

Therapeutic histone deacetylase inhibition with vorinostat or romidepsin significantly reduces ventricular sodium current density and NaV1.5 protein expression. A slight positive shift in the voltage activation curve, presumably the result of lysine acetylation, decreases the inactivation rate at low activating potentials but does not increase the late sodium current amplitude.
Keywords: trichostatin A, vorinostat, romidepsin, sodium current, gap junctions
Abstract
Histone deacetylase (HDAC) inhibitors are small molecule anticancer therapeutics that exhibit limiting cardiotoxicities including QT interval prolongation and life-threatening cardiac arrhythmias. Because the molecular mechanisms for HDAC inhibitor-induced cardiotoxicity are poorly understood, we performed whole cell patch voltage-clamp experiments to measure cardiac sodium currents (INa) from wild-type neonatal mouse ventricular or human-induced pluripotent stem cell-derived cardiomyocytes treated with trichostatin A (TSA), vorinostat (VOR), or romidepsin (FK228). All three pan-HDAC inhibitors dose dependently decreased peak INa density and shifted the voltage activation curve 3- to 8-mV positive. Increases in late INa were not observed despite a moderate slowing of the inactivation rate at low activating potentials (<−40 mV). Scn5a mRNA levels were not significantly altered but NaV1.5 protein levels were significantly reduced. Immunoprecipitation with anti-NaV1.5 and Western blotting with anti-acetyl-lysine antibodies indicated that NaV1.5 acetylation is increased in vivo after HDAC inhibition. FK228 inhibited total cardiac HDAC activity with two apparent IC50s of 5 nM and 1.75 μM, consistent with previous findings with TSA and VOR. FK228 also decreased ventricular gap junction conductance (gj), again consistent with previous findings. We conclude that pan-HDAC inhibition reduces cardiac INa density and NaV1.5 protein levels without affecting late INa amplitude and, thus, probably does not contribute to the reported QT interval prolongation and arrhythmias associated with pan-HDAC inhibitor therapies. Conversely, reductions in gj may enhance the occurrence of triggered activity by limiting electrotonic inhibition and, combined with reduced INa, slow myocardial conduction and increase vulnerability to reentrant arrhythmias.
NEW & NOTEWORTHY
Therapeutic histone deacetylase inhibition with vorinostat or romidepsin significantly reduces ventricular sodium current density and NaV1.5 protein expression. A slight positive shift in the voltage activation curve, presumably the result of lysine acetylation, decreases the inactivation rate at low activating potentials but does not increase the late sodium current amplitude.
histone deacetylase (HDAC) inhibitors represent a novel class of small molecule therapeutics with indications for the treatment of numerous human disorders including cancer, neurodegenerative diseases, type 2 diabetes, and autoimmunity (20, 22, 24, 28, 58). HDAC inhibitors are divided into four major chemical subgroups, the hydroxamic acid derivatives, benzamides, cyclic peptides, and short chain fatty acids (50). The naturally occurring prototypical hydroxamic acid and cyclic peptide HDAC inhibitors trichostatin A (TSA) and romidepsin (depsipeptide, FK228) inhibit all 11 Zn2+-dependent human HDAC isoforms (1–11) with high (nM-μM) affinity and are considered to be pan-HDAC inhibitors (5). In contrast, the benzamide derivatives like entinostat (MS-275) and mocetinostat (MGCD-0103) are high affinity inhibitors of only the class I HDACs (HDAC1, 2, 3, and 8). Class IIb inhibitors target HDAC6 with relative specificity and class IIa (HDACs 4, 5, 7, and 9) are under development (11, 34). Four pan-HDAC inhibitors have received Food and Drug Administration (FDA) approval since 2006, all for the treatment of cutaneous and/or peripheral T-cell lymphoma (CTCL and/or PTCL) or multiple myeloma (MM) (14). Cardiac arrhythmias have been associated with the clinical use of HDAC inhibitors since 2006 and the reported global incidence of HDAC inhibitor-induced adverse cardiac side effects is 28.6% (17, 50, 55, 57). Two of the FDA approved HDAC inhibitors, romidepsin (Istodax) and panobinostat (LBH589, Farydak), carry warnings for severe ECG changes and cardiac arrhythmias including ventricular tachycardias, although grade 3 or higher QT interval prolongations and ventricular fibrillation have occasionally occurred with vorinostat (SAHA, VOR, Zolinza) and belinostat (PXD101, Beleodaq) (14, 15, 36).
We previously reported that TSA and VOR dose dependently decreased connexin43 (Cx43) expression and electrical coupling between ventricular cardiomyocytes (71). The Duchenne muscular dystrophic (mdx) mouse model exhibits a hyperacetylated protein state resulting in acetylated Cx43, a decrease in NaV1.5 cardiac sodium channel protein expression, and an increased incidence of restraint-induced cardiac arrhythmias (8, 9). The Nε-lysine acetylated Cx43 dissociates from ventricular gap junctions, which may account for the stress-induced increased arrhythmogenesis in mdx mice (9). Treatment of mdx mice with VOR reduced the incidence of stress-induced arrhythmias and improved Cx43 localization to the intercalated disc. Conversely, mdx mice exhibited a lower incidence of arrhythmias than wild-type mice when treated with aconitine, a sodium channel opener, and VOR administration increased the incidence of these arrhythmias in mdx mice, suggesting functional differences in the cardiac sodium current between control and mdx mice (8). Since loss of Cx43 myocardial gap junctions is associated with reductions in cardiac sodium current (INa) carried by the NaV1.5 channel protein, we chose to study the effect of pan-HDAC inhibition on cardiac INa (25, 46). In this study we report our findings of TSA, VOR, and FK228 effects on the cardiac sodium current (INa). This is the first reported study to examine the effects of pan-HDAC inhibition on cardiac INa in wild-type cardiomyocytes and these three HDAC inhibitors all decreased INa density in a dose-dependent manner. Additionally, a slight 3- to 8-mV positive shift in the steady-state voltage-dependent activation curve was observed with high doses of TSA and VOR without a concomitant shift in the inactivation curve. No increases in late INa were observed, suggesting that the only contribution of pan-HDAC inhibition to INa-dependent cardiac arrhythmias may be attributed to decreased excitability and slowed conduction, especially when combined with decreased myocardial gap junction coupling.
MATERIALS AND METHODS
Cell culture.
Newborn C57BL/6 mice were anesthetized with isoflurane and the hearts excised in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol (IACUC #263) was approved by the Institutional Animal Care and Use Committee (IACUC) of SUNY Upstate Medical University. The atria and ventricles were dissociated separately and cultured in Medium 199 (M199) supplemented with 10% FBS as previously described (71). Neonatal mouse ventricular myocytes (NMVMs) were cultured at low (≈2 × 10+5 cells/35 mm culture dish) or high (≈10+7 cells/35 mm culture dish) density for patch-clamp and real-time PCR (RT-PCR) or Western blot procedures, respectively. Cryopreserved human-induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CM, iCell cardiomyocytes) were purchased from Cellular Dynamics (CDI, Madison, WI) and plated and grown in culture media and prepared for electrophysiological experiments per manufacturer's instructions (37).
HDAC inhibitors.
Trichostatin A (TSA) was purchased from Calbiochem or ENZO Life Sciences and 1 mg was dissolved in DMSO and stored at −20°C. Vorinostat (VOR) and romidepsin (FK228) were purchased from Selleck Chemicals, dissolved in DMSO, and stored at −20°C. The 100-mM DMSO stock solutions of TSA, VOR, and FK228 were diluted to the desired experimental test concentrations in M199 or maintenance media and applied to NMVM or hiPSC-CM cultures overnight for 18–24 h before experimental procedures. Final DMSO levels were <0.005% (vol/vol).
Whole cell patch clamping.
Single whole cell patch electrode voltage-clamp experiments were performed on NMVMs and hiPSC-CMs using conventional procedures with an Alembic Instruments VE-2, Axopatch 1D, or Axopatch 200B patch-clamp amplifier, Digidata 1320A or 1440 A/D converter, and pClamp8.2 or 10.1 software (Molecular Devices). Voltage-gated sodium currents (INa) were elicited from a holding potential (Vh) of −120 mV during voltage steps from −90 to +50 mV in 5-mV increments for 150 ms using reduced NaCl solutions (Table 1). For the INa inactivation protocol, Vh was −120 mV and the prepulse voltage increased from −130 to −30 mV in +5-mV increments for 150 ms followed by a 30-ms activation step to −40 mV. In some experiments, 30 μM tetrodotoxin (TTX) was added to the bath to verify that the inward current was TTX sensitive. Gap junction conductance (gj) measurements were obtained using conventional dual whole cell patch-clamp procedures as previously described (64, 71).
Table 1.
Electrophysiological solutions
| Composition, mM | Normal Saline | Pipette Solution | Low Na+ Saline | INa Pipette Solution | hiPSC-CM Saline | CM Pipette Solution |
|---|---|---|---|---|---|---|
| NaCl | 140 | — | 20 | 5 | 30 | 5 |
| KCl | 1.3 | 140 | — | — | — | — |
| TMACl | — | — | 100 | 115 | — | — |
| TEACl | — | — | 20 | 20 | 110 | 135 |
| CsCl | — | — | — | — | 5 | 5 |
| NaH2PO4 | 1.0 | — | — | — | — | — |
| CaCl2 | 1.8 | 3.0 | 1.8 | 3.0 | 1.8 | 3.0 |
| MgCl2/(MgSO4) | (0.8) | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| Glucose | 5.5 | — | 5.5 | — | 5.5 | — |
| BaCl2 and CdCl2 | — | — | 0.1 and 0.1 | — | — | — |
| BAPTA/(EGTA) | — | 5.0 | — | 5.0 | — | (5) |
| HEPES* | 10 | 25 | 10 | 20 | 20 | 20 |
INa sodium current; hiPSC-CM, human-induced pluripotent stem cell-derived cardiomyocyte; TMACl, tetramethylammoniumCl; TEACl, tetramethylammoniumCl.
pH was titrated to 7.4 with 1 N NaOH, KOH, TEAOH, or CsOH.
Real-time PCR.
Cellular RNA was extracted from cultured NMVMs and 10-μl RT-PCR reactions were carried out on 384-well plates using LightCycler 480 Real-Time PCR System (Roche) using previously published procedures (71). All results were normalized to Gapdh and control samples. Cycle threshold (CT) values were determined by the apparatus and the quality of the PCR product was confirmed by analyzing the melt curve. Since HDAC inhibition is known to alter housekeeping gene expression (45), the control and VOR Gapdh CT and relative (to control) expression level values were analyzed statistically and found to not be significantly different (paired t-test, P > 0.25). Primer sets for murine Cdh2, Gja1, Scn5a, and Slc8a, and Cdh2 were designed to span exon-intron regions of the gene of interest. The murine forward and reverse primer sequences were as follows: Cdh2, 5′-TATGTGATGACGGTCACTGC-3′ and 5′-GAAAGGCCATAAGTGGGATT-3′; Gapdh, 5′- TGCCACTCAGAAGACTGTGG-3′ and 5′-AGGAATGGGAGTTGCTGTTG-3′; Gja1, 5′-GAGAGCCCGAACTCTCCTTT-3′ and 5′-TGGAGTAGGCTTGGACCTTG-3′; Scn5a, 5′-CTTCACCAACAGCTGGAACA-3′ and 5′-GACATCATGAGGGCGAAGAG-3′; and Slc8a, 5′-TGAATCTTGCACGCTCATTA-3′ and 5′-CCAGGCACATCCAAAGTATC-3′.
Western blotting.
NMVMs were cultured for 3 days in 200 μM bromodeoxyuridine (BrdU)/M199 media supplemented with 10% fetal FBS and 10/10 U/μg penicillin/streptomycin. TSA, VOR, or 3 ml of control BrDU/M199 media was added overnight. On the fourth day, the NMVM cultures were harvested, lysed, and 15-μg protein samples underwent immunoblot procedures as previously described using EC Western Blot Detection Reagents (Bio-Rad Laboratories, Hercules, CA; Ref. 71). The rabbit polyclonal anti-NaV1.5 (cat. no. ASC-005) primary antibody was purchased from Alomone Labs (Jerusalem, Israel), the mouse monoclonal anti-α-tubulin antibody (cat. no. T8203, clone AA13) was purchased from Sigma-Aldrich (St. Louis, MO), and the rabbit polyclonal anti-acetylated-α-tubulin and HDAC1 (cat. no. BML-SA452 and BML-SA401) antibodies were purchased from Enzo Life Sciences (Farmingdale, NY).
Immunoprecipitation.
Six adult C57BL/6 mice were intraperitoneally injected with either 10 mg/kg TSA (1 mg/ml in sterile saline: 5% DMSO, n = 3) or saline:DMSO (10 μl/gm body weight, n = 3) daily for 5 consecutive days and killed 4–6 h after the last injection as approved by the SUNY Upstate Medical University IACUC. The hearts were excised under ketamine/xylazine anesthesia and atria and ventricles were separated, and lysed in 1% Triton X-100 extraction buffer (50 mM Tris pH 8.0, 150 mM NaCl, 0.02% sodium azide, 1.0 mM PMSF, 1 μg Aprotinin, 1% Triton X-100, 1 mM Na3VO4, and 50 mM NaF) with protease inhibitors (Roche). The ventricular heart lysates were subjected to immunoprecipitation (IP) and Western blot procedures using primary anti-NaV1.5 and anti-acetyl-lysine (Ac-K, Abcam, cat. no. ab21623) rabbit polyclonal antibodies and the Dynabeads Protein G (Thermo Fisher Scientific) immunoprecipitation procedures. The primary antibody was bound to the protein G magnetic beads according to manufacturer's instructions and the target antigen (NaV1.5) was immunoprecipitated in an IP buffer containing 1% Triton X-100, 0.5% NP-40, 20 mM HEPES, 50 mM NaCl, and protease inhibitors (Roche), pH 7.4, using a magnet. The immunoprecipitate was washed with washing buffer, eluted with elution buffer, and subjected to SDS-PAGE electrophoresis and Western blot procedures with anti-Ac-K and anti-NaV1.5 antibodies as previously described. The NaV1.5 immunoprecipitation experiments were performed in triplicate.
HDAC activity assay.
Aliquots of 6 × 10+5 NMVMs per well (96-well plate) were grown in 200 μl of 200 μM BrdU/M199, exchanged daily. Cell wells were incubated with 2,000 pmoles of the acetylated Fluor-de-Lys substrate for 6 h during continuous (overnight) HDAC inhibition (71). Cell, media, and standard curve deacetylated substrate sample wells were developed according to manufacturer's directions (BML-AK503 HDAC fluorometric cellular activity assay kit; Enzo Life Sciences) and background subtracted relative fluorescence unit (RFU) counts were acquired with a BIO-TEK Synergy H1 plate reader (360-nm excitation, 460-nm emission).
Statistics.
Averaged values are presented as the mean ± SE. Statistical analyses were performed with the normality and one-way ANOVA tests using the Bonferroni method in Origin 8.6. Statistical comparisons of the late (100 ms) INa datasets were performed using the F-test comparison of two datasets function in Origin7.5.
RESULTS
Effects of pan-HDAC inhibition on cardiac Na+ current density.
To examine the effect of pan-HDAC inhibition on functional cardiac INa, cultured NMVMs were incubated overnight with 50 or 100 nM TSA, 1 or 5 μM VOR, or 10 nM FK228 and whole cell patch-clamp experiments were performed the next day (18–24 h later). To activate INa, NMVM membrane potential (Vm) was depolarized between −90 and +60 mV in 150 ms and 5-mV increments from a Vh of −120 mV. Examples of INa current traces elicited from control, 100 nM TSA-, 1 μM VOR-, and 10 nM FK228-treated NMVMs are illustrated in Fig. 1, A–D. Peak INa density decreased from control values of −36.2 ± 2.8 to −13.7 ± 2.5 pA/pF with 100 nM TSA, −17.6 ± 1.7 pA/pF with 5 μM VOR, and −15.1 ± 2.0 pA/pF with 10 nM FK228 (Figs. 1, F–H). Cellular input capacitance values were not significantly changed from control values (22.5 ± 1.4 pF, n = 20) except at the highest concentrations of TSA (100 nM; 15.3 ± 0.7 pF, n = 17) and VOR (5 μM; 16.2 ± 0.7 pF, n = 24) tested. The results are consistent with a pan-HDAC inhibitor-induced reduction in peak cardiac INa irrespective of whether the HDAC inhibitor belongs to the hydroxamate or cyclic peptide chemical group of HDAC inhibitors.
Fig. 1.

Reduction of cardiac sodium current INa by pan-histone deacetylase (HDAC) inhibition. Families of cardiac Na+ current traces are shown for 10-mV depolarizing voltage-clamp steps from −90 to +50 mV under control (A), 100 nM trichostatin A (TSA; B), 5 μM vorinostat (VOR; C), and 10 nM romidepsin (FK228; D). E: diagram of the Vm pulse protocol for activating INa. F–H: INa current density (pA/pF)-Vm relationships under control conditions and after an 18- to 24-h treatment with 50 or 100 nM TSA (F), 1 or 5 μM VOR (G), and 10 nM FK228 (H) illustrate dose-dependent decreases in peak INa with pan-HDAC inhibitor treatments.
Persistent activation of INa, caused by mutations in the NaV1.5 sodium channel protein that inhibit inactivation (e.g., ΔKPQ) or the α-sodium channel toxins like aconitine, is known to cause cardiac arrhythmias via induction of early afterdepolarizations (EADs) (8, 65). Thus we measured the steady state current values at 100 ms for all control, 100 nM TSA, 1 μM VOR, and 10 nM FK228 experiments. Linear regression analyses of the whole cell current measured at the 100 ms time point of the voltage-clamp steps from −120 mV to −65 through 0 mV for all four experimental groups revealed no significant differences in the slopes (+0.37, +0.24, +0.34, and +0.44 pA/mV for control, TSA, VOR, and FK228, respectively) of all four datasets. The y-intercepts were significantly (P < 0.05) more negative (19 pA inward) for TSA and significantly more positive (6 pA outward) for FK228 relative to Control and VOR values. Taken together, there is no evidence of an increase in the late INa current in ventricular cardiomyocytes with either of the clinically approved HDAC inhibitors tested.
INa activation and inactivation.
A slight positive shift in the activation voltage is apparent in the INa current-voltage relationships in Fig. 1, F–H, so the peak sodium conductances (gNa) were calculated by dividing the normalized peak INa density values by (Vm − ENa) for each I–V curve in Fig. 1 and normalized by dividing by the average maximum gNa (ḡNa) for each experimental group. The calculated Nernst potential for Na+ (ENa) was +35 mV and the experimental reversal potential (Erev) values were within 5 mV or ± 1 pA/pF of ENa for all experimental groups. The Vm-dependent activation curves for TSA, VOR, and FK228 (Fig. 2, A–C) illustrate a +3- to +8-mV shift in the half activation voltage (V½act) for all three pan-HDAC inhibitor experimental groups. Since INa activation was slightly affected by the HDAC inhibitor treatments, INa inactivation was examined using a prepulse protocol illustrated in Fig. 2E. The highest inhibitory concentrations of TSA (100 nM) and VOR (5 μM) tested shifted the control INa half-inactivation voltage (V½inact) of −83 mV by less than ±1 mV (Fig. 2D). The effect of FK228 on INa inactivation was not examined. The small positive shift in the INa activation curves without an accompanying change in the inactivation curve decreases the Vm-dependent “window” for persistent INa activation (Fig. 2F).
Fig. 2.

Shift of INa activation by pan-HDAC inhibition. A–C: the apparent shift in Vm-dependent activation present in the I–V curves in Fig. 1, F–H, was examined by calculating the Na+ conductance (gNa) and normalizing the data to the maximum gNa (ḡNa) for each experimental group and plotting the activation curves for TSA (A), VOR (B), and FK228 (C) relative to the control activation curve. The data points were fitted with a Boltzmann function with a half-activation voltage (V½act) of −49.8 mV for control, −46.6 and −47.0 mV for 50 and 100 nM TSA, −46.0, and −42.3 mV for 1 and 5 μM VOR, and −46.2 mV for 10 nM FK228, respectively. D: Vm-dependent inactivation was assessed using a prepulse protocol illustrated in E and no shift in the half-inactivation voltage (V½inact) of −83.0 ± 0.5 mV was observed between control, 100 nM TSA, and 5 μM VOR conditions. F: comparison of the overlap between the gNa inactivation and activation curves under control and 5 μM VOR conditions illustrating the decrease in the Vm-dependent “window” for persistent activation of INa.
The time dependence of INa inactivation was also examined by curve-fitting the decaying phase of the INa traces with a first-order exponential function. The Vm-dependent inactivation time constants (τh) were significantly slower in the −60- to −45-mV range for 100 nM TSA and 1 μM VOR but not for 10 nM FK228 (Fig. 3, A–C). There was no difference in the inactivation rates between control and HDAC inhibitor-treated ventricular cardiomyocytes within the −40- to −10-mV range, suggesting that the slow τh values at low activating voltages seen with pan-HDAC inhibition result from the positive shift in the activation curves (Fig. 2, A–C) and that INa inactivation rates were otherwise unaffected.
Fig. 3.
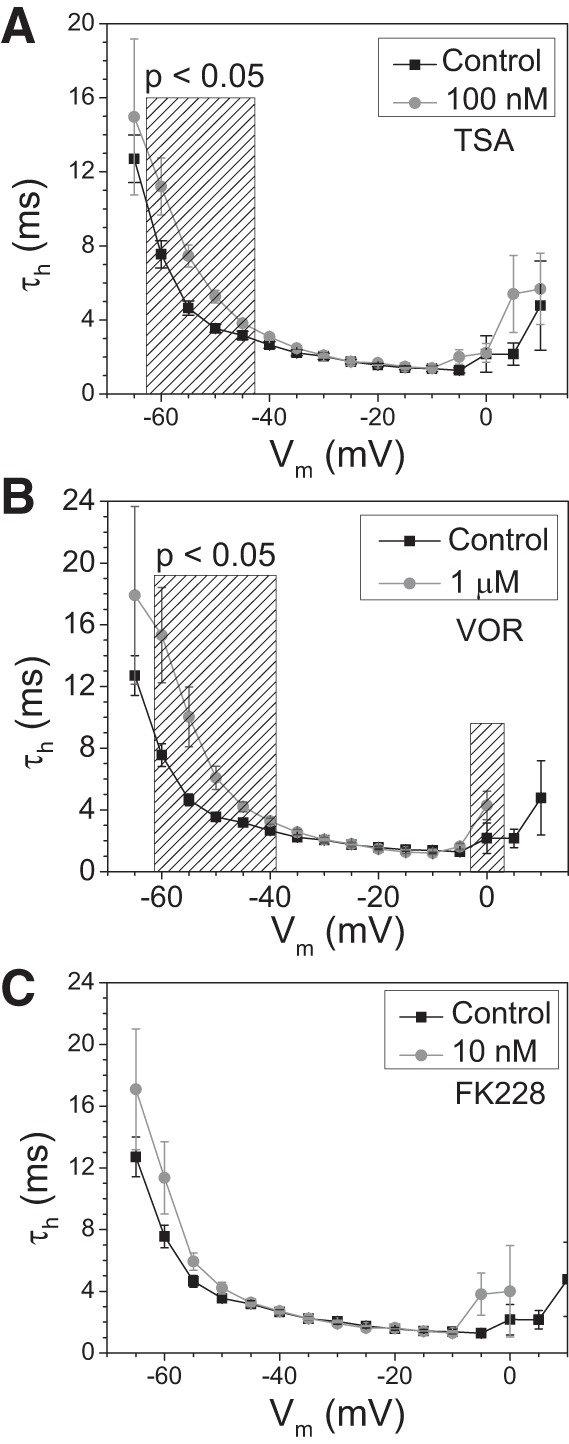
INa inactivation time constants. A–C: the average Vm-dependent inactivation time constants (τh) from the control, 100 nM TSA (A), 1 μM VOR (B), and 10 nM FK228 (C) INa current traces illustrate only a slight slowing of the inactivation rates at low activating potentials of −60 to −40 mV with no slowing of inactivation at fully activated potentials > −40 mV.
hiPSC-CMs.
To assess whether the pan-HDAC inhibitor-induced decrease in cardiac INa occurs in human ventricular myocytes, hiPSC-CMs were treated with 1 μM VOR and INa whole cell patch-clamp experiments were performed (Fig. 4). In hiPSC-CMs, the recommended human therapeutic dose of VOR reduced cardiac INa density by 52% relative to control experiments, indicating that NMVMs are a valid experimental model for studying the effects of HDAC inhibitors on cardiac INa. The mean whole cell current, measured between 100 and 150 ms, did not reveal any increase in late inward currents in the VOR hiPSC-CM group relative to the control group, consistent with our findings in NMVMs suggesting that late INa was not induced by clinically relevant HDAC inhibitor treatments.
Fig. 4.

Effect of pan-HDAC inhibition on human INa. A and B: examples of a family of INa traces obtained from a human-induced pluripotent stem cell-derived cardiomyocyte (hiPSC-CM) under control (A) and 1 μM VOR (B) conditions. C: INa current density (pA/pF)-Vm relationships under control conditions and 1 μM VOR conditions illustrate a significant reduction in INa density after pan-HDAC inhibitor treatment, verifying previous results obtained from neonatal mouse ventricular myocyte (NMVM) cultures.
NaV1.5 expression.
Experiments in NMVMs and hiPSC-CMs consistently indicate that pan-HDAC inhibition reduced INa density, but the molecular basis for the decrease in cardiac INa is unknown. The shift in the INa activation curve is not sufficient to decrease peak INa levels and only slowed the inactivation kinetics at low activating potentials (less than −40 mV). To test the hypothesis that pan-HDAC inhibition may be decreasing the NaV1.5 protein expression level, real-time PCR and Western blot experiments were performed on NMVM cultures under control and pan-HDAC inhibitor-treated conditions. The relative Scn5a mRNA abundance was less than that observed for Gja1, Cdh2, or Slc8 and all were <50% of Gapdh levels (Fig. 5A). As previously reported, the Gja1 and Cdh2 mRNA levels were significantly reduced by 1 μM VOR (71), but Scn5a and Slc8a mRNA levels were not significantly different from control values (Fig. 5B). 100 nM TSA did reduce Scn5a mRNA levels by 50% (n = 2) but did not reach statistical significance (P > 0.05). However, Western blot analyses, performed in triplicate, revealed that NaV1.5 protein levels were significantly reduced at all concentrations of TSA and VOR tested. The decrease in INa density correlates closely (within 15–25% deviation from unity) with the reduction in NaV1.5 protein levels for both TSA and VOR, suggesting that the molecular basis for the pan-HDAC inhibitor-induced decrease in cardiac INa is the dose-dependent reduction in NaV1.5 protein expression. Real-time PCR results with 10 nM FK228 exhibited qualitatively similar abundances for the Chd2, Gja1, Scn5a, and Slc8a genes and a significant reduction in their mRNA levels relative to control values for all except the Scn5a gene transcript levels (Fig. 5E).
Fig. 5.

Effects of pan-HDAC inhibition on NaV1.5 expression. A: the abundance of mRNA levels were measured by RT-PCR from high density NMVM cultures under control conditions for the Cx43 (Gja1), NaV1.5 (Scn5a), N-cadherin (Cdh2), and the sodium-calcium exchanger (NCX, Slc8a) genes relative to Gapdh. B: the relative mRNA levels for these same cardiac genes after 1 μM VOR treatment were compared with control values and only Gja1 and Cdh2 levels were significantly reduced as previously reported. The Gapdh levels were not significantly different from control values (P > 0.25, paired t-test). C: representative Western blots for NaV1.5 protein from TSA- and VOR-treated NMVM samples. HDAC1 was used as loading control and acetylated α-tubulin (Ac-α-t) as an indicator of increased protein acetylation. The experiments were performed in triplicate. D: densitometric scans of the Western blot data were performed to quantify the NaV1.5 protein levels at low and high doses of TSA and VOR. NaV1.5 protein levels were significantly reduced at both doses of TSA and VOR. E: the relative mRNA levels for the Slc8a, Gja1, and Cdh2 genes were significantly reduced by 10 nM FK228 treatment compared with control values but the Scn5a levels were not significantly affected.
Inhibition of cardiac HDAC activity by romidepsin.
To correlate the decreases of cardiac INa and Cx43 mRNA expression levels with the HDAC inhibitory activity of FK228, the total NMVM HDAC activity was measured using a fluorimetric HDAC activity assay in the presence of increasing [FK228] to detect the amount of deacetylated Fluor-de-Lys substrate using previously published procedures (71). As reported for TSA and VOR, inhibition of total cardiac HDAC activity by FK228 exhibited two affinities with the higher affinity site possessing at least twice the capacity of the lower affinity site (Fig. 6A). The IC50 values for FK228 were 5.4 ± 0.6 nM and 1.75 ± 0.47 μM with capacities of 63.5 and 27.7% of total HDAC activity, respectively. Thus, 10 nM FK228 corresponds to twice the IC50 of the high affinity HDAC inhibitory site and is far below the threshold concentration for the secondary low affinity site previously associated with significant reductions in Cx43 expression and function (71). Since TSA and VOR reduced cardiac gj in a dose-dependent manner, we assessed the dose-dependent effects of FK228 on initial gj values of NMVM cell pairs (Fig. 6B). Consistent with our previous findings, FK228 concentrations >1 nM significantly decreased gj.
Fig. 6.
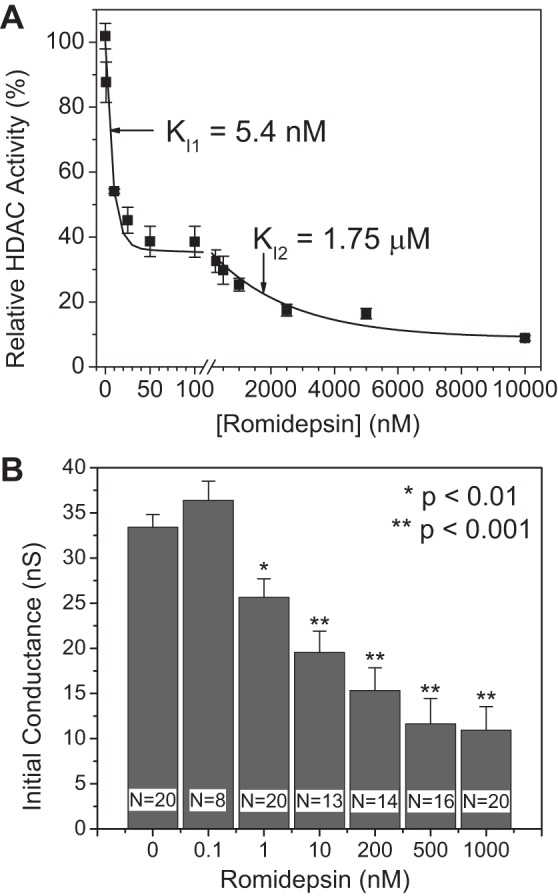
Romidepsin inhibition of cardiac HDAC activity and gap junction conductance. A: total HDAC activity was measured in NMVM cultures exposed to increasing concentrations of FK228 using the deacetylated Fluor-de-Lys fluorescence method. All data were normalized to the background subtracted maximum relative fluorescence of the control well. The data from three experiments were averaged and fitted with a second-order exponential decaying function and equilibrium inhibition constants (Ki) were calculated from the decay constant for each exponential component. B: the gap junction conductance (gj) was measured in NMVM cell pairs from control and FK228-treated culture dishes and a dose-dependent decrease in gj was observed, consistent with published results with TSA and VOR (71).
DISCUSSION
This study consistently found that pan-HDAC inhibition reduced peak INa density and NaV1.5 protein levels (Figs. 1, 4, and 5). The only other functional alteration of cardiac INa observed with pan-HDAC inhibition was a slight (3–8 mV) shift in the Vm-dependent activation curve and a subsequent slowing of the inactivation time constants at low activating potentials (< −40 mV). Scn5a mRNA levels were not decreased by 1 μM VOR yet there was a 40% reduction in NaV1.5 protein levels (Fig. 5, B–D). The 400 mg daily maximum recommended human therapeutic dose of VOR correlates to a maximum plasma concentration (Cmax) value equal to 1.2 μM (320 ng/ml) (29). Reduced NaV1.5 protein levels and INa density without a concomitant decrease in Scn5a mRNA levels have also been reported in mdx mice (16). TTX-sensitive sodium channel isoforms NaV1.3, NaV1.4, NaV1.2, NaV1.1, and NaV1.6 (in order of abundance) have been detected in neonatal and adult mouse and rat ventricular cardiomyocytes (21, 27, 38, 39). These neuronal and skeletal muscle NaV isoforms, along with the β1- and β3-auxiliary subunits, are primarily located in the transverse tubules whereas the predominantly more abundant cardiac NaV1.5 along with the β1-, β2-, and β4-subunits preferentially localize to the intercalated disc. The noncardiac NaV isoforms may account for up to 30% of cardiac INa in neonatal rat ventricular myocytes (NRVMs) but account for <10% of total cardiac INa in isolated adult mouse ventricular myocytes (21, 27). We did not assay for noncardiac NaV or β-subunit isoforms in the present study and will consider these transcripts and proteins in future experiments, but the close correlation between the decrease in NaV1.5 protein levels and peak INa density in our experiments (Figs. 1 and 5) is consistent with the interpretation that the decrease in peak cardiac INa observed with pan-HDAC inhibition is due to dose-dependent reductions in NaV1.5 protein levels.
The precise molecular basis for the decrease in NaV1.5 protein expression remains to be determined by future investigations, though downregulation of Scn5a expression cannot be ruled out at higher inhibitory concentrations of TSA and VOR. Possible mechanisms for the downregulation of cardiac INa include variations in the Scn5a transcripts that do not equally translate into functional NaV1.5 protein, microRNA regulation of Scn5a transcripts, increased NaV1.5 degradation by Nedd4-2-mediated ubiquitination, and reduced expression of the MOG1 protein that directly interacts with and increases the functional surface expression of NaV1.5 channels (10, 52, 63, 69). The surface expression and localization of NaV1.5 is also modulated by direct interactions with α-actinin-2, ankyrin-G, glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L), SAP-97, and tyrosine-phosphorylated β1 (pYb1) and indirect protein-protein interactions with Cx43, N-cadherin (Ncad), the syntrophin-dystrophin complex including calcium/calmodulin-dependent serine kinase (CASK), and plakophilin-2 (PKP2), all of which can result in functional reductions in NaV1.5 expression (12, 25, 35, 41, 46, 48, 51, 52, 54, 68, 74). Thus our findings of reduced Ncad and Cx43 protein levels observed with TSA, VOR, and FK228 are consistent with decreases in INa (71).
Besides the many possible direct and indirect protein interactions with NaV1.5, numerous posttranslational modifications alter NaV1.5 function including glycosylation, phosphorylation by numerous kinases (e.g., PKA, PKC, CaMKII, and Fyn), ubiquitination, S-nitrosylation, and methylation (1, 3, 7, 52, 63). Nε-lysine acetylation directly or indirectly impacts protein methylation, neddylation, ubiquitylation, and phosphorylation either by mutually exclusive posttranslational modification (PTM) of lysine residues or lysine residues within concensus protein kinase phosphorylation sites (72). NH2-terminal acetylation of NaV1.5 was recently discovered in end stage human heart failure and is associated with specific protein degradatory pathways that likely results in the downregulation of cardiac INa (2, 4, 23, 42). NH2-terminal (Nt- or Nα) acetylation is irreversibly catalyzed by ribosome-associated NH2-terminal acetyltransferases (NATs) and is distinct from the Nε-lysine acetylation reaction catalyzed by the histone acetyltransferases (HATs) and reversed by the HDACs and sirtuins (SIRTs, class III NAD+-dependent HDACs) (2, 73). Classical HDAC inhibition will not affect NAT or SIRT activities, although HDAC6 and SIRT2 colocalize to form the tubulin deactylase complex (47). Whether or not NaV1.5 is a substrate for Nε-lysine acetylation is currently unknown, so we subjected mouse and human NaV1.5 sequences to Nε-lysine acetylation site analysis using the PHOSIDA website (18). This bioinformatic analysis predicted two high probability (>90%) cytoplasmic acetylation sites near the domain II and IV S4 segments known to be involved in Vm-dependent activation (Fig. 7A). To determine if NaV1.5 is acetylated, we injected six adult C57BL/6 mice with 10 mg/kg TSA or DMSO/saline control intraperitoneal injections for 5 days and immunoprecipitated the ventricular heart lysates with anti-NaV1.5 antibodies. Subsequent immunoblotting for acetylated protein with an anti-acetyl-lysine (Ac-K) antibody revealed an increase in acetylated NaV1.5 protein with TSA treatments (Fig. 7, B and C). The human and murine NaV1.5 proteins possess 94% sequence identity; hence we hypothesize that acetylation of K830 and K1644 (K1641 in the human sequence) may account for the small depolarizing shift in the INa activation curve and the slowing of the inactivation rate at low activation potentials seen with pan-HDAC inhibition (Figs. 2 and 3). This postulated acetylation-dependent shift of NaV1.5 activation decreased the Vm-dependent window for prolonged activation of INa (window current) and is unlikely to account for the observed decrease in results in INa density, which correlates closely with the decreased levels of NaV1.5 protein. Identification of the actual NaV1.5 acetylation sites will require future liquid chromatography-electron spray ionization tandem mass spectroscopic analyses of IP-enriched and/or SDS-PAGE proteolytic NaV1.5 peptide digests (32).
Fig. 7.

A: putative Nε-lysine acetylation sites on the NaV1.5 Na+ channel subunit. The full length mouse NaV1.5 amino acid sequence was analyzed for possible lysine acetylation (Ac-K) sites using the prediction algorithm available at the PHOSIDA Posttranslational Modification database (http://www.phosida.org; Ref. 18). Eight putative acetylation sites (probability >90%) were identified and mapped onto the membrane topological model for the NaV1.5 channel protein. Six of the eight predicted NaV1.5 Ac-K sites (K158, K175, K767, K863, K1362, and K1617) mapped to transmembrane or extracellular domain locations and were discounted as possible acetylation sites (lined circle). Two possible Ac-K sites, K830 and K1644, mapped to cytoplasmic domains located near the cytoplasmic COOH-terminal side of the repeat domain II and IV S4 domains (star circle). We hypothesize that acetylation of these 2 sites, which are conserved in the human NaV1.5 sequence, may account for the slight positive voltage shift (< 10 mV) in the cardiac INa activation curve associated with pan-HDAC inhibitor treatments (Fig. 2, A-C). B, left: an example of an immunoprecipitation (IP) performed on saline:DMSO (control, C) or TSA-injected (T) mouse heart ventricular lysates with anti-NaV1.5-protein G magnetic beads and immunoblotted (IB) with an anti-acetyl-lysine (Ac-K) antibody to detect protein acetylation. TSA injection for 5 days caused a dramatic increase in the acetylated NaV1.5 band. Rabbit IgG controls for control (IgGc) and TSA heart samples (IgGt) are shown in lanes 3 and 4. B, right: lanes 1 and 2 are the NaV1.5 immunoprecipitate input controls for the control and TSA heart lysates and the lanes 3 and 4 are the rabbit IgG controls for each sample. C: Normalized mean Ac-K NaV1.5 band densities for the upper [high molecular weight (MW), upper arrow] and lower (lower MW, lower arrow) from 3 control:TSA immunoprecipitation experiments as illustrated in B. Each Ac-K TSA band density was divided the control band density for each experiment and this TSA/control band ratio was normalized by dividing by the NaV1.5 input TSA/control band ratio.
There are nearly 500 NaV1.5 mutations associated with cardiac arrhythmias including long QT (LQT3) syndrome, Brugada syndrome (BrS), cardiac conduction disease (CCD), sick sinus syndrome (SSS), and atrial fibrillation (AF) (13, 40, 51, 52, 65, 75). NaVβ1–4 subunits also contribute to LQT and AF cardiac arrhythmias (43, 66, 67). Approximately 75% of these mutations cause loss of NaV1.5 function except those involved in LQT and some AF arrhythmias. Gain-of-function NaV1.5 mutations typically increase activation or decrease inactivation, resulting in a net increase of a persistent (sustained) or late INa that prolongs action potential duration and enhances the occurrence of early afterdepolarizations (EADs) (43, 65). For example, expression of the NaV1.5F1486del LQT mutation increased late INa and produced EADs in NRVMs (59). Recently, increased expression of NaV1.1 and NaV1.3 isoforms in Scnb1−/− mice and ischemic heart failure dog hearts produced increases in late INa and arrhythmias, thus indicating a role for neuronal isoforms in cardiac arrhythmogenesis (33, 44). However, we found no evidence for pan-HDAC inhibitor-induced increase in late INa in NMVMs or hiPSC-CMs. By contrast, loss-of-function mutations shorten the cardiac refractory period and slow conduction velocity leading to arrhythmias including BrS, SSS, and CCD (6, 56, 61, 67). Peak INa density and gap junction conductance (gj) are primary determinants of myocardial conduction velocity and reductions in both of these factors will slow conduction and increase the vulnerability to reentrant arrhythmias (30, 62). BrS arrhythmias frequently develop in the right ventricle, which is more susceptible to conduction slowing secondary to NaV1.5 dysfunction (49, 51).
All three pan-HDAC inhibitors we have tested thus far caused significant reductions in gj and INa, consistent with an increased susceptibility to cardiac arrhythmias due to slowed or discontinuous propagation (71). Reduced gap junction coupling also enhances the ability of triggered afterdepolarizations to initiate an arrhythmia including LQT-induced Torsades de Pointes (TdP) arrhythmias by modulating the source-sink interactions of an ectopic focus with the surrounding normal myocardium (53, 70). Enhancement of late INa currents and blockade of repolarizing delayed outward K+ currents account for >90% of all congenital and drug-induced LQT syndromes (26). However, the IC50 for VOR blockade of the rapid delayed outward rectifying K+ current (IKr) formed by the hERG and MiRP1 channel and accessory proteins is 300 μM, orders of magnitude higher than the recommended therapeutic dose of 1 μM (29). Thus direct hERG blockade is unlikely to account for the observed QT interval prolongation and TdP arrhythmias occasionally observed with this HDAC inhibitor (36, 50). Two recent studies have concluded that the cardiotoxic effects of HDAC inhibitors likely results from transcriptional changes in genes affecting ion channel function owing to their delayed onset of cardiotoxicity and lack of acute ion channel blockade (31, 60). Owing to the present and recent findings, identification of the ionic mechanisms responsible for HDAC inhibitor-induced QT interval prolongation and TdP arrhythmias merits further investigation. In conclusion, pan-HDAC-induced reductions in peak cardiac INa and NaV1.5 protein levels and shifted the activation curve positive, possibly due to direct acetylation of two S4 segment adjacent cytoplasmic lysine residues. These effects on INa should decrease cardiac excitability, refractoriness, and conduction velocity in a manner that will increase myocardial vulnerability to reentrant arrhythmias but not LQT-associated TdP arrhythmias that occasionally occur with pan-HDAC inhibitory treatments.
GRANTS
The work was supported by National Heart, Lung, and Blood Institute Grant HL-042220, the Joseph C. Georg Fund of the Central New York Community Foundation/Upstate Medical University Foundation, and the Hendricks Fund of the Health Science Center Foundation at Syracuse (to R. D. Veenstra).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Q.X. and R.D.V. conception and design of research; Q.X., D.P., and X.Z. performed experiments; Q.X., D.P., X.Z., and R.D.V. analyzed data; Q.X., D.P., X.Z., and R.D.V. interpreted results of experiments; Q.X., D.P., X.Z., and R.D.V. prepared figures; Q.X., D.P., X.Z., and R.D.V. edited and revised manuscript; Q.X., D.P., X.Z., and R.D.V. approved final version of manuscript; R.D.V. drafted manuscript.
ACKNOWLEDGMENTS
We thank Li Gao for technical assistance during the performance of this work.
Present address of Q. Xu: Dept. of Psychiatry, Faculty of Medicine, The University of British Columbia, 2255 Wesbrook Mall, Vancouver, BC V6T 1Z3, Canada.
REFERENCES
- 1.Ahern GP, Hsu SF, Klyachko VA, Jackson MB. Induction of persistent sodium current by exogenous and endogenous nitric oxide. J Biol Chem 275: 28810–28815, 2000. [DOI] [PubMed] [Google Scholar]
- 2.Arnesen T. Towards a functional understanding of protein N-terminal acetylation. PLoS Biol 9: e1001074, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beltran-Alvarez P, Pagans S, Brugada R. The cardiac sodium channel is post-translationally modified by arginine methylation. J Proteome Res 10: 3712–3719, 2011. [DOI] [PubMed] [Google Scholar]
- 4.Beltran-Alvarez P, Tarradas A, Chiva C, Pérez-Serra A, Batlle M, Pérez-Villa F, Schulte U, Sabidó E, Brugada R, Pagans S. Identification of N-terminal protein acetylation and arginine methylation of the voltage-gated sodium channel in end-stage heart failure human heart. J Mol Cell Cardiol 76: 126–129, 2014. [DOI] [PubMed] [Google Scholar]
- 5.Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, Mazitschek R. Chemical phylogenetics of histone deacetylases. Nat Chem Biol 6: 238–243, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, Potenza D, Moya A, Borggrefe M, Breithardt G, Ortiz-Lopez R, Wang Z, Antzelevitch C, O'Brien RE, Schulze-Bahr E, Keating MT, Towbin JA, Wang Q. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 392: 293–296, 1998. [DOI] [PubMed] [Google Scholar]
- 7.Cheng J, Valdivia CR, Vaidyanathan R, Balijepalli RC, Ackerman MJ, Makielski JC. Caveolin-3 suppresses late sodium current by inhibiting nNOS-dependent S-nitrosylation of SCN5A. J Mol Cell Cardiol 61: 102–110, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colussi C, Berni R, Rosati J, Straino S, Vitale S, Spallotta F, Baruffi S, Bocchi L, Delucchi F, Rossi S, Savi M, Rotili D, Quaini F, Macchi E, Stilli D, Musso E, Mai A, Gaetano C, Capogrossi MC. The histone deacetylase inhibitor suberoylanilide hydroxamic acid reduces cardiac arrhythmias in dystrophic mice. Cardiovasc Res 87: 73–82, 2010. [DOI] [PubMed] [Google Scholar]
- 9.Colussi C, Rosati J, Straino S, Spallotta F, Berni R, Stilli D, Rossi S, Musso E, Macchi E, Mai A, Sbardella G, Castellano S, Chimenti C, Frustaci A, Nebbioso A, Altucci L, Capogrossi MC, Gaetano C. Nε-lysine acetylation determines dissociation from gap junctions and lateralization of connexin 43 in normal and dystrophic heart. Proc Natl Acad Sci USA 108: 2795–2800, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daimi H, Lozano-Velasco E, Haj Khelil A, Chibani JB, Barana A, Amorós I, González de la Fuente M, Caballero R, Aranega A, Franco D. Regulation of SCN5A by microRNAs: miR-219 modulates SCN5A transcript expression and the effects of flecainide intoxication in mice. Heart Rhythm 12: 1333–1342, 2015. [DOI] [PubMed] [Google Scholar]
- 11.Dasmahapatra G, Patel H, Friedberg J, Quayle SN, Jones SS, Grant S. In vitro and in vivo interactions between the HDAC6 inhibitor ricolinostat (ACY1215) and the irreversible proteasome inhibitor carfilzomib in non-Hodgkin lymphoma cells. Mol Cancer 13: 2886–2897, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Eichel CA, Beuriot A, Chevalier M, Rougier JS, Louault F, Dilanian G, Amour J, Coulombe A, Abriel H, Hatem SN, Balse E. Lateral membrane-specific MAGUK CASK down-regulates NaV1.5 channel in cardiac myocytes. Circ Res 119: 544–556, 2016. [DOI] [PubMed] [Google Scholar]
- 13.Ellinor PT, Nam EG, Shea MA, Milan DJ, Ruskin JN, MacRae CA. Cardiac sodium channel mutation in atrial fibrillation. Heart Rhythm 5: 99–105, 2008. [DOI] [PubMed] [Google Scholar]
- 14.Food and Drug Administration. FDA Reference ID: 3043460 (2011), 3536712 (2014), 3643827 (2014), and 3699607 (2015).
- 15.Foss F, Advani R, Duvic M, Hymes KB, Intragumtornchai T, Lekhakula A, Shpilberg O, Lerner A, Belt RJ, Jacobsen ED, Laurent G, Ben-Yehuda D, Beylot-Barry M, Hillen U, Knoblauch P, Bhat G, Chawla S, Allen LF, Pohlman B. A Phase II trial of Belinostat (PXD101) in patients with relapsed or refractory peripheral or cutaneous T-cell lymphoma. Br J Haematol 168: 811–819, 2015. [DOI] [PubMed] [Google Scholar]
- 16.Gavillet B, Rougier JS, Domenighetti AA, Behar R, Boixel C, Ruchat P, Lehr HA, Pedrazzini T, Abriel H. Cardiac sodium channel Nav1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ Res 99: 407–414, 2006. [DOI] [PubMed] [Google Scholar]
- 17.Giles F, Fischer T, Cortes J, Garcia-Manero G, Beck J, Ravandi F, Masson E, Rae P, Laird G, Sharma S, Kantarjian H, Dugan M, Albitar M, Bhalla K. A phase I study of intravenous LBH589, a novel cinnamic hydroxamic acid analogue histone deacetylase inhibitor, in patients with refractory hematologic malignancies. Clin Cancer Res 12: 4628–4635, 2006. [DOI] [PubMed] [Google Scholar]
- 18.Gnad F, Gunawardena J, Mann M. PHOSIDA 2011: the posttranslational modification database. Nucleic Acid Res 39, Suppl 1: D253–D260, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gryder BE, Sodji QH, Oyelere AK. Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed. Future Med Chem 4: 505–524, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implication for disease and therapy. Nat Rev Genetics 10: 32–42, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haufe V, Camacho JA, Dumaine R, Günther B, Bollensdorff C, von Banchet GS, Benndorf K, Zimmer T. Expression pattern of neuronal and skeletal muscle voltage-gated Na+ channels in the developing mouse heart. J Physiol 564: 683–696, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heerboth S, Lapinska K, Snyder N, Leary M, Rollinson S, Sarkar S. Use of epigenetic drugs in disease: an overview. Genet Epigenet 6: 9–19, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hwang CS, Shemorry A, Varshavsky A. N-terminal acetylation of cellular proteins creates specific degradation signals. Science 327: 973–977, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iyer A, Fairlie DP, Brown L. Lysine acetylation in obesity, diabetes and metabolic disease. Immunol Cell Biol 90: 39–46, 2012. [DOI] [PubMed] [Google Scholar]
- 25.Jansen JA, Noorman M, Musa H, Stein M, de Jong S, van der Nagel R, Hund TJ, Mohler PJ, Vos MA, van Veen TA, de Bakker JM, Delmar M, van Rijen HV. Reduced heterogeneous expression of Cx43 results in decreased Nav1.5 expression and reduced sodium current that accounts for arrhythmia vulnerability in conditional Cx43 knockout mice. Heart Rhythm 9: 600–607, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kannankeril P, Roden DM, Darbar D. Drug-induced long QT syndrome. Pharmacol Rev 62: 760–781, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaufmann SG, Westenbroek RE, Zechner C, Maass AH, Bischoff S, Muck J, Wischmeyer E, Scheuer T, Maier SK. Functional protein expression of multiple sodium channel alpha- and beta-subunit isoforms in neonatal cardiomyocytes. J Mol Cell Cardiol 48: 261–269, 2010. [DOI] [PubMed] [Google Scholar]
- 28.Kazantsev AG, Thompson LM. Therapeutic application of histone deaceytlase inhibitors for central nervous system disorders. Nat Rev Drug Disc 7: 854–868, 2008. [DOI] [PubMed] [Google Scholar]
- 29.Kerr JS, Galloway S, Lagrutta A, Armstrong M, Miller T, Richon VM, Andrews PA. Nonclinical safety assessment of the histone deacetylase inhibitor vorinostat. Intl J Toxicol 29: 3–19, 2010. [DOI] [PubMed] [Google Scholar]
- 30.Kléber AG, Rudy Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol Rev 84: 431–488, 2003. [DOI] [PubMed] [Google Scholar]
- 31.Kopljar I, Gallacher DJ, De Bondt A, Cougnaud L, Vlaminckx E, Van den Wyngaert I, Lu HR. Functional and transcriptional characterization of histone deacetylase inhibitor-mediated cardiac adverse effects in human induced pluripotent stem cell-derived cardiomyocytes. Stem Cells Transl Med 5: 602–612, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, Silva JC, Skinner ME, Lombard DB. Mass spectrometry-based detection of protein acetylation. Methods Mol Biol 1077: 81–104, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin X, O'Malley H, Chen C, Auerbach D, Foster M, Shekhar A, Zhang M, Coetzee W, Jalife J, Fishman GI, Isom L, Delmar M. Scn1b deletion leads to increased tetrodotoxin-sensitive sodium current, altered intracellular calcium homeostasis and arrhythmias in murine hearts. J Physiol 593: 1389–1407, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lobera M, Madauss KP, Pohlhaus DT, Wright QG, Trocha M, Schmidt DR, Baloglu E, Trump RP, Head MS, Hofmann GA, Murray-Thompson M, Schwartz B, Chakravorty S, Wu Z, Mander PK, Kruidenier L, Reid RA, Burkhart W, Turunen BJ, Rong JX, Wagner C, Moyer MB, Wells C, Hong X, Moore JT, Williams JD, Soler D, Ghosh S, Nolan MA. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat Chem Biol 9: 319–325, 2013. [DOI] [PubMed] [Google Scholar]
- 35.London B, Michalec M, Mehdi H, Zhu X, Kerchner L, Sanyal S, Viswanathan PC, Pfahnl AE, Shang LL, Madhusudanan M, Baty CJ, Lagana S, Aleong R, Gutmann R, Ackerman MJ, McNamara DM, Weiss R, Dudley SC Jr. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation 116: 2260–2268, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lynch DR, Washam JB, Newby LK. QT interval prolongation and torsades de pointes in a patient undergoing treatment with vorinostat: A case report and review of the literature. Cardiol J 19: 434–438, 2012. [PubMed] [Google Scholar]
- 37.Ma J, Guo L, Fiene SJ, Anson BD, Thomson JA, Kamp TJ, Kolaja KL, Swanson BJ, January CT. High purity human-induced pluripotent stem cell-derived cardiomyocytes: electrophysiological properties of action potentials and ionic currents. Am J Physiol Heart Circ Physiol 301: H2006–H2017, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maier SK, Westenbroek RE, McCormick KA, Curtis R, Scheuer T, Catterall WA. Distinct subcellular localization of different sodium channel alpha and beta subunits in single ventricular myocytes from mouse heart. Circulation 109: 1421–1427, 2004. [DOI] [PubMed] [Google Scholar]
- 39.Maier SK, Westenbroek RE, Schenkman KA, Feigl EO, Scheuer T, Catterall WA. An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc Natl Acad Sci USA 99: 4073–4078, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Makiyama T, Akao M, Shizuta S, Doi T, Nishiyama K, Oka Y, Ohno S, Nishio Y, Tsuji K, Itoh H, Kimura T, Kita T, Horie M. A novel SCN5A gain-of-function mutation M1875T associated with familial atrial fibrillation. J Am Coll Cardiol 52: 1326–1334, 2008. [DOI] [PubMed] [Google Scholar]
- 41.Malhotra JD, Thyagarajan V, Chen C, Isom LL. Tyrosine-phosphorylated and nonphosphorylated sodium channel beta1 subunits are differentially localized in cardiac myocytes. J Biol Chem 279: 40748–40754, 2004. [DOI] [PubMed] [Google Scholar]
- 42.Marionneau C, Abriel H. Regulation of the cardiac Na+ channel NaV1.5 by post-translational modifications. J Mol Cell Cardiol 82: 36–47, 2015. [DOI] [PubMed] [Google Scholar]
- 43.Medeiros-Domingo A, Kaku T, Tester DJ, Iturralde-Torres P, Itty A, Ye B, Valdivia C, Ueda K, Canizales-Quinteros S, Tusié-Luna MT, Makielski JC, Ackerman MJ. SCN4B-encoded sodium channel beta4 subunit in congenital long-QT syndrome. Circulation 116: 134–142, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mishra S, Reznikov V, Maltsev VA, Undrovinas NA, Sabbah HN, Undrovinas A. Contribution of sodium channel neuronal isoform Nav1.1 to late sodium current in ventricular myocytes from failing hearts. J Physiol 593: 1409–1427, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mogal A, Abdulkadir SA. Effects of histone deacetylase inhibitor (HDACi); trichostatin-A (TSA) on the expression of housekeeping genes. Mol Cell Probes 20: 81–86, 2006. [DOI] [PubMed] [Google Scholar]
- 46.Noorman M, Hakim S, Kessler E, Groeneweg JA, Cox MG, Asimaki A, van Rijen HV, van Stuijvenberg L, Chkourko H, van der Heyden MA, Vos MA, de Jonge N, van der Smagt JJ, Dooijes D, Vink A, de Weger RA, Varro A, de Bakker JM, Saffitz JE, Hund TJ, Mohler PJ, Delmar M, Hauer RN, van Veen TA. Remodeling of the cardiac sodium channel, connexin43, and plakoglobin at the intercalated disk in patients with arrhythmogenic cardiomyopathy. Heart Rhythm 10: 412–419, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell 11: 437–444, 2003. [DOI] [PubMed] [Google Scholar]
- 48.Petitprez S, Zmoos AF, Ogrodnik J, Balse E, Raad N, El-Haou S, Albesa M, Bittihn P, Luther S, Lehnart SE, Hatem SN, Coulombe A, Abriel H. SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels Nav1.5 in cardiomyocytes. Circ Res 108: 294–304, 2011. [DOI] [PubMed] [Google Scholar]
- 49.Postema PG, van Dessel PF, de Bakker JM, Dekker LR, Linnenbank AC, Hoogendijk MG, Coronel R, Tijssen JG, Wilde AA, Tan HL. Slow and discontinuous conduction conspire in Brugada syndrome: a right ventricular mapping and stimulation study. Circ Arrhythm Electrophysiol 1: 379–386, 2008. [DOI] [PubMed] [Google Scholar]
- 50.Rasheed WK, Johnstone RW, Price HM. Histone deacetylase inhibitors in cancer therapy. Expert Opin Invest Drugs 16: 659–678, 2007. [DOI] [PubMed] [Google Scholar]
- 51.Remme CA, Bezzina CR. Sodium channel (dys)function and cardiac arrhythmias. Cardiovasc Ther 28: 287–294, 2010. [DOI] [PubMed] [Google Scholar]
- 52.Rook MB, Evers MM, Vos MA, Bierhuizen MF. Biology of cardiac sodium channel Nav1.5 expression. Cardiovasc Res 93: 12–23, 2012. [DOI] [PubMed] [Google Scholar]
- 53.Ruan L, Quan X, Li L, Bai R, Ni M, Xu R, Zhang C. Increasing gap junction coupling supresses ibutilide-induced torsades de pointes. Exp Ther Med 7: 1279–1284, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sato PY, Musa H, Coombs W, Guerrero-Serna G, Patiño GA, Taffet SM, Isom LL, Delmar M. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ Res 105: 523–526, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schiattarella GG, Sannino A, Toscano E, Cattaneo F, Trimarco B, Esposito G, Perrino C. Cardiovascular effects of histone deacetylase inhibitors epigenetic therapies: Systematic review of 62 studies and hypotheses for future research. Int J Cardiol 219: 396–403, 2016. [DOI] [PubMed] [Google Scholar]
- 56.Schott JJ, Alshinawi C, Kyndt F, Probst V, Hoorntje TM, Hulsbeek M, Wilde AA, Escande D, Mannens MM, Le Marec H. Cardiac conduction defects associate with mutations in SCN5A. Nat Genet 23: 20–21, 1999. [DOI] [PubMed] [Google Scholar]
- 57.Shah MH, Binkley P, Chan K, Xiao J, Arbogast D, Collamore M, Farra Y, Young D, Grever M. Cardiotoxicity of histone deacetylase inhibitor depsipeptide in patients with metastatic neuroendocrine tumors. Clin Cancer Res 12: 3997–4003, 2006. [DOI] [PubMed] [Google Scholar]
- 58.Shakespear MR, Halili MA, Irvine KM, Fairlie DP, Sweet MJ. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol 32: 335–343, 2011. [DOI] [PubMed] [Google Scholar]
- 59.Song W, Xiao Y, Chen H, Ashpole NM, Piekarz AD, Ma P, Hudmon A, Cummins TR, Shou W. The human Nav1.5 F1486 deletion associated with long QT syndrome leads to impaired sodium channel inactivation and reduced lidocaine sensitivity. J Physiol 590: 5123–5139, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Spence S, Deurinck M, Ju H, Traebert M, McLean L, Marlowe J, Emotte C, Tritto E, Tseng M, Shultz M, Friedrichs GS. Histone deacetylase inhibitors prolong cardiac repolarization through transcriptional mechanisms. Toxicol Sci 153: 39–54, 2016. [DOI] [PubMed] [Google Scholar]
- 61.Tan HL, Bink-Boelkens MT, Bezzina CR, Viswanathan PC, Beaufort-Krol GC, van Tintelen PJ, van den Berg MP, Wilde AA, Balser JR. A sodium-channel mutation causes isolated cardiac conduction disease. Nature 409: 1043–1047, 2001. [DOI] [PubMed] [Google Scholar]
- 62.Tse G, Yeo JM. Conduction abnormalities and ventricular arrhythmogenesis: The roles of sodium channels and gap junctions. Int J Cardiol Heart Vasc 9: 75–82, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van Bemmelen MX, Rougier JS, Gavillet B, Apothéloz F, Daidié D, Tateyama M, Rivolta I, Thomas MA, Kass RS, Staub O, Abriel H. Cardiac voltage-gated sodium channel Nav1.5 is regulated by Nedd4-2 mediated ubiquitination. Circ Res 95: 284–291, 2004. [DOI] [PubMed] [Google Scholar]
- 64.Veenstra RD. Voltage clamp limitations of dual whole cell recordings of gap junction current and voltage recordings. I. Conductance measurements. Biophys J 80: 2231–2247, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 80: 805–811, 1995. [DOI] [PubMed] [Google Scholar]
- 66.Watanabe H, Darbar D, Kaiser DW, Jiramongkolchai K, Chopra S, Donahue BS, Kannankeril PJ, Roden DM. Mutations in sodium channel β1- and β2-subunits associated with atrial fibrillation. Circ Arrhythm Electrophysiol 2: 268–75, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Watanabe H, Koopmann TT, Le Scouarnec S, Yang T, Ingram CR, Schott JJ, Demolombe S, Probst V, Anselme F, Escande D, Wiesfeld AC, Pfeufer A, Kääb S, Wichmann HE, Hasdemir C, Aizawa Y, Wilde AA, Roden DM, Bezzina CR. Sodium channel β1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J Clin Invest 118: 2260–2268, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Willis BC, Ponce-Balbuena D, Jalife J. Protein assemblies of sodium and inward rectifier potassium channels control cardiac excitability and arrhythmogenesis. Am J Physiol Heart Circ Physiol 308: H1463–H1473, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wu L, Yong SL, Fan C, Ni Y, Yoo S, Zhang T, Zhang X, Obejero-Paz CA, Rho HJ, Ke T, Szafranski P, Jones SW, Chen Q, Wang QK. Identification of a new co-factor, MOG1, required for the full function of cardiac sodium channel Nav 1.5. J Biol Chem 283: 6968–6978, 2008. [DOI] [PubMed] [Google Scholar]
- 70.Xie Y, Sato D, Garfinkel A, Qu Z, Weiss JN. So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys J 99: 1408–1415, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu Q, Lin X, Andrews L, Patel D, Lampe PD, Veenstra RD. Histone deacetylase inhibition reduces cardiac connexin43 expression and gap junction communication. Front Pharmacol 4: 44, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang XJ, Grégoire S. Metabolism, cytoskeleton and cellular signalling in the grip of protein Nepsilon - and O-acetylation. EMBO Rep 8: 556–562, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol 9: 206–218, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ziane R, Huang H, Moghadaszadeh B, Beggs AH, Levesque G, Chahine M. Cell membrane expression of cardiac sodium channel Na(v)1.5 is modulated by alpha-actinin-2 interaction. Biochemistry 49: 166–178, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zimmer T, Haufe V, Blechschmidt S. Voltage-gated sodium channels in the mammalian heart. Glob Cardiol Sci Pract 2014: 449–463, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]