

Abstract
Background
Current approaches for post-market medical device safety surveillance are limited in their ability to produce timely and accurate assessments of adverse event rates.
Methods and Results
The DELTA (Data Extraction and Longitudinal Trend Analysis) network study was a multicenter prospective observational study designed to evaluate the safety of devices used during percutaneous coronary interventions (PCI). All adult patients undergoing PCI from January 2008 through December 2012 at five participating Massachusetts sites were included. A safety alert was triggered if the cumulative observed adverse event rates for the study device exceeded the upper 95% confidence interval of the event rates of propensity-matched control cohort. Pre-specified sensitivity analyses were developed to validate any identified safety signal.
A total of 23,805 consecutive PCI procedures were evaluated. Two out of 24 safety analyses triggered safety alerts. Patients receiving Perclose vascular closure device (VCD) experienced an increased risk of minor vascular complications (relative risk [RR] 4.14; p <0.01) and any vascular complication (RR: 2.06; p = 0.01) as compared with propensity-matched patients receiving alternative VCD; a result primarily driven by relatively high event rates at one participating center. Sensitivity analyses based on alternative risk adjustment methods confirmed the a pattern of increased rate of complications at one of the five participating sites in their use of Perclose VCD.
Conclusions
The DELTA network study demonstrates that distributed automated prospective safety surveillance has the potential of providing near real-time assessment of safety risks of newly approved medical devices.
Keywords: safety, vascular closure devices, quality monitoring
Background
Assuring the safety of medical devices after FDA approval (“post-market surveillance”) is crucial to protect public health. Device malfunction can result in serious injury and even fatality.1–3 In contrast to medications, implantable medical devices pose unique challenges to effective post-marketing safety surveillance. These challenges include the complex interactions among devices, concurrently administered medications, existing patient co-morbidities, and the impact of operator learning in the safe and effective use of the device. Post-marketing device safety surveillance is further complicated because the approval of a medical device often triggers rapid and widespread use in ‘real-world’ patient populations. These populations are more diverse and complex than those studied in the controlled settings of clinical trials and premarket evaluations.4–6
Post-approval surveillance strategies in the United States rely on a combination of mandatory and voluntary adverse event reporting systems including ‘MedSun’, ‘MedWatch’, ‘MAUDE’, ‘MDR’ and ‘FAERS’7–10 as well as FDA mandated post-approval studies after initial market release.11 While these reporting systems are vital in identifying unexpected and unique adverse events, they suffer from a dependence on voluntary, and inconsistent, reporting of adverse events by physicians and hospitals. Consequently, event under-reporting and lack of available “denominator data” regarding the total number of devices implanted result.12 Post-approval studies have historically had low execution rates due to a variety of challenges, and frequently do not cover a broad range of longer-term outcomes.13–15 These limitations are well known – the FDA recently released a roadmap for strengthening the national medical device post-market surveillance system, which includes developing additional capacity in automated signal detection and surveillance methodologies, and deploying prospective surveillance networks.15–17
We developed a set of automated surveillance tools, denoted DELTA (Data Extraction and Longitudinal Trend Analysis System), and validated the methods and informatics infrastructure within a variety of retrospective data sources from clinical databases18–20 as well as randomized controlled clinical trials.21 Prospective use of DELTA has not been studied.
We sought to assess the operating characteristics of DELTA when utilized prospectively. We deployed this tool set utilizing advanced data de-identification and sharing methods, and a variety of robust statistical techniques for continuous surveillance of clinical registries to identify low-frequent safety signals.22 To our knowledge, prospective execution of this type of automated approach to medical device surveillance has not previously been described. The DELTA Network Study (DELTA-NS) is a multicenter, prospective, observational research study utilizing a distributed network architecture for data collection, designed to evaluate the safety of new cardiovascular devices used during percutaneous coronary intervention (PCI).23 Participating institutions used the National Cardiovascular Data Registry (NCDR) CathPCI data collection instrument24 to facilitate secure, de-identified, clinical data submission to a central DELTA server for prospective safety analyses. The primary objective of the study was to monitor the safety of devices used during PCI procedures for higher than expected adverse event rates. We monitored newly approved drug-eluting coronary stents (DES), coronary embolic protection devices (EPD), and vascular closure devices (VCD) in patients undergoing PCI.
Methods
A detailed description of the DELTA network study design has been published previously.23 In brief, the DELTA surveillance network was developed as a two-tiered network of secure database servers, with remote DELTA agents installed at five independent centers performing PCI in Massachusetts. Each remote DELTA agent was responsible for de-identification of all personal health information, as well as HIPAA compliant and secure information transfer to a central DELTA server located in the Partners Healthcare Research computing center. The central DELTA server conducted the prospective safety analyses, generating safety alerts and other messages that were transmitted via e-mail to the DELTA agent at the participating site. In addition to full de-identification of personal health information, information sent to the central DELTA server was stripped of physician and center identifiers to preserve complete anonymity of participating physicians.
Eligibility Criteria and Data Collection
All patients aged 18 or older who underwent PCI at one of the participating centers between January 1, 2010 and December 31, 2012 were included in the study. The participation of each site was reviewed and approved by each local Institutional Review Board (IRB), while the overall study was reviewed and approved by the Partners Healthcare IRB. Informed consent requirements were waived on the basis of minimal risk to individual patients through secondary research use of their de-identified clinical data.
All participating sites utilized their NCDR certified CathPCI data submission software that was used for state-mandated quarterly submission of PCI dataset files to the Massachusetts Data Analysis Center (Mass-DAC). The same data format was used to submit information to the DELTA-NS. Clinical information was entered, per the previously used center-specific workflow, by trained data managers or their designees, and uploaded to the local DELTA agent on a quarterly basis. Since the same dataset format was, by design, used by DELTA as was used to submit datasets to Mass-DAC, data management teams only entered data once. Retrospective data for PCI’s performed between January 1, 2008 and December 31, 2009 were pooled with the prospectively collected data to increase specific device exposure sample size and to facilitate propensity matched analyses.
Device Exposures and Adverse Outcomes
We evaluated the acute post-procedural safety of three types of medical devices used routinely in interventional cardiology practice. These included: 1) two new DES (Abbott Vascular Xience™ stent and Medtronic Endeavor™ stent), 2) two new EPD (Boston Scientific FilterWire™ and EV3 Spider™) and 3) four VCD (St. Jude Medical AngioSeal™, Access Mynx™, Abbott Perclose Proglide™ and Abbott Vascular StarClose™). Distinct products, as listed in the NCDR CathPCI intracoronary devices and vascular closure devices tables that represented only incremental changes in device design, were grouped into device “families” in order to increase sample size, and therefore analytic power. The implant or active component of the device was the same within each device family, with only minor differences in the device delivery system appearance.
Each medical device was evaluated for risk-adjusted adverse outcomes specific to the device type (i.e. DES, VCD or EPD). The adverse events for stents and EPD included in-hospital death, in-hospital post-procedural myocardial infarction (MI), and a composite of major adverse cardiac events (MACE) including in-hospital death, post-procedural MI, stroke or unplanned coronary revascularization. For the VCDs, adverse events included in-hospital minor vascular complications (including groin bleeding, hematoma >5cm, pseudoaneurysm, and arteriovenous fistula), major vascular complications (including retroperitoneal hemorrhage, limb ischemia and any surgical or interventional repair), and any vascular complication. Based on historical adverse event rates from the Massachusetts statewide statistics20, we determined that the analyses would have one-sided power to detect a 50% increase in composite adverse events, using a 10% alpha error, ranging from 72% for EV3 Spider EPD to 99% for the Xience DES, and 10 of the 18 device-outcome analyses would have a power of greater than 85%.
Propensity Score Matching and Statistical Methods
A propensity matched comparison population was developed for each device-outcome analysis, based on previously published risk factors for the outcome of interest as well as factors considered by domain experts to potentially influence the selection of one device versus another. A complete list of variables included in the propensity match analysis of each class of device has been published previously23 and is provided in the appendix. Propensity scores were developed using non-parsimonious logistic regression models developed with the device of interest (exposure) used as the dependent variable, adjusting for the specified variables. Matching cases were randomly selected in a 1:1 ratio, within 6 months of the date of the exposed case, using a fixed propensity score caliper width of 0.05 as previously validated20,23,25.
Adverse-event rates were calculated quarterly for the propensity score–matched cohorts for each device-outcome analysis. The cumulative number of events per quarter was used to calculate a difference of proportions between the two groups by the Wilson method25. If the confidence intervals (CIs) of point estimates of the difference of proportions did not cross zero, a statistically significant difference between the comparison and exposed groups was detected. Safety alerts were triggered if the cumulative event rates in the exposed group exceeded the upper confidence limit of the event rates in the comparison group by greater than 20% (selected to represent a clinically meaningful difference in safety profile), using 95% confidence intervals, corrected for type I error inflation using the O’Brien–Fleming alpha-spending method. Importantly, the propensity matched cohort comparison, as employed in this study, inherently compares the relative safety of one device to that of a comparator device in a population of similar patients, and cannot be used to directly assess absolute safety relative to expected performance or prior experience.
Any device-outcome analysis that generated two or more consecutive safety alerts during cumulative monitoring was considered a “sustained alert” and prompted detailed sensitivity analyses, using alternative risk adjustment methods, as previously described23, in order to verify, or refute, the adverse safety signal identified using the propensity-matched analysis. Procedures for the propensity score model and statistical analysis were performed by the integrated open-source Observational Cohort Event Analysis and Notification System (OCEANS) (http://sourceforge.net/projects/oceans/) developed to provide biostatistics processing support for automated surveillance. All algorithms used have been validated against SAS version 9.1 (Cary, NS). All statistical tests were 2-sided, with p< 0.05 considered statistically significant for all comparisons.
Results
A total of 23,333 PCI cases were performed between January 1, 2008 and December 31, 2012 at the five participating sites. Among the 8 devices studied, three adverse outcomes were monitored for each device, resulting in 24 separate simultaneous prospective safety analyses; each repeated on a quarterly basis throughout the study. Two analyses within the Perclose VCD analysis triggered sustained safety alerts. The remaining 7 devices (including the Xience DES, Endeavor DES, Filterwire EPD, Spiderwire EPD, Angio-Seal VCD, Mynx VCD and StarClose VCD) did not trigger any safety alerts throughout the study.
Tables 1, 2 and 3 summarize the results of the safety analyses performed and the concurrent matched comparison populations chosen for each device safety analysis. The proportion of exposures successfully matched using the propensity-matching algorithm ranged from 40.2% for the Xience DES to 99.9% for the Mynx. The relatively low match rate for the Xience DES was due to the use of Xience as the predominant DES within the participating sites, resulting in relatively fewer alternative DES to use as controls in the propensity match.
Table 1.
Drug Eluting Stents: Summary of safety monitoring results.
| Endeavor | Xience | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Device Type | Drug eluting stent | Drug eluting stent | ||||
| Manufacturer | Medtronic | Abbott Vascular | ||||
| Surveillance dates | 1/1/2008–12/30/2012 | 7/1/2008a–12/31/2012 | ||||
| Total device exposure cases | 1,459 | 9,374 | ||||
| Number of quarterly analyses | 20 | 18 | ||||
| Mean exposures per study period (Quarter) | 73 | 521 | ||||
| Comparator group | Alternative drug eluting stent | Alternative drug eluting stent | ||||
| Number of cases matched | 1,443 | 3,768 | ||||
| Proportion of cases matched | 98.9% | 40.2% | ||||
|
| ||||||
| Outcomes monitored | In Hospital Death | Post procedure myocardial infarction (MI) | Major adverse cardiac events (MACE) | In Hospital Death | Post procedure myocardial infarction (MI) | Major adverse cardiac events (MACE) |
|
| ||||||
| Observed Event Rate % (within propensity matched cohort) | 0.76% | 3.47% | 5.06% | 0.72% | 2.36% | 3.72% |
| Relative risk (95% CI) | 1.83 (0.68–4.94) | 1.32 (0.87–1.99) | 1.33 (0.94–1.87) | 1.17 (0.67–2.04) | 1.09 (0.80–1.46) | 1.17 (0.92–1.48) |
| Safety Alerts Triggered | None | None | None | None | None | None |
| Time to first safety alert | NAb | NAb | NAb | NAb | NAb | NAb |
Abbreviations: CI, Confidence Interval
Xience device received FDA approval July 2, 2008
No repeated or sustained safety signal generated during the study
Table 2.
Embolic Protection Devices: Summary of safety monitoring results.
| FilterWire | SpiderWire | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Device Type | Embolic protection device | Embolic protection device | ||||
| Manufacturer | Boston Scientific | ev3Inc | ||||
| Surveillance dates | 1/1/2008–12/31/2012 | 1/1/2008–12/31/2012 | ||||
| Total device exposure cases | 350 | 452 | ||||
| Number of quarterly analyses | 20 | 20 | ||||
| Mean exposures per study period (Quarter) | 18 | 23 | ||||
| Comparator group | Alternative embolic protection device, not Filterwire | Alternative embolic protection device, not Spiderwire | ||||
| Number of cases matched | 328 | 352 | ||||
| Proportion of cases matched | 93.7% | 77.9% | ||||
|
| ||||||
| Outcomes monitored | In Hospital Death | Post procedure myocardial infarction (MI) | Major adverse cardiac events (MACE) | In Hospital Death | Post procedure myocardial infarction (MI) | Major adverse cardiac events (MACE) |
|
| ||||||
| Observed Event Rate % (within propensity matched cohort) |
2.13% | 4.88% | 6.40% | 0.57% | 4.83% | 5.40% |
| Relative risk (95% CI) | 2.33 (0.61–8.95) | 1.00 (0.51–1.97) | 1.00 (0.56–1.80) | 0.25 (0.05–1.17) | 0.89 (.47–1.69) | 0.73 (0.41–1.30) |
| Safety Alerts Triggered | None | None | None | None | None | None |
| Time to first safety alert | NAa | NAa | NAa | NAa | NAa | NAa |
Abbreviations: CI, Confidence Interval
No repeated or sustained safety signal generated during the study
Table 3.
Vascular Closure Devices: Summary of safety monitoring results.
| AngioSeal | Mynx | Perclose | Starclose | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||
| Device Type | Vascular Closure Device | Vascular Closure Device | Vascular Closure Device | Vascular Closure Device | ||||||||
| Manufacturer | St. Jude | Access | Abbott | Abbott | ||||||||
| Surveillance dates | 1/1/2008–12/31/2012 | 1/1/2008–12/30/2012 | 1/1/2008–12/31/2012 | 1/1/2008–12/31/2012 | ||||||||
| Total device exposure cases | 5,734 | 717 | 2,539 | 951 | ||||||||
| Number of quarterly analyses | 20 | 20 | 20 | 20 | ||||||||
| Mean exposures per study period (Quarter) | 287 | 36 | 127 | 48 | ||||||||
| Comparator group | Alternative vascular closure device, not AngioSea | Alternative vascular closure device, not Mynx | Alternative vascular closure device, not Perclose | Alternative vascular closure device, not Starclose | ||||||||
| Number of cases matched | 4,028 | 716 | 2,489 | 947 | ||||||||
| Proportion of cases matched | 70.2% | 99.9% | 98.0% | 99.6% | ||||||||
|
| ||||||||||||
| Outcomes monitored | Minor Vascular Complication |
Major Vascular Complication |
Any Vascular Complication |
Minor Vascular Complication |
Major Vascular Complication |
Any Vascular Complication |
Minor Vascular Complication |
Major Vascular Complication |
Any Vascular Complication |
Minor Vascular Complication |
Major Vascular Complication |
Any Vascular Complication |
|
| ||||||||||||
| Observed Event Rate % (within propensity matched cohort) |
0.22% | 0.55% | 0.70% | 1.12% | 0.70% | 1.54% | 1.17% | 0.48% | 1.41% | 0.42% | 0.32% | 0.74% |
| Relative risk (95% CI) | 0.27 (0.13–0.57) | 1.38 (0.72–2.16) | 0.67 (0.41–1.07) | 2.67 (0.71–10.0) | 1.67 (0.40–6.95) | 2.20 (0.77–6.30) | 4.14 (1.82–9.44) | 1.09 (0.48–2.47) | 2.06 (1.16–3.67) | 0.79 (0.22–2.97) | 0.50 (0.13–1.99) | 0.78 (0.29–2.08) |
| Safety Alerts Triggered | None | None | None | None | None | None | Yes | None | Yes | None | None | None |
| Time to first safety alert (in months) | NAa | NAa | NAa | NAa | NAa | NAa | 30 | NAa | 33 | NAa | NAa | NAa |
Abbreviations: CI, Confidence Interval
No repeated or sustained safety signal generated during the study
Exploration of Safety Alerts
While there were no sustained safety alerts for any device studied within the DES or EPD device type analyses, there were sustained alerts triggered for the Perclose VCD, as well as high relative event rates noted for the Mynx VCD, though this analysis did not reach statistical threshold for triggering a sustained alert. The Perclose family of VCD was used in 2,539 procedures, of which 2,489 (98%) were successfully matched to comparison procedures receiving alternative VCD (Table 3). A safety alert for the Perclose family of VCD related to an increased risk of minor vascular complications was first triggered at Q3 2010 (after accruing 1,575 cases), and continued through completion of the study (total exposures = 2,539 cases). The safety alerts included an increased risk of both minor vascular complications (Figure 1) and any vascular complication (Figure 2). By the end of the study period, the relative risk of minor vascular complications was 4.14 (p value < 0.001) with the use of the Perclose VCD compared with propensity-matched patients receiving alternative VCDs. Similarly, the relative risk of any vascular complication was 2.06 (p value = 0.01) with Perclose VCD versus alternative VCD; a result driven by the increased risk of minor vascular complications observed. Baseline disparities between patients receiving Perclose VCD and those receiving alternative VCD were eliminated for most covariates after the propensity match was applied (see Table 4), though differences remained in the proportion of patients with prior peripheral arterial disease, patients presenting with STEMI, and those undergoing left main PCI. There were no significant differences between Perclose VCD as compared with alternative VCD with respect to the rate of major vascular complications.
Figure 1. Propensity-matched analysis of the incidence of minor vascular complications following use of Perclose vascular closure device.
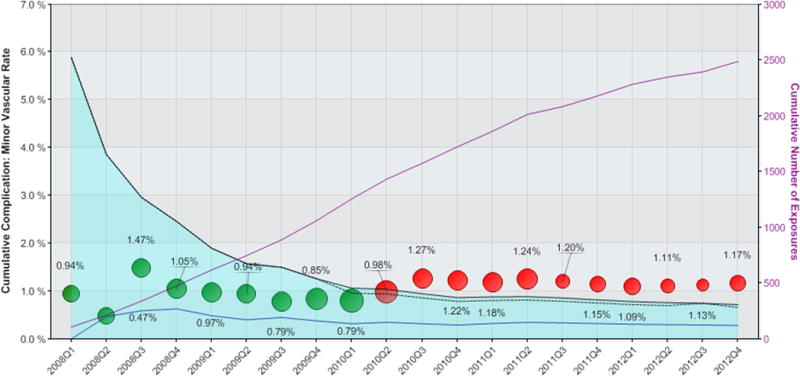
The cumulative rate of minor vascular complications for patients receiving the Perclose VCD are shown as circles, where the size of the circle is proportional to the number of Perclose vascular closure devices used during the calendar quarter. The light blue shaded area represents the 95% confidence interval, corrected for multiple comparisons, with black dashed lines representing the uncorrected 95% confidence limits. The solid blue line indicates mean event rates of matched patients receiving alternative vascular closure device Green circles indicate cumulative observed event rates for patients receiving Perclose fall within propensity-matched 95% confidence intervals. Red circles indicate safety alerts were triggered due to cumulative observed event rates exceeding the upper 95% confidence limits. The solid purple line denotes cumulative number of exposures as shown on the right-sided vertical axis.
Figure 2. Propensity-matched analysis of the incidence of any vascular complication following use of Perclose vascular closure device.
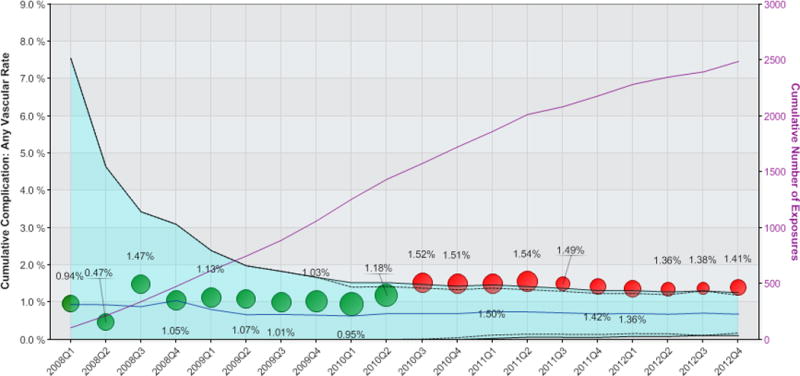
The cumulative rate of any vascular complication for patients receiving the Perclose VCD are shown as circles, where the size of the circle is proportional to the number of Perclose vascular closure devices used during the calendar quarter. The light blue shaded area represents the 95% confidence interval, corrected for multiple comparisons, with black dashed lines representing the uncorrected 95% confidence limits. The solid blue line indicates mean event rates of matched patients receiving alternative vascular closure device. Green circles indicate cumulative observed event rates for patients receiving Perclose fall within propensity-matched 95% confidence intervals. Red circles indicate safety alerts were triggered due to cumulative observed event rates exceeding the upper 95% confidence limits. The solid purple line denotes cumulative number of exposures as shown on the right-sided vertical axis.
Table 4.
Perclose propensity match covariate distributions.
| Covariate | Total Study Population | Propensity Matched | Unmatched Cases | |||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Perclose n = 2,539 |
Alternative VCD n = 7,414 |
Standard Differencea | Perclose n = 2,489 |
Alternative VCD n = 2,489 |
Standard Differencea | Perclose n = 50 |
Standard Differencea | |
| Age≥70 years | 39.54 | 34.23 | 11.02 | 39.71 | 36.20 | 6.63 | 46.00 | 13.35 |
| Women | 25.88 | 23.95 | 4.44 | 25.87 | 23.62 | 5.22 | 26.00 | 0.29 |
| Patient Height, mean (SD), cm | 170.9 (9.9) | 171.5 (10.0) | 2.85 | 170.9 (9.8) | 171.5 (9.9) | 2.71 | 171.9 (10.9) | 4.77 |
| Patient Weight, mean (SD), kg | 84.5 (18.0) | 86.4 (18.7) | 5.09 | 84.5 (18.0) | 86.2 (18.7) | 4.64 | 83.8 (20.7) | 1.96 |
| History of Diabetes | 34.53 | 32.33 | 4.67 | 34.58 | 32.90 | 3.56 | 32.00 | 5.48 |
| Hypertension | 84.43 | 80.81 | 9.56 | 84.31 | 80.86 | 9.11 | 90.00 | 17.06 |
| Chronic Lung Disease | 15.37 | 12.18 | 9.27 | 15.23 | 12.91 | 6.69 | 22.00 | 17.45 |
| Current Dialysis | 2.21 | 1.70 | 3.66 | 2.09 | 1.61 | 3.58 | 8.00 | 27.26 |
| History of Peripheral Artery Disease | 13.96 | 9.54 | 13.75 | 13.92 | 8.27 | 18.07 | 16.00 | 5.85 |
| NSTEMI on Presentation | 23.63 | 22.15 | 3.53 | 23.22 | 24.15 | 2.17 | 44.00 | 45.09 |
| STEMI on Presentation | 14.34 | 15.35 | 2.85 | 14.18 | 18.04 | 10.51 | 22.00 | 20.41 |
| Emergent Procedure | 16.98 | 16.31 | 1.79 | 16.63 | 19.69 | 7.93 | 34.00 | 40.76 |
| Left main percutaneous cardiac intervention | 5.00 | 2.10 | 15.79 | 4.58 | 2.61 | 10.59 | 26.00 | 62.34 |
| Bivalirudin use | 44.51 | 49.29 | 9.59 | 45.08 | 47.69 | 5.24 | 16.00 | 66.54 |
| GlycoProtein IIb/IIIa antagonist use | 16.03 | 21.03 | 12.89 | 16.27 | 19.37 | 8.09 | 4.00 | 41.53 |
| Number of Vessels Treated, mean (SD) | 1.3 (0.6) | 1.3 (0.6) | 4.17 | 1.3 (0.6) | 1.3 (0.6) | 5.94 | 1.4 (0.6) | 3.03 |
| Intra-aortic balloon pump | 2.40 | 1.01 | 10.75 | 2.09 | 1.25 | 6.59 | 18.00 | 54.89 |
| FluoroTime, minutes | 257 (19.1) | 21.7 (15.1) | 12.06 | 25.5 (18.9) | 22.1 (15.3) | 9.91 | 51.7 (28.9) | 56.60 |
Abbreviations: SD, standard deviation;
Absolute standarized difference (percentile) in covariate means and proportions, before and after matching
Pre-specified sensitivity analyses were performed per study protocol to further explore the safety signals detected for Perclose VCD. A multiple logistic regression predictive model for the risk of minor vascular complications, based on the cohort of all non–Perclose VCD used in 2008, was developed and applied prospectively to the entire cohort of patients receiving Perclose VCD. We observed no significant difference between the alerting behavior using the logistic regression predictive model versus the original propensity match analysis; thereby supporting the findings of the primary propensity analysis. Examination of the incidence of minor vascular complications according to participating site was conducted and indicated that the increased risk for minor vascular complications was driven exclusively by a substantially increased risk at one of the five participating centers. Further subgroup analysis demonstrated that, among the patients in whom a VCD was used, the outlier institution treated a similar proportion of patients who had PAD as compared to the other participating centers (12.8% vs. 10.6%, p=0.14), while the outlier center used Perclose significantly less often than alternative VCDs in these PAD patients (69.2% vs. 95.3%, p<0.001).
Regarding the Mynx family of VCD, a non-significant trend towards increased rate of any vascular complication was observed in Q3 2010 (after accruing 687 cases), with a relative risk of 2.20 as compared with alternative VCD (p value = 0.13). However, there was limited utilization of the Mynx device in clinical practice at the participating sites and use declined considerably after 2010, which made this analysis underpowered to confirm the presence of a sustained safety signal in the use of the Mynx device.
Discussion
The DELTA Network Study employed a novel approach of prospective post-marketing safety surveillance of recently approved medical devices through continuous sharing and transmission of de-identified clinical data by a network of independent medical centers, along with automated, near real-time comparative safety analysis. The existing NCDR CathPCI data collection instrument was used to minimize the need for customization of the local database interface which permitted seamless integration of the DELTA surveillance system into each institution’s quality monitoring procedures.
No safety alerts were identified for either the two recently approved DES (Abbott Vascular Xience™ stent and Medtronic Endeavor™ stent) or the two commonly used EPD (Boston Scientific FilterWire™ and EV3 Spider™). However, increased rates of minor and any vascular complications (but not major vascular complications) with use of Perclose VCD compared with propensity-matched alternative VCD were observed. The findings were sustained after temporal trend analyses, and application of alternative risk models via multivariate logistic regression adjustment. Further exploration of the rates of vascular complications in the use of Perclose at the participating sites enabled identification of a single ‘outlier’ site that had an unusually high rate of vascular complications with use of Perclose VCD. This outlier site was notified of the safety signal and has implemented additional device training and quality improvement initiatives intended to improve the safety in the use of the device. The identification of a single outlier site as the driving factor in generating the safety signal for Perclose VCD likely indicates that safety concern was likely a result of case/patient selection or operator training at the outlier site, rather than an indication of intrinsic safety concerns for the Perclose device itself. In addition, the identification of a single outlier site highlights the critical importance of pre-specification of sensitivity, subgroup, and secondary endpoint analyses, in order to provide the most robust and valid interpretation of any safety signals generated by the primary analyses during a prospective safety study.
Putting these findings into context, it is important to recognize that, although there are few comparative data regarding the safety of various VCD, a previous meta-analysis as well as multiple large observational registry studies have found the Perclose VCD to be safe with no increase in the incidence of vascular complications compared with alternative VCD or manual compression26. While a safety signal was identified in this analysis, it is more likely that this signal represents residual confounding due to case selection or training at the single, outlier, center, rather than an intrinsic safety concern with the Perclose device itself. In addition, the current analysis was not designed to account for the experience of specific operators in the use of particular devices. The impact of device specific learning has been shown to significantly influence VCD clinical outcomes27 and differences in experience between providers at the participating centers cannot be excluded as potentially impacting the results observed. Additionally, the DELTA-NS identified a non-significant trend towards increased rate of vascular complications with use of the Mynx family of VCD, relative to propensity matched patients receiving alternative VCD. This analysis was underpowered due to the significant decline in the use of the Mynx device during the course of the study (see Figure 3). One possible explanation for the dramatic reduction in the use of the Mynx device was the observation of the higher than anticipated vascular complication in patients receiving the device on the part of the treating interventional cardiologists at the participating centers, leading to decreased utilization of this device.
Figure 3. Utilization of the Mynx VCD as a proportion of total VCD use during the study.
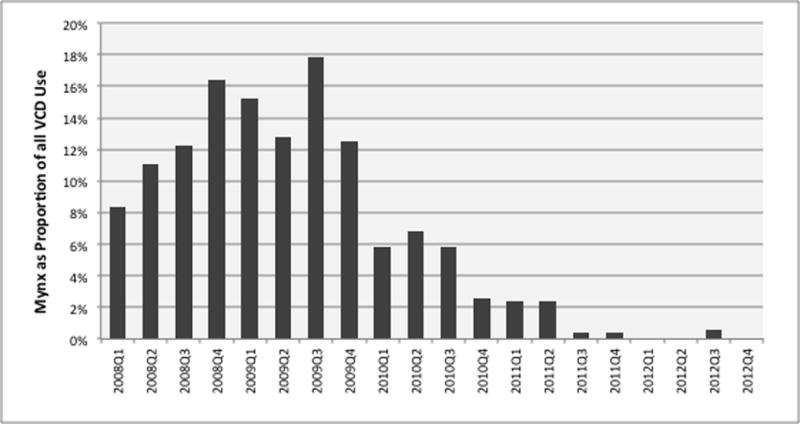
Among the study sites, Mynx VCD utilization peaked at 17.8% of all VCD implants in the third quarter of 2009, followed by a sudden and dramatic decline in utilization to a rate of <1% of all VCD by 2012.
A strategy of prospective device safety surveillance through continuous analysis of high-quality clinical registries, as validated by the DELTA network study, has the potential to identify low-frequency safety signals through the simultaneous monitoring of numerous high-risk devices by pooling detailed clinical data from multiple institutions. Such a strategy represents a substantial change from current approaches to the routine monitoring of post-market safety of medical devices. Current device safety surveillance methods employed by public health officials principally rely on passive reporting of adverse events by patients, physicians, manufacturers and health care organizations. These passive surveillance systems suffer from event under-reporting and lack denominator data regarding comprehensive exposure12,15. Other more recent mechanisms for monitoring device safety, including federally mandated post-approval clinical registries frequently have limited, or no, control populations and often lack statistical power to detect very low-frequency safety signals12–15. The DELTA-NS approach may therefore complement future prospective post-marketing surveillance approaches evaluating the safety and efficacy of medical devices through the monitoring of routinely collected clinical data. Although such automated safety surveillance tools will initially depend on detailed clinical registries, future efforts should also be directed towards the routine monitoring of clinical data extracted from EHR.
Automated prospective surveillance of medical devices must overcome several important challenges to be used as an effective surveillance tool. Timely submissions of complete data by all participating sites with prompt adjudication and validation of alerts is essential. Any safety signal identified through automated safety surveillance must be interpreted with caution and explored in-depth by detailed sensitivity analyses and other rigorous epidemiologic explorations. Such analyses are critical to investigate potential residual confounders of the observed outcomes, and confirm that the preliminary safety alerts triggered by propensity score matching, are likely ‘true’ safety signals. In addition, it is important that alert driven sensitivity analyses, subgroup analyses, and secondary endpoints be pre-defined in a clear and written safety surveillance analytic plan, in order to minimize the risks associated with multiple comparisons or the temptation of unstructured data exploration.
Study Limitations
By virtue of being a non-randomized prospective observational study, the DELTA NS suffers from biases inherent to observational studies. Given the dependence on the NCDR CathPCI data collection instrument for data acquisition and submission, the scope of the clinical factors that were available for analysis was limited. Version 4 of CathPCI does not include vascular sheath size as a covariate in the dataset, and therefore an imbalance in the use of large bore devices (>8 French) among centers or VCD populations cannot be excluded, though unlikely due to the extremely rare use of such sheath sizes in contemporary practice at the participating sites. Similarly, the dataset did not identify non-standard VCD deployment such as the “preclose” technique using Perclose VCD28 which could have confounded the observed event rates. Likewise, the adherence of participating sites to the comprehensive reporting of post-procedure clinical events is essential to minimize ascertainment bias, as it is for all clinical registry based studies. Also, for this study, clinical events were monitored only through the time of discharge, limiting the scope of any safety comparisons for the studied devices. Thus, potentially important late safety concerns such as stent thrombosis (DES devices) or access site infections (VCD) could not be assessed. In addition, the collection of post-PCI biomarkers is variable across participating centers, thereby limiting the potential value of surveillance for outcomes, such as post-procedure myocardial infarction, based on analysis of this laboratory result. While the dataset used in this analysis was larger than many observational studies of interventional cardiology devices, it was significantly smaller than studies of national or regional registries, and suffers from a lack of generalizability due to the small number of participating centers. Finally, since this was a non-randomized study in which patients might have been treated with multiple devices during a procedure, the ability to attribute a particular clinical outcome event to an exposure may have been confounded by simultaneous exposures to other devices or treatment decisions.
While propensity score matching were used to reduce the potential confounding between the population of patients exposed to a particular device and patients receiving alternative devices, the effectiveness of the matching process is inherently limited. In this study, there remained significant residual imbalance between the study cohorts within several covariates including a history of peripheral arterial disease as well as increased fluoroscopy times, potentially indicating more complex procedures for patients receiving Perclose as compared to other VCD. These imbalances may have biased the results toward observing a higher rate of vascular complications in patients receiving Perclose. Potentially mitigating this bias, there were fewer Perclose patients treated for STEMI which would have been expected to be associated with reduced vascular access site complications. In addition, propensity score matching led to the exclusion of variable number of exposures due to inability to find adequate matched cohorts, thereby, limiting any comparative statements that can be made about device safety in these patient populations. Such inability to identify adequate matched controls for exposed cases may reflect either limited sample size or might imply truly different populations of patients were receiving the device of interest, making comparisons with alternative devices more challenging. The comparison of cohorts of propensity matched cases results in a comparison of the relative safety of one device to another, rather than an absolute assessment of safety as compared to prior experience or clinical trial experience. Thus, propensity matched cohort analyses, as used in this study, could miss the identification of an adverse safety signal if the comparator group itself experienced a higher rate of adverse events than would have been initially expected. Finally, since the DELTA network was a pilot multicenter prospective surveillance study, it may have had insufficient power to detect a safety signal at the thresholds for alerting selected.
Conclusion
The DELTA-NS demonstrates the successful implementation of an automated prospective medical device safety surveillance study conducted within a network of participating clinical centers sharing routinely collected clinical registry data in near real-time. Such a strategy may offer substantial advantages over existing strategies employed by U.S. regulatory agencies. Early identification of clinical safety concerns through such automated surveillance could further support public health officials to identify the need for additional device-specific training, refinement of patient selection criteria, or initiation of comprehensive epidemiologic exploration into inherent device failures, in order to reduce medical device related-risks to future patients.
Supplementary Material
Acknowledgments
Funding
This research was supported through research grants from the National Library of Medicine (R01 LM008142) and the U.S. Food and Drug Administration (HHSF Contract 223200830058C). In addition, Dr. Resnic and Dr. Normand’s efforts were funded, in part, by Contract HHSF223201110172C (MDEpiNet Methodology Center) and grant 1U01 FD004493-01 (MDEpiNet Medical Counter Measures Study), both from the U.S. Food and Drug Administration.
Footnotes
Subject Codes:
[23] Catheter-based coronary and valvular interventions: other
[24] Catheter-based coronary interventions: stents
[100] Health policy and outcome research
Disclosures
FSR serves as a consultant to St. Jude Medical, Inc. a medical device manufacturer. RC is employed by Boston Advanced Analytics which develops and maintains software supporting the DELTA system.
References
- 1.Hauser RG, Kallinen LM, Almquist AK, Gornick CC, Katsiyiannis WT. Early failure of a small-diameter high-voltage implantable cardioverter-defibrillator lead. Heart Rhythm. 2007;4:892–896. doi: 10.1016/j.hrthm.2007.03.041. [DOI] [PubMed] [Google Scholar]
- 2.Steele GD, Fehring TK, Odum SM, Dennos AC, Nadaud MC. Early failure of articular surface replacement total hip arthroplasty. J Arthroplasty. 26:14–18. doi: 10.1016/j.arth.2011.03.027. [DOI] [PubMed] [Google Scholar]
- 3.Maisel WH. Semper fidelis–consumer protection for patients with implanted medical devices. N Engl J Med. 2008;358:985–987. doi: 10.1056/NEJMp0800495. [DOI] [PubMed] [Google Scholar]
- 4.Shah JS, Maisel WH. Recalls and safety alerts affecting automated external defibrillators. JAMA. 2006;296:655–660. doi: 10.1001/jama.296.6.655. [DOI] [PubMed] [Google Scholar]
- 5.Maisel WH. Unanswered questions–drug-eluting stents and the risk of late thrombosis. N Engl J Med. 2007;356:981–984. doi: 10.1056/NEJMp068305. [DOI] [PubMed] [Google Scholar]
- 6.Rosen CJ. The rosiglitazone story–lessons from an FDA advisory committee meeting. N Engl J Med. 2007;357:844–846. doi: 10.1056/NEJMp078167. [DOI] [PubMed] [Google Scholar]
- 7.Kessler DA. Introducing MedWatch. A new approach to reporting medication and device adverse effects and product problems. JAMA. 1993;269:2765–2768. doi: 10.1001/jama.269.21.2765. [DOI] [PubMed] [Google Scholar]
- 8.Mehran R, Leon MB, Feigal DA, Jefferys D, Simons M, Chronos N, Fogarty TJ, Kuntz RE, Baim DS, Kaplan AV. Post-market approval surveillance: A call for a more integrated and comprehensive approach. Circulation. 2004;109:3073–3077. doi: 10.1161/01.CIR.0000134694.78653.B6. [DOI] [PubMed] [Google Scholar]
- 9.FDA. Manufacturer and User Facility Device Experience Database – (MAUDE) Available at: http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/PostmarketRequirements/ReportingAdverseEvents/ucm127891.htm.
- 10.FDA. FDA Adverse Event Reporting System (FAERS) Available at: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Surveillance/AdverseDrugEffects/default.htm.
- 11.FDA. FDA Medical Devices Post-Approval Studies. Available at: http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/PostmarketRequirements/PostApprovaStudies/default.htm.
- 12.O’Shea JC, Kramer JM, Califf RM, Peterson ED. Part i: Identifying holes in the safety net. Am Heart J. 2004;147:977–984. doi: 10.1016/j.ahj.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 13.Vidi VD, Matheny ME, Resnic FS. Post-marketing device safety surveillance. Contemp Clin Trials. 2011;32:307–308. doi: 10.1016/j.cct.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 14.Normand SL, Hatfield L, Drozda J, Resnic FS. Postmarket surveillance for medical devices: America’s new strategy. BMJ. 2012;345:e6848. doi: 10.1136/bmj.e6848. [DOI] [PubMed] [Google Scholar]
- 15.Resnic FS, Normand SL. Postmarketing surveillance of medical devices–filling in the gaps. N Engl J Med. 2012;366:875–877. doi: 10.1056/NEJMp1114865. [DOI] [PubMed] [Google Scholar]
- 16.FDA. Strengthening our national system for medical device postmarket surveillance. 2012 Available at: http://www.fda.gov/downloads/AboutFDA/CentersOffices/CDRH/CDRHReports/UCM301924.pdf.
- 17.FDA. Strengthening our national system for medical device postmarket surveillance: Update and next steps. 2013 Available at: http://www.fda.gov/downloads/MedicalDevices/Safety/CDRHPostmarketSurveillance/UCM348845.pdf.
- 18.Arora N, Matheny ME, Sepke C, Resnic FS. A propensity analysis of the risk of vascular complications after cardiac catheterization procedures with the use of vascular closure devices. Am Heart J. 2007;153:606–611. doi: 10.1016/j.ahj.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 19.Hauser RG, Mugglin AS, Friedman PA, Kramer DB, Kallinen L, McGriff D, Hayes DL. Early detection of an underperforming implantable cardiovascular device using an automated safety surveillance tool. Circ Cardiovasc Qual Outcomes. 2012;5:189–196. doi: 10.1161/CIRCOUTCOMES.111.962621. [DOI] [PubMed] [Google Scholar]
- 20.Resnic FS, Gross TP, Marinac-Dabic D, Loyo-Berrios N, Donnelly S, Normand SL, Matheny ME. Automated surveillance to detect post-procedure safety signals of approved cardiovascular devices. JAMA. 2010;304:2019–2027. doi: 10.1001/jama.2010.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matheny ME, Morrow DA, Ohno-Machado L, Cannon CP, Sabatine MS, Resnic FS. Validation of an automated safety surveillance system with prospective, randomized trial data. Med Decis Making. 2009;29:247–256. doi: 10.1177/0272989X08327110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matheny ME, Ohno-Machado L, Resnic FS. Monitoring device safety in interventional cardiology. J Am Med Inform Assoc. 2006;13:180–187. doi: 10.1197/jamia.M1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vidi VD, Matheny ME, Donnelly S, Resnic FS. An evaluation of a distributed medical device safety surveillance system: The DELTA network study. Contemp Clin Trials. 2011;32:309–317. doi: 10.1016/j.cct.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cannon CP, Battler A, Brindis RG, Cox JL, Ellis SG, Every NR, Flaherty JT, Harrington RA, Krumholz HM, Simoons ML, Van De Werf FJ, Weintraub WS, Mitchell KR, Morrisson SL, Anderson HV, Cannom DS, Chitwood WR, Cigarroa JE, Collins-Nakai RL, Gibbons RJ, Grover FL, Heidenreich PA, Khandheria BK, Knoebel SB, Krumholz HL, Malenka DJ, Mark DB, McKay CR, Passamani ER, Radford MJ, Riner RN, Schwartz JB, Shaw RE, Shemin RJ, Van Fossen DB, Verrier ED, Watkins MW, Phoubandith DR, Furnelli T. American college of cardiology key data elements and definitions for measuring the clinical management and outcomes of patients with acute coronary syndromes. A report of the american college of cardiology task force on clinical data standards (acute coronary syndromes writing committee) J Am Coll Cardiol. 2001;38:2114–2130. doi: 10.1016/s0735-1097(01)01702-8. [DOI] [PubMed] [Google Scholar]
- 25.Austin PC. Optimal caliper widths for propensity-score matching when estimating differences in means and differences in proportions in observational studies. Pharm Stat. 2011;10:150–161. doi: 10.1002/pst.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nikolsky E, Mehran R, Halkin A, Aymong ED, Mintz GS, Lasic Z, Negoita M, Fahy M, Krieger S, Moussa I, Moses JW, Stone GW, Leon MB, Pocock SJ, Dangas G. Vascular Complications associated with arteriotomy closure devices in patients undergoing percutaneous coronary procedures: A meta-analysis. J Am Colll Cardiol. 2004;44:1200–1209. doi: 10.1016/j.jacc.2004.06.048. [DOI] [PubMed] [Google Scholar]
- 27.Resnic FS, Wang TY, Arora N, Vidi V, Dai D, Ou FS, Matheny ME. Quantifying the learning curve in the use of a novel vascular closure device: an analysis of the NCDR (National Cardiovascular Data Registry) CathPCI registry. JACC: Card Interv. 2012;5:82–89. doi: 10.1016/j.jcin.2011.09.017. [DOI] [PubMed] [Google Scholar]
- 28.Greise DP, Reents W, Diegeler A, Kerber S, Babin-Ebell J. Simple, effective and safet vascular access site closure with the double-proglide preclose technique in 162 patients receiving transfemoral transcatheter aortic valve implantation. Catheter Cardiovasc Interv. 2013;82:E734–E741. doi: 10.1002/ccd.25053. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.