nat Cell Biol. 2006;8:640–642. doi: 10.1038/ncb1417. [DOI] [PubMed] [Google Scholar]
- 142.JOW, Kawamura K, Tada Y, Ohmori H, Kimura H, Sakiyama S, Tagawa M. DNA polymerase kappa, implicated in spontaneous and DNA damage-induced mutagenesis, is overexpressed in lung cancer. Cancer Res. 2001;61:5366–5369. [PubMed] [Google Scholar]
- 143.Johnson RE, Washington MT, Prakash S, Prakash L. Fidelity of human DNA polymerase eta. J Biol Chem. 2000;275:7447–7450. doi: 10.1074/jbc.275.11.7447. [DOI] [PubMed] [Google Scholar]
- 144.Lee DH, Pfeifer GP. Translesion synthesis of 7,8-dihydro-8-oxo-2′-deoxyguanosine by DNA polymerase eta in vivo. Mutat Res. 2008;641:19–26. doi: 10.1016/j.mrfmmm.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Patra A, Nagy LD, Zhang Q, Su Y, Muller L, Guengerich FP, Egli M. Kinetics, structure, and mechanism of 8-oxo-7,8-dihydro-2′-deoxyguanosine bypass by human DNA polymerase eta. J Biol Chem. 289:16867–16882. doi: 10.1074/jbc.M114.551820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.McIlwraith MJ, Vaisman A, Liu Y, Fanning E, Woodgate R, West SC. Human DNA polymerase eta promotes DNA synthesis from strand invasion intermediates of homologous recombination. Mol Cell. 2005;20:783–792. doi: 10.1016/j.molcel.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 147.Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, Hanaoka F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature. 1999;399:700–704. doi: 10.1038/21447. [DOI] [PubMed] [Google Scholar]
- 148.Srivastava AK, Han C, Zhao R, Cui T, Dai Y, Mao C, Zhao W, Zhang X, Yu J, Wang QE. Enhanced expression of DNA polymerase eta contributes to cisplatin resistance of ovarian cancer stem cells. Proc Natl Acad Sci (U S A) 2015;112:4411–4416. doi: 10.1073/pnas.1421365112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Nair DT, Johnson RE, Prakash S, Prakash L, Aggarwal AK. Replication by human DNA polymerase-iota occurs by Hoogsteen base-pairing. Nature. 2004;430:377–380. doi: 10.1038/nature02692. [DOI] [PubMed] [Google Scholar]
- 150.Tissier A, McDonald JP, Frank EG, Woodgate R. pol iota, a remarkably error-prone human DNA polymerase. Genes Dev. 2000;14:1642–1650. [PMC free article] [PubMed] [Google Scholar]
- 151.Yang J, Chen Z, Liu Y, Hickey RJ, Malkas LH. Altered DNA polymerase iota expression in breast cancer cells leads to a reduction in DNA replication fidelity and a higher rate of mutagenesis. Cancer Res. 2004;64:5597–5607. doi: 10.1158/0008-5472.CAN-04-0603. [DOI] [PubMed] [Google Scholar]