

Abstract
Tofacitinib is an oral Janus kinase inhibitor. Tofacitinib metabolism is primarily mediated by cytochrome P450 3A4. This phase 1 randomized, open‐label, 2‐way crossover study (NCT01137708) evaluated the effect of tofacitinib 30 mg twice daily on the single‐dose pharmacokinetics of combination oral contraceptives ethinylestradiol (EE) and levonorgestrel (LN). EE and LN were administered as a single Microgynon 30® tablet (30 μg EE and 150 μg LN) to 19 healthy women. In the presence of tofacitinib, the area under the curve from time zero to infinity (AUC∞) increased by 6.6% and 0.9% for EE and LN, respectively. Maximal plasma concentrations decreased by 10.4% for EE and increased by 12.2% for LN when coadministered with tofacitinib. The 90% confidence intervals for the adjusted geometric mean ratios for AUC∞ fell within the 80%–125% region for both EE and LN. Mean half‐life was similar in the presence and absence of tofacitinib: 13.8 and 13.3 hours, respectively, for EE; 25.9 and 25.4 hours, respectively, for LN. Tofacitinib had no clinically relevant net inhibitory or inductive effect on the pharmacokinetics of EE and LN. Therefore, there is no evidence to suggest dose adjustments of oral contraceptive drugs containing EE or LN when coadministered with tofacitinib.
Keywords: drug–drug interactions, ethinylestradiol, levonorgestrel, oral contraceptives, tofacitinib
Tofacitinib is an oral Janus kinase (JAK) inhibitor. In cellular settings under which JAKs signal in pairs, tofacitinib preferentially inhibits signaling by heterodimeric receptors associated with JAK1 and/or JAK3, with functional selectivity over receptors that signal via pairs of JAK2.1 JAK1/JAK3 inhibition leads to blockade of signaling through the common gamma chain‐containing receptors for several cytokines, including interleukin (IL)‐2, IL‐4, IL‐7, IL‐9, IL‐15, and IL‐21.1 These cytokines are integral to lymphocyte activation, proliferation, and function. Therefore, inhibition of JAK1/JAK3 by tofacitinib may result in therapeutic modulation of immune and inflammatory responses. The efficacy and safety of tofacitinib has been reported in 6 phase 3 and 2 long‐term extension studies in patients with active moderate to severe rheumatoid arthritis (RA).2, 3, 4, 5, 6, 7, 8 Tofacitinib is in clinical development for the treatment of several inflammatory diseases including psoriasis,9 psoriatic arthritis, and inflammatory bowel disease.10
The plasma pharmacokinetics (PK) of tofacitinib are characterized by rapid absorption and elimination.11 Steady‐state concentrations during twice‐daily dosing are achieved within 24–48 hours. Clearance of tofacitinib is mainly nonrenal (70%), with the remaining 30% renally excreted. Tofacitinib is primarily metabolized by cytochrome P450 (CYP) 3A4 (approximately 55%), with a limited contribution from CYP2C19. In vitro experiments evaluating tofacitinib as an inhibitor of 7 major CYP isoenzymes (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4) using human liver microsomes revealed low potential for interaction (half maximal inhibitory concentration >100 times the maximum plasma concentration [Cmax] of a 10 mg twice‐daily dose in patients with RA).11
Considering the major contribution of CYP3A4 to the metabolism of tofacitinib, the drug–drug interaction (DDI) potential with respect to coadministration with other drugs that are metabolized by CYP3A4 was evaluated. An in vivo evaluation of the highly sensitive CYP3A4 substrate midazolam (study A3921059; NCT00902460) demonstrated no change in midazolam total exposure (area under the curve from time zero extrapolated to infinite time [AUC∞]) and Cmax when coadministered with tofacitinib, indicating that tofacitinib had no inhibitory or inductive effect on CYP3A4.12
Oral contraceptives (OCs), usually in the form of fixed combinations of estrogen and progestin steroids, are metabolized by intestinal sulfation and glucurodination via uridine diphospho‐glucuronosyltransferase (UGT), and hepatic oxidation via CYP3A4.13 Because OCs are often coadministered with anti‐inflammatory therapies, the study was performed to inform physicians, clinicians, and regulators of the potential or lack thereof of a PK DDI between tofacitinib and OCs. Based on the available in vitro and in vivo data, the objective of this study was to confirm that tofacitinib had no inhibitory or inductive effect on the PK of the combination OCs, ethinylestradiol (EE), and levonorgestrel (LN).
Methods
Subjects
Eligible subjects were healthy adult female volunteers aged 18–55 years with a body mass index (BMI) of 17.5–30 kg/m2 and total body weight >50 kg (110 pounds). Exclusion criteria were evidence or history of clinically significant disease; evidence of clinically significant infections within the past 3 months; any infection within the past 7 days; history of disseminated herpes simplex infection or recurrent/disseminated herpes zoster; evidence of active, latent, or inadequately treated infection with Mycobacterium tuberculosis; personal or family history of hereditary immunodeficiency; being pregnant or nursing, and being of childbearing potential and unwilling/unable to use an acceptable method of nonhormonal contraception (as defined in the study protocol) from at least 14 days prior to the first dose of study medication until 4 weeks after receiving the last dose of study medication; any medical reason that would contraindicate the administration of OCs (as per label); and history of discontinued use of OCs for medical reasons. The use of CYP3A4 inhibitors/inducers, inhibitors of tubular secretion of creatinine, prescription or nonprescription drugs, vitamins, and dietary supplements within 28 days or 5 half‐lives (t1/2; whichever was longer) before the first dose of study medication was prohibited. Subjects who used tobacco‐ or nicotine‐containing products in excess of the equivalent of 5 cigarettes per day were excluded.
Clinical study design
This was a phase 1 randomized, open‐label, 2‐way crossover study (A3921071; NCT01137708) conducted at a single center in Belgium (Pfizer Clinical Research Unit, Brussels, Belgium) to assess the effect of oral tofacitinib 30 mg twice daily for 11 days on the single‐dose PK of combination EE/LN OCs (administered orally as a single Microgynon 30® tablet containing 30 μg EE and 150 μg LN) in healthy female volunteers.
Food and Drug Administration (FDA) guidance for drug interaction studies recommends administration of the highest dose of the precipitant at the shortest dosing interval.14 However, inducers can take several days to exert their effects on enzyme activity; thus, multiple‐dose administration of the investigational drug for a minimum of 10 days is generally recommended. Based on this guidance, the current study investigated tofacitinib 30 mg twice daily for 11 days. The dose level was chosen based on the highest anticipated exposure attainable in the different patient populations and for the different indications for which tofacitinib is being developed.
Subjects were randomized to 1 of 2 sequences, which each consisted of 2 treatment periods (Figure 1). In treatment A, subjects received a single Microgynon 30® tablet on day 1. In treatment B, subjects received tofacitinib 30 mg twice daily on days 1–11 and a single Microgynon 30® tablet administered simultaneously with the morning dose of tofacitinib on day 10. Treatment A was immediately followed by treatment B with no washout in sequence 1, whereas in sequence 2, subjects received treatment B first, followed by a washout period of at least 10 days, then treatment A.
Figure 1.
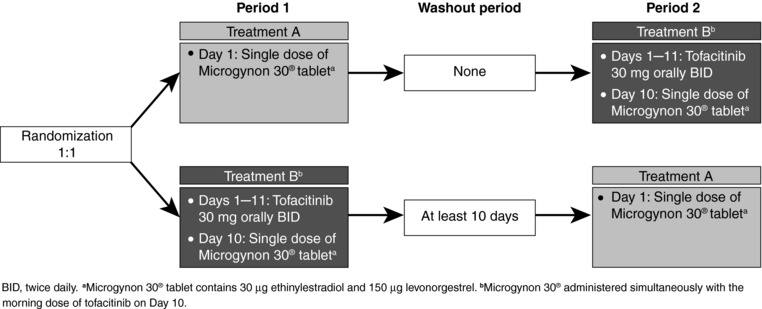
Study Design.
The final protocol, any amendments, and informed consent documentation were reviewed and approved by the institutional review board (IRB) and Independent Ethics Committee of the investigational center participating in the study. The IRB for this study was Comite d'Ethique de l'hôpital Erasme, Route de Lennik 808, 1070 Bruxelles (Brussels), Belgium. This study was conducted in compliance with the ethical principles originating in or derived from the Declaration of Helsinki and in compliance with all International Conference on Harmonization Good Clinical Practice Guidelines. All subjects provided written informed consent.
Analytical Methodology
To assess PK end points, blood samples for EE and LN PK analyses were collected predose and at 0.5, 1, 1.5, 2, 4, 6, 8, 12, 24, 36, and 48 hours postdose on day 1 or day 10 in treatment periods A and B, respectively. Blood samples for tofacitinib PK analyses were collected 2 hours postdose on day 9 of treatment B.
Analytical method for EE and LN in human plasma
Plasma samples with K3‐ethylenediaminetetraacetic acid were assayed using validated high‐performance liquid chromatography–tandem mass spectrometry (LC/MS/MS) methods by Covance Bioanalytical Services (Shanghai, China). Human plasma samples were stored at approximately ‐20°C until analysis and assayed within 926 days of established stability data generated during validation. EE, LN, and their internal standards (ethinylestradiol‐d4 and norgestrel‐d6) were extracted from 500 μL of plasma by liquid–liquid extraction with n‐butyl chloride. The organic phase was separated from the aqueous layer in a dry ice/acetone bath decanted into a clean tube, and evaporated to dryness. The residue was derivatized with dansyl chloride. Samples were injected onto a C18 column with Ultra Pressure Liquid Chromatography. The mobile phase was a gradient containing 0.05% of formic acid in acetonitrile/water and 0.05% formic acid in methanol/water. The detection was by an API 5000 (Sciex) with ionization of positive ion electrospray. The multiple reaction monitoring ion transition was m/z 530→171 for EE; m/z 534→171 for the EE internal standard, ethinylestradiol‐d4; m/z 313→245 for LN; and m/z 319→245 for the LN internal standard, norgestrel‐d6.
Selectivity of the method was acceptable toward endogenous compounds, potential interferences, and possible impurities of the internal standard, as illustrated by the chromatograms of a blank plasma sample with and without internal standard. In addition, selectivity of the method was demonstrated in the presence of tofacitinib at 600 ng/mL in human plasma.
Calibration standard responses were linear over the ranges of 2.50–500 pg/mL (EE) and 50.0–10 000 pg/mL (LN) using weighted (1/concentration2) linear regression. The lower limit of quantitation was 2.50 pg/mL for EE and 50.0 pg/mL for LN. The between‐day assay accuracy, expressed as percent relative error (%RE), ranged from ‐1.1% to 1.9% for EE and ‐0.3% to 1.9% for LN for the low, medium, and high concentrations (2.5, 7.50, and 80 pg/mL, respectively, for EE; 50, 150, and 1600 pg/mL, respectively, for LN). Assay precision, expressed as the between‐day percent coefficient of variation (%CV) of the quality control (QC) samples was <14.7% for EE and <6.0% for LN for the low, medium, and high QC samples.
Analytical method for tofacitinib in human plasma
Human plasma samples in sodium heparin were analyzed for tofacitinib concentrations at BASi Laboratories (West Lafayette, Indiana) using a validated, sensitive, and specific high‐performance LC/MS/MS method. Human plasma specimens were stored at approximately ‐20°C until analysis and assayed within 693 days of established stability data generated during validation. Tofacitinib was extracted from 300 μL of human plasma by 96‐well solid‐phase extraction plate with [13C15N] CP‐690,550 as the internal standard. The sample was eluted with 13% ammonium hydroxide in methanol and evaporated to dryness, then reconstituted with 50% methanol in water. The reconstituted sample was injected onto an HPLC column (Synergi Polar‐RP 4 Micro column; Phenomenex Inc, Torrance, California), with a mobile phase of 40% 10 mM ammonium acetate and 60% methanol (with 0.05% formic acid). Detection was performed by a Sciex API 4000 in the positive ion mode. The multiple reaction monitoring ion transition was m/z 313.4→173.2 for tofacitinib and m/z 316.3→173.1 for the internal standard.
Selectivity of the method was acceptable toward endogenous compounds, potential interferences, and possible impurities of the internal standard, as illustrated by the chromatograms of a blank plasma sample with and without internal standard. Selectivity of the method was also demonstrated in the presence of EE and LN at 150 pg/mL and 10 000 pg/mL, respectively, in human sodium heparin plasma.
Calibration standard responses were linear over the range of 0.100–350 ng/mL using a weighted (1/concentration2) quadratic regression. The lower limit of quantitation was 0.100 ng/mL for tofacitinib. Samples were analyzed in 1 analytical run. The assay accuracy, expressed as %RE for QC concentrations, ranged from 0.5% to 4.7% for the low, medium, and high QC samples.
Pharmacokinetic parameters
PK parameters of EE and LN were estimated for each subject for each treatment using noncompartmental analysis of concentration–time data. The primary PK measure was the AUC∞. Secondary PK measures included area under the plasma concentration–time profile from time zero to the time of the last quantifiable concentration (AUClast), Cmax, the time point at which Cmax was observed (Tmax), and apparent terminal elimination, t½. PK parameters were determined using an internally developed and validated software system, eNCA (electronic noncompartmental analysis).
Statistical Analyses
A sample size of 18 completers (9 per treatment sequence) was required to provide 95% power so that the 90% CI for the ratios of the 2 treatments for AUC∞ for both EE and LN was within the acceptance region of 80% and 125% assuming a true ratio of 1.0. Consequently, the study had at least 90% power overall to demonstrate a lack of effect in the presence and absence of tofacitinib (ie, equivalence in AUC∞) for both EE and LN.
Natural log‐transformed AUC∞, AUClast, and Cmax of EE and LN were analyzed separately using a mixed‐effects model with sequence, period, and treatment as fixed effects and subject within sequence as a random effect. Adjusted mean differences and corresponding 90% CIs were obtained from the model that were exponentiated to provide estimates of the ratio and 90% CIs of adjusted geometric means (calculated as Microgynon 30® coadministered with tofacitinib/ Microgynon 30® alone). Statistical calculations were performed using SAS.
A lack of effect of tofacitinib on OC total exposure would be concluded if the 90% CIs for the ratio of adjusted geometric mean for AUC∞ of both EE and LN fell wholly within 80% to 125%.
Safety
All adverse events (AEs), their severity, and their relationship to the study drug were recorded. Vital signs (supine blood pressure, pulse rate, and oral temperature) were measured at screening, immediately prior to the OC dose, and at follow‐up. A complete physical examination was carried out on day 0 and also at the end of period 2, at the discretion of the investigator. A 12‐lead electrocardiogram was performed at screening and at follow‐up for treatment sequence 1 or on day 3 of period 2 for treatment sequence 2. Clinical laboratory parameters were determined from blood and urine samples obtained at screening, on day 0, and at the end of study. The clinical laboratory sample analyses were performed by IBC (Brussels, Belgium).
Results
Subject Disposition and Demographics
A total of 20 female volunteers were screened and randomized (10 to each treatment sequence). All 20 subjects received tofacitinib 30 mg twice daily alone or coadministered with a single dose of Microgynon 30®, and 19 of the 20 subjects received treatment with Microgynon 30® alone. All but 1 subject completed the study. Eighteen subjects were white, 1 was black, and 1 was reported to be “other.” The mean age was 33.5 years (range, 19–50 years), the mean height was 164 cm (range, 151–176 cm), the mean weight was 63.9 kg (range, 52–75 kg), and the mean BMI was 23.7 kg/m2 (range, 20.1–29.0 kg/m2).
Pharmacokinetics
For both EE and LN, mean plasma concentration–time profiles in the presence and absence of tofacitinib were similar (Figure 2A,B).
Figure 2.
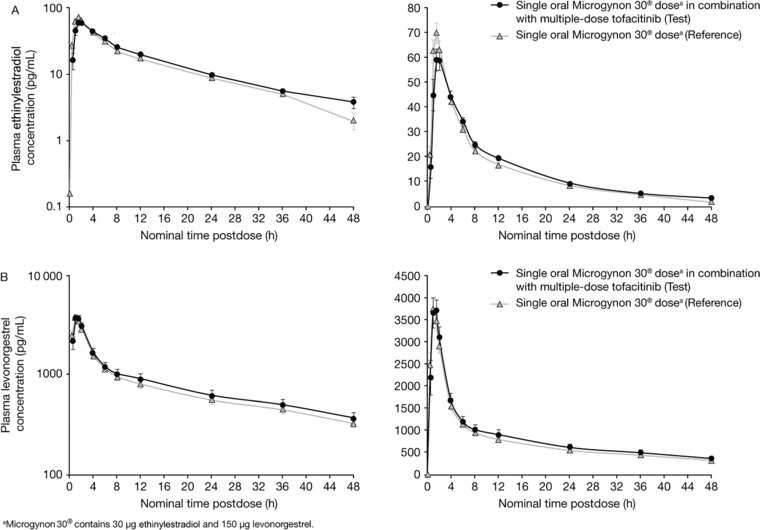
Mean ± Standard Error (A) Ethinylestradiol and (B) Levonorgestrel Concentration–Time Profiles on Semilog and Linear Scales.
Mean t1/2 was similar for EE with tofacitinib and EE alone (13.8 and 13.3 hours, respectively) and similar for LN with tofacitinib and LN alone (25.9 and 25.4 hours, respectively); see Table 1. Median Tmax values were identical for both EE treatments (1.5 hours) and for both LN treatments (1.0 hours); see Table 1.
Table 1.
Descriptive Summary of PK Parameters Following a Single Oral Microgynon 30® Dose Alone and in Combination With Multiple‐Dose Tofacitinib.
| Parameter (Units)a | Microgynon 30®b + Tofacitinib | Microgynon 30®b Alone |
|---|---|---|
| Ethinylestradiol | n = 20 | n = 19 |
| AUClast (pg·h/mL) | 707.7 (149.0) | 661.1 (137.9) |
| AUC∞ (pg·h/mL) | 776.8 (144.4) | 731.4 (154.8) |
| Cmax (pg/mL) | 64.31 (19.5) | 71.32 (17.7) |
| Tmax (h) | 1.5 (1.0–4.0) | 1.5 (1.0–2.1) |
| t1/2 (h) | 13.8 (2.3) | 13.3 (2.2) |
| Levonorgestrel | n = 20 | n = 19 |
| AUClast (pg·h/mL) | 39 970 (18 564) | 36 760 (11 604) |
| AUC∞ (pg·h/mL) | 45 910 (13 994) | 47 290 (15 583) |
| Cmax (pg/mL) | 4362 (1050.6) | 3906 (954.5) |
| Tmax (h) | 1.0 (0.5–2.0) | 1.0 (1.0–2.0) |
| t1/2 (h) | 25.9 (3.8) | 25.4 (4.1) |
AUC∞, area under the plasma concentration–time profile from time zero to infinity; AUClast, area under the plasma concentration–time profile from time zero to the time of the last quantifiable concentration; CI, confidence interval; Cmax, maximum plasma concentration; Tmax, time to Cmax; PK, pharmacokinetics; t1/2, apparent terminal elimination half‐life. aParameters reported as arithmetic mean (standard deviation) for all except Tmax, which is median (range). bMicrogynon 30® contains 30 μg of ethinylestradiol and 150 μg of levonorgestrel.
The ratios of adjusted geometric means of AUC∞ were 106.55% (90% CI, 98.91%–114.78%) for EE and 100.87% (90% CI, 94.73%–107.42%) for LN (Table 2). For both EE and LN, 90% CIs for the ratios of adjusted geometric means for AUC∞ fell wholly within the acceptance region (80%–125%), and similar results were observed for Cmax (Table 2).
Table 2.
Statistical Comparisons of PK Parameters Following a Single Oral Microgynon 30® Dose Alone and in Combination With Multiple‐Dose Tofacitinib.
| Adjusted Geometric Mean | ||||
| Ratio (Test/Reference) | 90% CI for | |||
| Test | Reference | of Adjusted | Ratio of Adjusted | |
| Parameter (Units) | (Microgynon 30®a + Tofacitinib) | (Microgynon 30®a Alone) | Geometric Meansb | Geometric Meansb |
| Ethinylestradiol | ||||
| AUC∞ (pg·h/mL) | 762.8 | 715.9 | 106.55 | 98.91–114.78 |
| AUClast (pg·h/mL) | 691.4 | 647.3 | 106.81 | 98.30–116.06 |
| Cmax (pg/mL) | 61.51 | 68.64 | 89.62 | 81.98–97.97 |
| Levonorgestrel | ||||
| AUC∞ (pg·h/mL) | 45 000 | 44 610 | 100.87 | 94.73–107.42 |
| AUClast (pg·h/mL) | 36 580 | 34 970 | 104.60 | 96.63–113.22 |
| Cmax (pg/mL) | 4242 | 3781 | 112.19 | 105.30–119.53 |
AUC∞, area under the plasma concentration–time profile from time zero to infinity; AUClast, area under the plasma concentration–time profile from time zero to the time of the last quantifiable concentration; CI, confidence interval; Cmax, maximum plasma concentration; PK, pharmacokinetics. aMicrogynon 30® contains 30 μg of ethinylestradiol and 150 μg of levonorgestrel. bRatios (and 90% CIs) are expressed as percentages.
The arithmetic mean of tofacitinib concentrations in PK samples obtained 2 hours postdose on day 9 of tofacitinib twice‐daily dosing was 215.2 (range, 143–300) ng/mL.
Safety
The only AEs to occur in ≥2 subjects during any treatment were nausea, headache, insomnia, and acne (Supplementary Table 1). All other reported AEs were represented by single occurrences under each treatment. The most frequently reported AE was acne, which was reported in 7 subjects during Microgynon 30® treatment: 4 during tofacitinib 30 mg twice daily alone and 13 during tofacitinib plus Microgynon 30® treatment. All the AEs reported in this study were mild in severity with the exception of 4, which were assessed as moderate (contusion, n = 2 [1 with Microgynon 30® alone, 1 with tofacitinib plus Microgynon 30®]; urinary tract infection, n = 1; and headache, n = 1 [both with tofacitinib plus Microgynon 30®]). The subject with a urinary tract infection discontinued the study because of this AE, which was considered a result of a bacterial infection, rather than the study drug. No other discontinuations because of AEs were reported. There were no deaths, serious AEs, temporary discontinuations, or dose reductions because of AEs in this study. No reportable laboratory abnormalities, changes in vital signs, or abnormal ECG findings were observed.
Discussion
Tofacitinib undergoes CYP3A4‐mediated metabolism; therefore, the potential for tofacitinib to alter the PK of the OCs EE and LN, which both undergo CYP3A4 metabolism, was evaluated. This study indicated that tofacitinib had no clinically relevant net inhibitive or inductive effect on the PK of the combination OCs EE and LN. When a single oral dose of EE and LN was administered alone or in combination with multiple‐dose tofacitinib, the 90% CIs for the ratios of adjusted geometric means for AUC∞ and Cmax were entirely within the predefined acceptance range (80%–125%). Most AEs were mild in severity, and there were no deaths, serious AEs, temporary discontinuations, or dose reductions because of AEs reported in this study. The study results support a lack of effect of tofacitinib on the PK of EE and LN.
The PK results reported here, showing no net inhibitory or inductive effect of tofacitinib on the PK of the combination OCs EE and LN, are consistent with a previous drug–drug interaction study, which reported no change in total exposure or Cmax of the highly sensitive CYP3A4 substrate midazolam when coadministered with tofacitinib 30 mg twice daily.12 Both the current OC study and the midazolam DDI study support the previous in vitro data reporting that tofacitinib does not modulate CYP3A4 activity.11, 15, 16
The dose of combination OC was selected from the choice of commercially available products. Microgynon 30® contains 30 μg of EE and 150 μg of LN and has been used in previous DDI studies.17, 18 DDI evaluations using both multiple and single doses of OC steroids have been carried out; single‐dose designs using the same steroids as in this study (EE and LN) include studies of vigabatrin (epilepsy), ABT‐761 (asthma), tenidap (rheumatoid arthritis), and Diflucan (fluconazole; antifungal; Pfizer unpublished data).19, 20, 21 The present study employed a single administered dose of the combination OC rather than repeat dosing; a single dose of Microgynon 30® was considered sufficient to confirm the lack of significant effect of tofacitinib on the PK of EE and LN, given the in vitro profile of tofacitinib and the previously reported lack of interaction between tofacitinib and the more sensitive CYP3A4 substrate, midazolam.12 In addition, repeat dosing of OC was not considered necessary, especially if steady‐state PK is predictable from single‐dose data, even if there is drug accumulation. Steady‐state PK is reportedly predictable from single‐dose data for EE.22, 23 However, the steady‐state PK of LN appears to be influenced by changes in sex hormone–binding globulin levels by coadministered EE, such that drug accumulation of LN appears to be greater than expected based on single‐dose data.22, 23
In conclusion, no dose adjustments are needed for OC drugs containing EE or LN when coadministered with tofacitinib. Together with evidence from previous studies with midazolam12 and in vitro data,11, 15, 16 these results indicate that tofacitinib is unlikely to affect the clearance of drugs metabolized by CYP3A4 enzymes.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Acknowledgments
The authors thank the A3921071 investigators and study team.
Funding and Declaration of Conflicting Interests
Sujatha Menon, Ronnie Wang, Christine W. Alvey, Haihong Shi, and Sriram Krishnaswami are employees and shareholders of Pfizer Inc. Richard Riese and Wendy Petit were employees and shareholders of Pfizer Inc at the time of the study. This study was sponsored by Pfizer Inc. Editorial support was provided by Anne Marie Reid, PhD, at Complete Medical Communications and was funded by Pfizer Inc.
References
- 1. Meyer DM, Jesson MI, Li X, et al. Anti‐inflammatory activity and neutrophil reductions mediated by the JAK1/JAK3 inhibitor, CP‐690,550, in rat adjuvant‐induced arthritis. J Inflamm (Lond). 2010;7:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Burmester GR, Blanco R, Charles‐Schoeman C, et al. Tofacitinib (CP‐690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised phase 3 trial. Lancet. 2013;381:451–460. [DOI] [PubMed] [Google Scholar]
- 3. Fleischmann R, Kremer J, Cush J, et al. Placebo‐controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med. 2012;367:495–507. [DOI] [PubMed] [Google Scholar]
- 4. Kremer J, Li ZG, Hall S, et al. Tofacitinib in combination with nonbiologic disease‐modifying antirheumatic drugs in patients with active rheumatoid arthritis: a randomized trial. Ann Intern Med. 2013;159:253–261. [DOI] [PubMed] [Google Scholar]
- 5. Lee EB, Fleischmann R, Hall S, et al. Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med. 2014;370:2377–2386. [DOI] [PubMed] [Google Scholar]
- 6. van der Heijde D, Tanaka Y, Fleischmann R, et al. Tofacitinib (CP‐690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve‐month data from a twenty‐four‐month phase III randomized radiographic study. Arthritis Rheum. 2013;65:559–570. [DOI] [PubMed] [Google Scholar]
- 7. van Vollenhoven RF, Fleischmann R, Cohen S, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med. 2012;367:508–519. [DOI] [PubMed] [Google Scholar]
- 8. Wollenhaupt J, Silverfield J, Lee EB, et al. Safety and efficacy of tofacitinib, an oral Janus kinase Inhibitor, for the treatment of rheumatoid arthritis in open‐label, longterm extension studies. J Rheumatol. 2014;41:837–852. [DOI] [PubMed] [Google Scholar]
- 9. Boy MG, Wang C, Wilkinson BE, et al. Double‐blind, placebo‐controlled, dose‐escalation study to evaluate the pharmacologic effect of CP‐690,550 in patients with psoriasis. J Invest Dermatol. 2009;129:2299–2302. [DOI] [PubMed] [Google Scholar]
- 10. Sandborn WJ, Ghosh S, Panes J, et al. Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. N Engl J Med. 2012;367:616–624. [DOI] [PubMed] [Google Scholar]
- 11. Dowty ME, Lin J, Ryder TF, et al. The pharmacokinetics, metabolism, and clearance mechanisms of tofacitinib, a Janus kinase inhibitor, in humans. Drug Metab Dispos. 2014;42:759–773. [DOI] [PubMed] [Google Scholar]
- 12. Gupta P, Alvey C, Wang R, et al. Lack of effect of Tofacitinib (CP‐690,550) on the pharmacokinetics of the CYP3A4 substrate midazolam in healthy volunteers: confirmation of in vitro data. Br J Clin Pharmacol. 2012; 74:109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang H, Cui D, Wang B, et al. Pharmacokinetic drug interactions involving 17alpha‐ethinylestradiol: a new look at an old drug. Clin Pharmacokinet. 2007;46:133–157. [DOI] [PubMed] [Google Scholar]
- 14. US Health and Human Services. Food and Drug Administration . Guidance for industry drug interaction studies—study design, data analysis, and implications for dosing and labeling draft guidance. 2006. www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072101.pdf. Accessed May 17, 2011.
- 15. Fahmi OA, Kish M, Boldt S, Obach RS. Cytochrome P450 3A4 mRNA is a more reliable marker than CYP3A4 activity for detecting pregnane X receptor‐activated induction of drug‐metabolizing enzymes. Drug Metab Dispos. 2010;38:1605–1611. [DOI] [PubMed] [Google Scholar]
- 16. Walsky RL, Obach RS. Validated assays for human cytochrome P450 activities. Drug Metab Dispos. 2004;32:647–660. [DOI] [PubMed] [Google Scholar]
- 17. Abel S, Russell D, Whitlock LA, Ridgway CE, Muirhead GJ. Effect of maraviroc on the pharmacokinetics of midazolam, lamivudine/zidovudine, and ethinyloestradiol/levonorgestrel in healthy volunteers. Br J Clin Pharmacol. 2008;65(Suppl 1):19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Friedrich C, Port A, Ring A, et al. Effect of multiple oral doses of linagliptin on the steady‐state pharmacokinetics of a combination oral contraceptive in healthy female adults: an open‐label, two‐period, fixed‐sequence, multiple‐dose study. Clin Drug Investig. 2011;31:643–653. [DOI] [PubMed] [Google Scholar]
- 19. Coates PE, Mesure R. An investigation into the effect of tenidap sodium on the pharmacokinetics of a combined oral contraceptive. Br J Clin Pharmacol. 1995;39(Suppl 1):47S–50S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bartoli A, Gatti G, Cipolla G, et al. A double‐blind, placebo‐controlled study on the effect of vigabatrin on in vivo parameters of hepatic microsomal enzyme induction and on the kinetics of steroid oral contraceptives in healthy female volunteers. Epilepsia. 1997;38:702–707. [DOI] [PubMed] [Google Scholar]
- 21. Wong SL, O'Dea RF, Dube LM, Awni WM. Effects of ABT‐761, a novel 5‐lipoxygenase inhibitor, on the pharmacokinetics of a single dose of ethinyl estradiol and levonorgestrel in healthy female volunteers. J Clin Pharmacol. 1998;38:642–648. [DOI] [PubMed] [Google Scholar]
- 22. Kuhnz W, al‐Yacoub G, Fuhrmeister A. Pharmacokinetics of levonorgestrel and ethinylestradiol in 9 women who received a low‐dose oral contraceptive over a treatment period of 3 months and, after a wash‐out phase, a single oral administration of the same contraceptive formulation. Contraception. 1992;46:455–469. [DOI] [PubMed] [Google Scholar]
- 23. Kuhnz W, Staks T, Jutting G. Pharmacokinetics of levonorgestrel and ethinylestradiol in 14 women during three months of treatment with a tri‐step combination oral contraceptive: serum protein binding of levonorgestrel and influence of treatment on free and total testosterone levels in the serum. Contraception. 1994;50:563–579. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.