

Abstract
Heparan sulfate‐specific endosulfatase‐2 (SULF‐2) can modulate the signaling of heparan sulfate proteoglycan‐binding proteins. The involvement of SULF‐2 in cancer growth varies by cancer type. The roles of SULF‐2 expression in the progression and prognosis of renal cell carcinomas (RCC) have not yet been fully clarified. In the present study, the expression levels of SULF‐2 mRNA and protein in 49 clinical RCC samples were determined by RT‐PCR and immunostaining. The existence of RCC with higher SULF‐2 expression and lower SULF‐2 expression compared to the adjacent normal kidney tissues was suggested. High SULF‐2 expression was correlated with an early clinical stage and less invasive pathological factors. Low SULF‐2 expression was correlated with an advanced stage and higher invasive factors. Three‐year cancer‐specific survival (CSS) for high SULF‐2 RCC and low SULF‐2 RCC were 100% and 71.4%, respectively (log‐rank P = 0.0019), with a significantly shorter CSS observed in low SULF‐2 RCC patients. The influence of SULF‐2 expression level on Wnt/VEGF/FGF signaling, cell viability and invasive properties was examined in three RCC cell lines, Caki‐2, ACHN and 786‐O, using a SULF‐2 suppression model involving siRNA or a SULF‐2 overexpression model involving a plasmid vector. High SULF‐2 expression enhanced Wnt signaling and Wnt‐induced cell viability, but not cell invasion. In contrast, low levels of SULF‐2 expression significantly enhanced both cell invasion and viability through the activation of VEGF/FGF pathways. RCC with lower SULF‐2 expression might have a higher potential for cell invasion and proliferation, leading to a poorer prognosis via the activation of VEGF and/or FGF signaling.
Keywords: Cancer progression, heparan sulfate proteoglycan binding growth factor, prognosis, renal cell carcinoma, Sulfatase‐2
Extracellular endosulfatases (SULF), which include Sulfatase‐1 (SULF‐1) and Sulfatase‐2 (SULF‐2, EC3.1.6.4), are secreted to the outside of cells and remove sulfate from heparan sulfate proteoglycan (HSPG).1 HSPG is one of the components of the extracellular matrix and acts as a co‐receptor for numerous protein ligands, including growth factors, cytokines, morphogens, proteases and matrix proteins.2, 3 The binding affinity of HSPG for these ligands depends mainly on its sulfation pattern.4, 5, 6 In fact, there have been reports that SULF‐2 positively regulates Wnt signaling7 and negatively regulates VEGF8 and FGF signaling9, 10 in several cell lines through modification of HSPG.
Although studies on the relationship between SULF and a number of malignancies have been recently reported, the conclusions drawn are dependent on specific cancer type. The expression levels of SULF‐2 in human neoplasms vary with cancer type.11 SULF‐2 is more highly expressed in cancer cells than in normal cells and enhances tumorigenesis in lung cancer12 and pancreatic cancer.13 In contrast, SULF are said to be tumor suppressors in breast cancer14 and myeloma.15 Furthermore, in hepatocellular carcinoma, SULF‐1 expression level displays a bimodal effect on prognosis.16 Therefore, the roles of SULF‐2 in the mechanisms of cancer progression appear to be complicated. Renal cell carcinoma (RCC) is the most common malignant tumor of the kidney,17 accounting for approximately 85% of all renal cancers.18 Although it is reported that SULF‐2 is one of the target genes of Von Hippel Lindau (VHL),19 which is known to be a tumor suppressor gene in clear cell RCC (ccRCC), little is known regarding the association between the SULF‐2 expression and the clinico‐pathological features or prognosis in patients with RCC. The results of the present study revealed the expression levels of SULF‐2 in RCC and demonstrated a correlation between SULF‐2 expression level and clinico‐pathological features. Moreover, we demonstrated that SULF‐2 contributed to cell proliferation and invasion in RCC cell lines and also showed that SULF‐2 levels might be indicative of prognosis in RCC patients.
Material and Methods
Patients
Forty‐nine patients (40 male, 9 female) with ccRCC were enrolled in this study. Age ranged from 39 to 87 years (mean age 64.6 years). Nephrectomy or partial nephrectomy was carried out for all the patients at the Fukushima Medical University Hospital (Table 1). Small pieces of tumor tissues containing normal renal tissues were excised from each patient, frozen within 15 min of resection and stored at −80°C in our department. The protocol for the present research was approved by the ethics board of Fukushima Medical University (Approval No. 2045).
Table 1.
Clinical background of 49 clear cell renal cell carcinoma patients
| Number (n) | |
| Male | 40 |
| Female | 9 |
| Age: mean (range) | 64.6 years (39–87 years) |
| Operation (n) | |
| Nephrectomy | 45 |
| Partial nephrectomy | 4 |
| Follow up time: median (range) | 37.9 months (5.6–73.4 months) |
| Death (n) | 9 |
Cell lines/recombinant human Wnt3a/VEGF/basic FGF
Human RCC cell lines ACHN, 786‐O and Caki‐2 were obtained from the American Type Culture Collection (Manassas, VA, USA) 3 months before the start of the experiments. ACHN cells were cultured in DMEM containing 5% MEM with 0.1 mmol/L non‐essential amino acids and 10% FBS. 786‐O and Caki‐2 cells were cultured in RPMI1640 medium with 10% FBS and McCoy's medium with 10% FBS, respectively. Recombinant human Wnt3a was purchased from R&D Systems, (Minneapolis, MN, USA) and recombinant human VEGF (AF‐100‐20) and recombinant human basic FGF (AF‐100‐18B) were purchased from PeproTech (Rocky Hill, NJ, USA).
RNA isolation and quantitative real‐time PCR
RNA isolation from clinical RCC samples was performed using ISOGEN (Nippon Gene, Tokyo, Japan) in accordance with the manufacturer's instructions. cDNA was constructed using the SuperScript III First‐strand Synthesis System for RT‐PCR (Invitrogen, Carlsbad, CA, USA). RNA isolation and cDNA construction from human RCC cell lines were performed using the TaqMan Fast Cells‐to‐CT Kit (Ambion/Life Technologies, Carlsbad, CA, USA). Each cell line was plated at a density of 8000 cells/well on a 96‐well culture plate. TaqMan PCR reagents for SULF‐2 (Hs01016476_m1) and cyclin D1 (Hs00765553_m1) were purchased from Applied Biosystems (Foster city, CA, USA). Quantitative real‐time PCR was carried out using the TaqMan Master Mix Reagent Kit protocol with a StepOne real‐time PCR system (Applied Biosystems). The data were standardized against β‐actin gene expression using Pre‐Developed TaqMan Assay reagents (Applied Biosystems). The expression level of SULF‐2 mRNA was determined by the ΔΔC t method based on the normal tissue from one patient as a control. All experiments were conducted at least three times.
Preparation of anti‐SULF‐2 polyclonal antibodies and immunohistochemical staining
Anti‐SULF‐2 polyclonal antibodies were produced by MBL, Japan. Rabbits were immunized with a peptide derived from the predicted sequence of HSulf‐2 (H2.1: NH2‐CFLSHHRLKGRFQRDRR‐COOH, where C denotes a cysteine residue added for coupling).20 The antibodies were purified by sequential passage of the rabbit sera through peptide affinity columns according to the manufacturer's instructions. Antisera and purified antibody titers were determined using ELISA. The expression of SULF‐2 protein was identified by western blot analysis for SULF‐2 using this antibody in the cell line study. Formalin‐fixed paraffin‐embedded samples from clinical RCC patients were examined by immunohistochemical staining with the purified SULF‐2 antibody. Coloring was performed with DAB. The nuclei of the section were counterstained with hematoxylin. The slides of stained specimens were evaluated by one pathologist from our university. The degree of staining was determined using a three‐grade evaluation system, − or + or ++. The criteria for evaluation were as follows. Strong uniform staining of the whole specimen was defined as ++ and an absence of staining of whole specimen as −. Partial non‐uniform staining was defined as +.
siRNA and transfection
Pre‐designed Silencer Select siRNA targeting SULF‐2 (SULF‐2 siRNA, s31805) was purchased from Ambion/Life Technologies. Negative control siRNA (NC siRNA) was designed using the siDirect program (RNAi, Tokyo, Japan, http://sidirect2.rnai.jp/). Three pmol of the SULF‐2 siRNA or the NC siRNA was added to 8000 cells of each human RCC cell line with Lipofectamine RNAiMax (Invitrogen).
Plasmid DNA and transfection
A cDNA clone encoding SULF‐2 (pCMV6‐SULF‐2: SC328022) and a negative control cDNA clone (pCMV6‐entry: PS100001) were purchased from ORIGENE (Rockville, MD, USA). One hundred ng of these plasmid DNAs was applied to 8000 cells of each RCC cell line with Lipofectamine LTX (Invitrogen).
Determination of Wnt3a/VEGF/basic FGF pathway activation
The stimulating factors were added to the cells cultured in FBS‐free medium at the defined concentrations. The concentrations of Wnt3a, VEGF and FGF were 100, 50 and 50 ng/mL, respectively. Activation of the Wnt signaling pathway was determined by western blot analysis of the β‐catenin protein and by RT‐PCR analysis of cyclinD1 mRNA. Wnt treatment was performed for 3 h prior to β‐catenin protein analysis and for 6 h prior to cyclinD1 mRNA analysis. Activation of the VEGF or FGF signaling pathway was determined by western blot analysis of phosphorylated ERK (p‐ERK) 3 h after the administration of the growth factors.
Western blotting analysis
Each human RCC cell line was plated at a density of 25 000 cells/well on a 24‐well cell culture plate and cultured for 24 h. After removing the medium, cellular proteins were extracted with RIPA buffer (Thermo Scientific, Waltham, MA, USA) and separated by SDS‐polyacrylamide gel electrophoresis. Anti‐β‐catenin antibody (Abcam, Cambridge, UK) and anti‐phosphorylated ERK antibody (Cell Signaling Technology, Beverly, MA, USA) were used as primary antibodies. Anti‐SULF‐2 (H2.1) antibody was produced by MBL as described in the above section. Antibody‐bound protein bands were visualized using ECL Advance Western detection reagents (GE Healthcare, Buckinghamshire, UK) and imaged using a ChemiDoc XRS plus system (BIO‐RAD, Hercules, CA, USA). Individual bands were quantified with Image Lab 2.0 software (BIO‐RAD), and normalized against the β‐actin band. Experiments were conducted at least three times.
Cell proliferation assay
The cellular proliferation of the human RCC cell lines was evaluated using an MTT assay (Cell Proliferation Kit I; Roche Diagnostics, Mannheim, Germany). Each human RCC cell line was plated at a density of 6000 cells/well on a 96‐well cell culture plate and was cultured for 24 h. After treatment for 48 h with Wnt/VEGF/basic FGF at the concentrations defined above, the MTT labeling reagent was added to the cells, which were then incubated for 4 h at 37°C. The spectrophotometric absorbance of the samples was measured using a Microtiter plate reader with 650 and 570‐nm filters. Experiments were conducted at least three times.
Cell invasion assay
The invasive capability of human RCC cell lines was evaluated using a BD BioCoat Matrigel Invasion Chamber (BD Bioscience, Franklin Lakes, NJ, USA). The number of cells that migrated through the transwell inserts with a polyethylene terephthalate membrane (8‐μm pore size) coated with a layer of BD Matrigel Basement Membrane Matrix was counted under a microscope. Following stimulation with Wnt/VEGF/basic FGF for 24 h, the cells were seeded on top of the transwell inserts with 500 μL FBS‐free medium, and the lower chamber was filled with 750 μL of medium containing 10% FBS as a chemoattractant. The number of cells seeded was 15 000 for Caki‐2 and ACHN cells, and 8000 of 786‐O cells. Experiments were conducted at least three times.
Statistical analysis
Clinical samples were analyzed using the Mann–Whitney U‐test. Comparisons between SULF‐2 expression and clinico‐pathological features were performed using the χ2‐test for categorical variables. Cancer specific survival (CSS) was estimated using the Kaplan–Meier method and curves were compared using the log‐rank test. Correlation of the SULF‐2 expression level with CSS was evaluated using Cox proportional hazard regression modeling. The results from experiments using the cell lines are expressed as means ± SD, and were analyzed using a two‐tailed paired t‐test. All statistical computations were carried out using SPSS version 22 (Nippon IBM, Tokyo, Japan) and P‐value < 0.05 was considered statistically significant.
Results
SULF‐2 expression and clinico‐pathological features in renal cell carcinomas
The expression levels of SULF‐2 in the clinical RCC samples were measured by RT‐PCR and immunohistochemical staining. The expression level of SULF‐2 mRNA in cancer tissue was compared to that in the normal kidney tissue in each patient. SULF‐2 was significantly more highly expressed in cancer tissues than in normal tissues in low‐stage cases, small cancers, low‐grade cases and cases without venous invasion (Table 2). Among 35 cases in which SULF‐2 mRNA expression was higher in the cancer tissue than in the normal tissue, 26 were positive for immunostaining (evaluated as + or ++), whereas nine cases were negative for immunostaining (evaluated as −). There were 14 cases in which SULF‐2 expression was lower in the cancer tissue than in the normal tissue, and all these cases were negative for immunostaining (Table 3). The patients were divided into the high SULF‐2 group and low SULF‐2 group based on SULF‐2 expression level. The high SULF‐2 group was defined as cases in which the SULF‐2 mRNA expression level was higher in the cancer tissue than in their own normal kidney tissue with positive immunostaining for SULF‐2. The low SULF‐2 group was defined as cases other than those above; that is, cases in which SULF‐2 mRNA expression was lower in the cancer tissues than in the normal tissues or cases that were negative for immunostaining for SULF‐2. As a result, the high SULF‐2 group included 26 cases and the low SULF‐2 group 23 cases. Representative immunostaining of clinical RCC samples is shown in Figure 1. As summarized in Table 4, 84% (22/26) of patients in the high SULF‐2 group were diagnosed as stage I–II and 78% (18/23) of those in the low SULF‐2 group were diagnosed as stage III–IV. Ninety‐two percent (24/26) of patients in the high SULF‐2 group were diagnosed as T1 or T2, and 69% (16/23) of those in the low SULF‐2 group were diagnosed as T3 or T4. Eighty‐four percent (22/26) of patients in the high SULF‐2 group were negative for venous invasion and 78% (18/23) of those in the low SULF‐2 group were positive for venous invasion. A high level of SULF‐2 expression was correlated with a less developed clinical stage and less invasive pathological features, including low T stage, negative venous invasion, and expansive growth pattern. In contrast, a low level of SULF‐2 expression was correlated with an advance clinical stage and highly invasive pathological features, including high T stage, positive venous invasion, and intermediate growth pattern. One of 26 patients in the high SULF‐2 group and 8 of 23 patients in the low SULF‐2 group died due to cancer progression at a median follow‐up time of 37.9 months (range, 5.6–73.4 months) from nephrectomy. CSS at 3 years was 100% and 71.4% in the high SULF‐2 group and low SULF‐2 group, respectively (log‐rank P = 0.0019). The low SULF‐2 group had significantly worse CSS compared to the high SULF‐2 group (HR = 13.23, 95% CI 1.63–107.62; P = 0.015) (Fig. 2).
Table 2.
Expression levels of SULF‐2 mRNA in clear cell renal cell carcinomas
| Number of patients (n) | Normal | Cancer | P‐value | |
|---|---|---|---|---|
| Mean ± SE (median) | ||||
| Clinical stage | ||||
| 1–2 | 27 | 0.77 ± 0.23 (0.35) | 40.3 ± 16.5 (3.3) | <0.001 |
| 3–4 | 22 | 31.9 ± 18.9 (1.21) | 6.6 ± 3.7 (1.2) | 0.33 |
| Pathological stage | ||||
| pT1–2 | 31 | 12.8 ± 11.9 (0.43) | 38.8 ± 14.4 (3.3) | <0.001 |
| pT3–4 | 18 | 16.9 ± 10.6 (0.52) | 1.5 ± 0.6 (1.03) | 0.5 |
| Histological grade | ||||
| G1–2 | 36 | 15.3 ± 10.8 (0.52) | 31.8 ± 12.6 (1.70) | <0.01 |
| G3 | 13 | 11.6 ± 10.5 (0.44) | 6.9 ± 5.2 (0.71) | 0.62 |
| Venous invasion | ||||
| v (−) | 27 | 0.88 ± 0.24 (0.43) | 42.7 ± 16.4 (4.0) | <0.001 |
| v (+) | 22 | 30.3 ± 18.0 (0.52) | 3.6 ± 2.1 (1.15) | 0.35 |
| Growthpatterna | ||||
| Expansive (INFa) | 32 | 17.1 ± 12.2 (0.50) | 35.4 ± 14.1 (1.75) | 0.01 |
| Intermediate (INFb) | 17 | 9.1 ± 7.9 (0.48) | 5.8 ± 4.0 (1.21) | 0.51 |
The patterns of tumor infiltration into the surrounding tissues were classified into the following three categories (General Rule for Clinical and Pathological Studies on Renal Cell Carcinoma, The 4th Edition. The Japanese Urological Association/The Japanese Society of Pathology/Japan Radiological Society): (i) INFa, expanding growth and a distinct border from the surrounding tissues; (ii) INFb, intermediate between INFa and INFc; and (iii) INFc, infiltrating growth and an indistinct border from the surrounding tissues.
Table 3.
The relationship between the level of SULF‐2 mRNA expression and the degree of immunostaining for SULF‐2 in clinical clear cell renal cell carcinoma patients
| mRNA expression level | ||
|---|---|---|
| Cancer >Normal (N = 35) | Cancer <Normal (N = 14) | |
| n | n | |
| Immunostaining grade | ||
| (++) | 11 | 0 |
| (+) | 15 | 0 |
| (−) | 9 | 14 |
Figure 1.
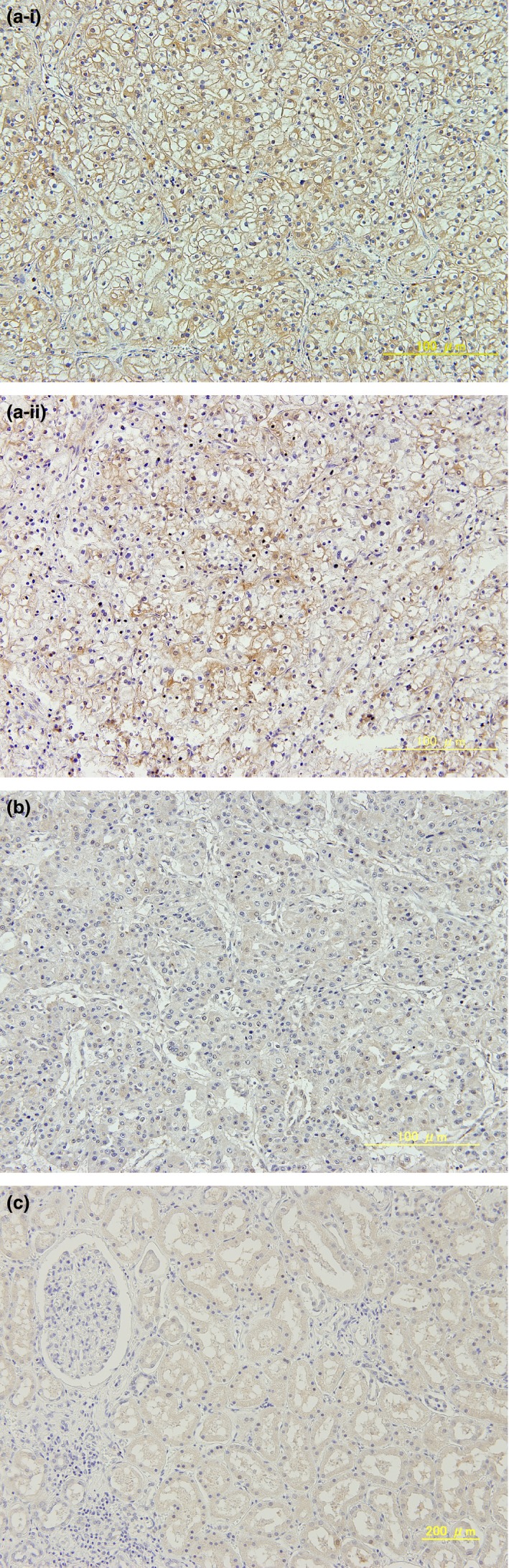
Immunostaining of clinical ccRCC tissues for SULF‐2. (a) Positive SULF‐2 expression in cancer tissues. (i) The expression level is judged as ++. (ii) The expression level is judged as +. (b) Negative SULF‐2 expression in cancer tissue judged as −. (c) Normal tissue. Images were obtained using a BX51 TRF microscope (Olympus, Tokyo, Japan).
Table 4.
Correlation of SULF‐2 expression and clinico‐pathological factors
| High SULF‐2 [N = 26] | Low SULF‐2 [N = 23] | P‐value | |
|---|---|---|---|
| N (%) | n (%) | ||
| Age: Mean ± SD | 64.3 ± 12.4 | 64.9 ± 10.4 | 0.417 |
| Gender | |||
| Male | 24 (92.3) | 16 (69.6) | 0.04 |
| Female | 2 (7.7) | 7 (30.4) | |
| Clinical stage | |||
| 1 | 20 (77.0) | 4 (17.4) | <0.001 |
| 2 | 2 (7.7) | 1 (4.3) | |
| 3 | 1 (3.8) | 7 (30.4) | |
| 4 | 3 (11.5) | 11 (47.8) | |
| Pathological tumor stage | |||
| T1 | 22 (84.6) | 6 (26.1) | <0.001 |
| T2 | 2 (7.7) | 1 (4.3) | |
| T3 | 2 (7.7) | 14 (60.9) | |
| T4 | 0 (0) | 2 (8.7) | |
| Histological grade | |||
| G1 | 5 (19.2) | 3 (13.0) | 0.764 |
| G2 | 15 (57.7) | 13 (56.5) | |
| G3 | 6 (23.1) | 7 (30.5) | |
| Venous invasion | |||
| v [−] | 22 (84.6) | 5 (21.7) | <0.001 |
| v [+] | 4 (15.4) | 18 (78.3) | |
| Growth pattern | |||
| Expansive (INFa) | 21 (80.8) | 11 (47.8) | 0.017 |
| Intermediate (INFb) | 5 (19.2) | 12 (52.2) | |
Figure 2.
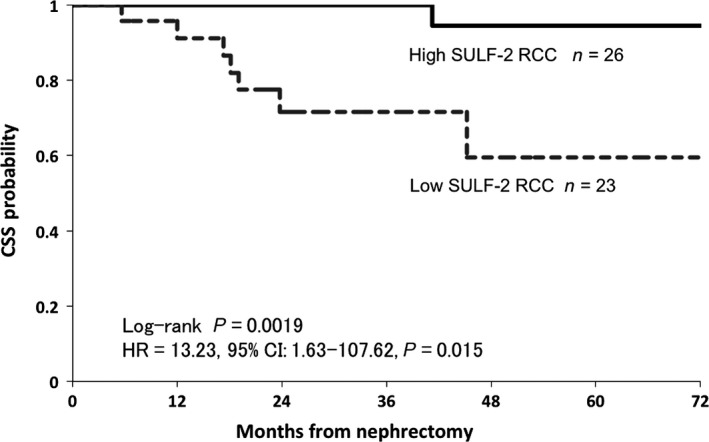
SULF‐2 expression level and cancer‐specific survival in clinical ccRCC cases. Kaplan–Meier curves for the cumulative cancer‐specific survival for ccRCC patients are shown. The survival rate in the high SULF‐2 group was significantly higher than that in the low SULF‐2 group (P = 0.0019).
SULF‐2 suppression and overexpression models in the renal cell carcinoma cell lines
The mRNA expression level of SULF‐2 was analyzed by RT‐PCR in three different RCC cell lines: Caki‐2, ACHN and 786‐O. The mRNA expression level of SULF‐2 was highest in the Caki‐2 cells and lowest in the 786‐O cells, with SULF‐2 expression in Caki‐2 cells being over 10 000 times higher than that in 786‐O cells, and SULF‐2 expression in ACHN cells over 100 times higher than that in 786‐O cells (Fig. S1a). Western blotting analysis for SULF‐2 using anti‐SULF‐2 (H2.1) antibody in these cell lines revealed protein bands of 132 and 72 kDa. The 132‐kDa species represents the precursors and the 72‐kDa species represents the products of the cleavage process.1, 20 SULF‐2 was detected in Caki‐2 and ACHN cells, but was almost undetectable in 786‐O cells (Fig. S1b). These results were in good agreement with the report that the protein expression level of SULF‐2 in 786‐O cells was very low.19 The Caki‐2 and ACHN cell lines were, therefore, regarded as high SULF‐2‐expressing cell lines and were used as a model system for analysis of the effects of SULF‐2 inhibition by transfection of SULF‐2 siRNA. The 786‐O cell line was regarded as a low SULF‐2‐expressing cell line and was used for the introduction of a SULF‐2 overexpression model by transfection of a plasmid vector encoding SULF‐2. Transfection of SULF‐2 siRNA into Caki‐2 and ACHN cells resulted in a 75–80% decrease in SULF‐2 mRNA compared to the control cells transfected with the NC siRNA. Transfection of the plasmid encoding SULF‐2 into 786‐O cells resulted in an approximately 2000‐fold increase in SULF‐2 expression compared to that in the control cells transfected with a negative control plasmid vector (Fig. S2).
High levels of SULF‐2 expression enhance Wnt signaling in renal cell carcinoma cell lines
We next analyzed whether the level of SULF‐2 expression affected Wnt signaling in RCC cell lines. Although Wnt‐induced β‐catenin was significantly increased 1.6‐fold (P = 0.002) and 2.4‐fold (P = 0.01) in Caki‐2 and ACHN cells transfected with NC siRNA, respectively, the increase was suppressed by SULF‐2 siRNA. Stimulation by Wnt in 786‐O cells transfected with the SULF‐2 plasmid vector significantly increased β‐catenin by 1.6‐fold (P = 0.001) (Fig. 3a,b).
Figure 3.
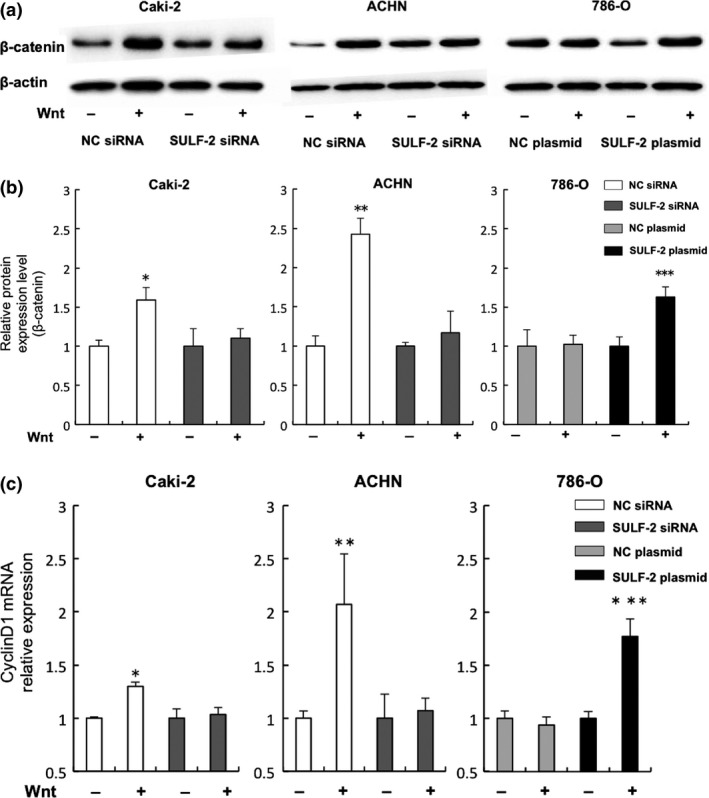
Analysis of the effect of cellular SULF‐2 level on the Wnt signaling pathway. Wnt3a signaling in the RCC cell lines was assayed by western blot analysis. Caki‐2 or ACHN cells were transfected with SULF‐2 siRNA or with a negative control (NC) siRNA. 786‐O cells were transfected with a SULF‐2 plasmid or an NC plasmid. (a, b) Activation of Wnt signaling following incubation with (+) or without (−) 100 ng/mL of Wnt3a was determined by β‐catenin expression level (a). Quantification of the relative β‐catenin levels of Wnt‐treated versus non‐treated cells (which were assigned a value of 1) is shown in (b). *P = 0.002 **P = 0.01 (compared with SULF‐2 siRNA‐transfected cells with Wnt stimulation). ***P = 0.001 (compared with NC plasmid‐transfected cells with Wnt stimulation). (c) The effect of treatment of the cells with (+) or without (−) 100 ng/mL Wnt3a on cyclin D1 mRNA expression was analyzed using quantitative RT‐PCR. The relative mRNA expression level of cyclinD1 of the RCC cell lines is shown. *P = 0.004, **P = 0.003 (compared with SULF‐2 siRNA‐transfected cells with Wnt stimulation) ***P < 0.001 (compared with NC plasmid‐transfected cells with Wnt stimulation).
Stimulation by Wnt in Caki‐2 and ACHN cells transfected with NC siRNA induced the increase of cyclin D1 mRNA expression 1.3‐fold (P = 0.004) and 2.0‐fold (P = 0.003), respectively, whereas almost no increase was observed in cells suppressed by SULF‐2 siRNA. Stimulation of 786‐O cells by Wnt led to a significant increase in cyclin D1, 1.8‐fold, in the SULF‐2 overexpressed cells (P < 0.001) (Fig. 3c). Wnt signaling was enhanced when the SULF‐2 level was high.
Low level of SULF‐2 expression correlates with the activation of VEGF or basic FGF signaling in renal cell carcinoma cell lines
The influence of SULF‐2 level on the VEGF and FGF pathways in the RCC cell lines was examined. When SULF‐2 was suppressed by siRNA, stimulation by VEGF induced significant increases in p‐ERK in Caki‐2 (2.0‐fold, P = 0.001) and ACHN (1.8‐fold, P < 0.001) cells. In 786‐O cells, VEGF did not induce an increase in p‐ERK when SULF‐2 was overexpressed by the plasmid vector, although a 1.6‐fold increase in p‐ERK was detected in the cells transfected with the NC plasmid vector (P < 0.001). Basic FGF treatment significantly increased p‐ERK, 2.0‐fold (P = 0.01) and 2.5‐fold (P = 0.01), in Caki‐2 and ACHN cells, respectively, when SULF‐2 was suppressed by siRNA. In 786‐O cells, the increase in p‐ERK by basic FGF stimulation was 2.5‐fold (P < 0.001) in the cells transfected with the NC plasmid vector, but no significant increase was observed when SULF‐2 was overexpressed (Fig. 4a–d). Both VEGF and FGF signaling pathways were enhanced when SULF‐2 expression was low.
Figure 4.
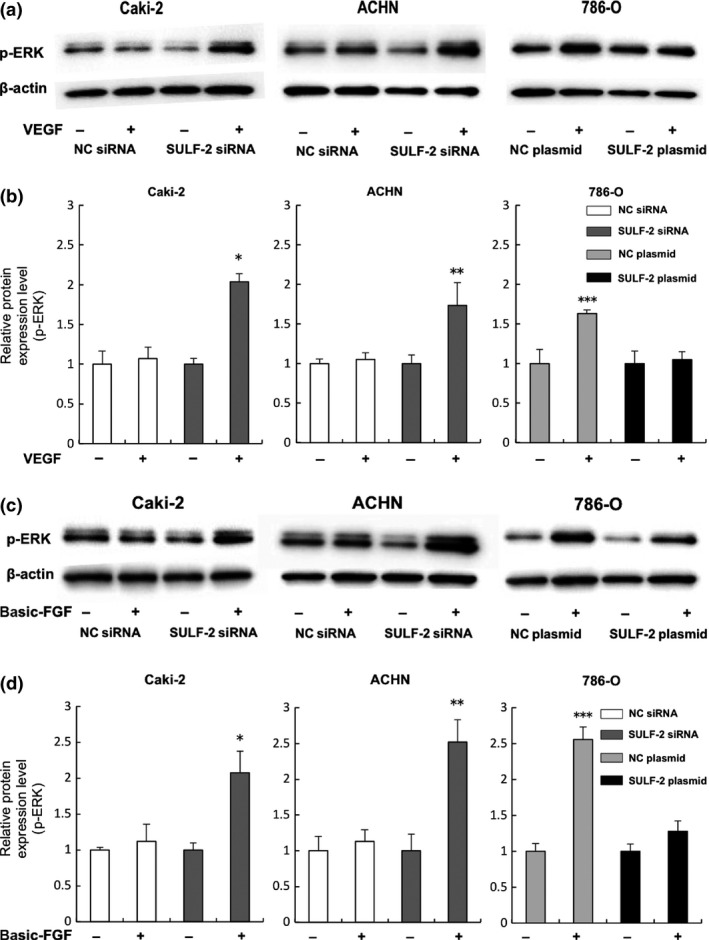
Analysis of the effect of cellular SULF‐2 levels on VEGF and FGF signaling pathways. VEGF (a, b) or basic FGF (c, d) signaling in the RCC cell lines was assayed by western blot analysis. Activation of VEGF (a, b) or basic FGF (c, d) signaling following incubation of the indicated cells with (+) or without (−) 50 ng/mL of VEGF or 50 ng/mL of basic FGF, respectively, was determined by p‐ERK expression level (a, c). Quantification of the relative p‐ERK levels of treated versus non‐treated cells (which were assigned a value of 1) is shown (b, d). For (b), *P = 0.001, **P < 0.001 (compared with NC siRNA‐transfected cells with VEGF stimulation), ***P < 0.001 (compared with SULF‐2 plasmid‐transfected cells with VEGF stimulation). For (d), *P = 0.01, **P = 0.01(compared with NC siRNA‐transfected cells with FGF stimulation), ***P < 0.001(compared with SULF‐2 plasmid‐transfected cells with FGF stimulation).
Effect of SULF‐2 on cell viability in vitro
The effect of the SULF‐2 expression on cell viability in the RCC cell lines was investigated using an MTT assay. No Wnt‐induced increase in cell viability was observed in SULF‐2‐suppressed Caki‐2 or ACHN cells, although significant increases were seen in the cells transfected with NC siRNA (P < 0.001, respectively). Wnt did not affect cell viability in 786‐O cells at any level of SULF‐2 expression. In contrast, VEGF‐induced cell viability was significantly increased in Caki‐2 and ACHN cells by 31% (P < 0.001) and 28% (P < 0.001), respectively, when SULF‐2 was suppressed by siRNA. In 786‐O, no significant increase was detected when SULF‐2 was overexpressed, even though VEGF significantly enhanced cell viability in the cells transfected with NC plasmid vector. Basic FGF did not induce a significant increase in cell viability in any of the cell lines (Fig. 5a–c).
Figure 5.
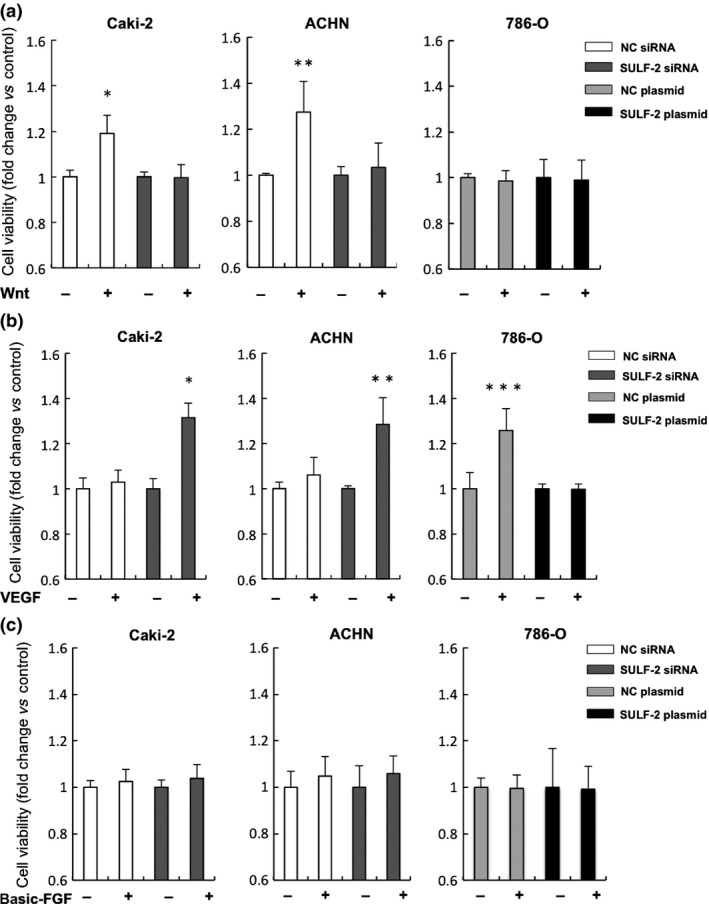
Analysis of the effect of cellular SULF‐2 levels on Wnt, VEGF or FGF regulation of cell viability. The effect of cellular SULF‐2 levels on Wnt (a), VEGF (b) or basic FGF (c) regulation of cell viability was determined by MTT assay. Fold change in cell viability compared to non‐treated control is shown. For (a), *P < 0.001, **P < 0.001 (compared with SULF‐2 siRNA‐transfected cells with Wnt stimulation); for (b), *P < 0.001, **P < 0.001 (compared with NC siRNA‐transfected cells with VEGF stimulation), ***P < 0.001(compared with SULF‐2 plasmid‐transfected cells with VEGF stimulation).
Low levels of SULF‐2 expression enhance cell invasion in vitro
The effect of SULF‐2 level on cell invasion was examined. Cells invading the BD Matrigel Basement Membrane Matrix are shown in Figure 6(a). No significant differences in the Wnt‐induced increase in cell invasion in relation to differences in SULF‐2 level were observed. Stimulation by VEGF increased the invasion of Caki‐2 and ACHN cells 3.6‐fold (P < 0.001) and three‐fold (P = 0.01), respectively, when SULF‐2 was suppressed. In 786‐O, the increase in invasion was suppressed to insignificant levels in SULF‐2‐overexpressed cells. Stimulation of Caki‐2 and ACHN cells by basic FGF increased the cell invasion eight‐fold (P = 0.02) and 15‐fold (P = 0.008), respectively, when SULF‐2 was suppressed. In 786‐O cells, the increase in cell invasion induced by basic FGF was suppressed when SULF‐2 was overexpressed (Fig. 6b–d). Cell invasion induced by VEGF or basic FGF stimulation was elevated when the level of SULF‐2 was low.
Figure 6.
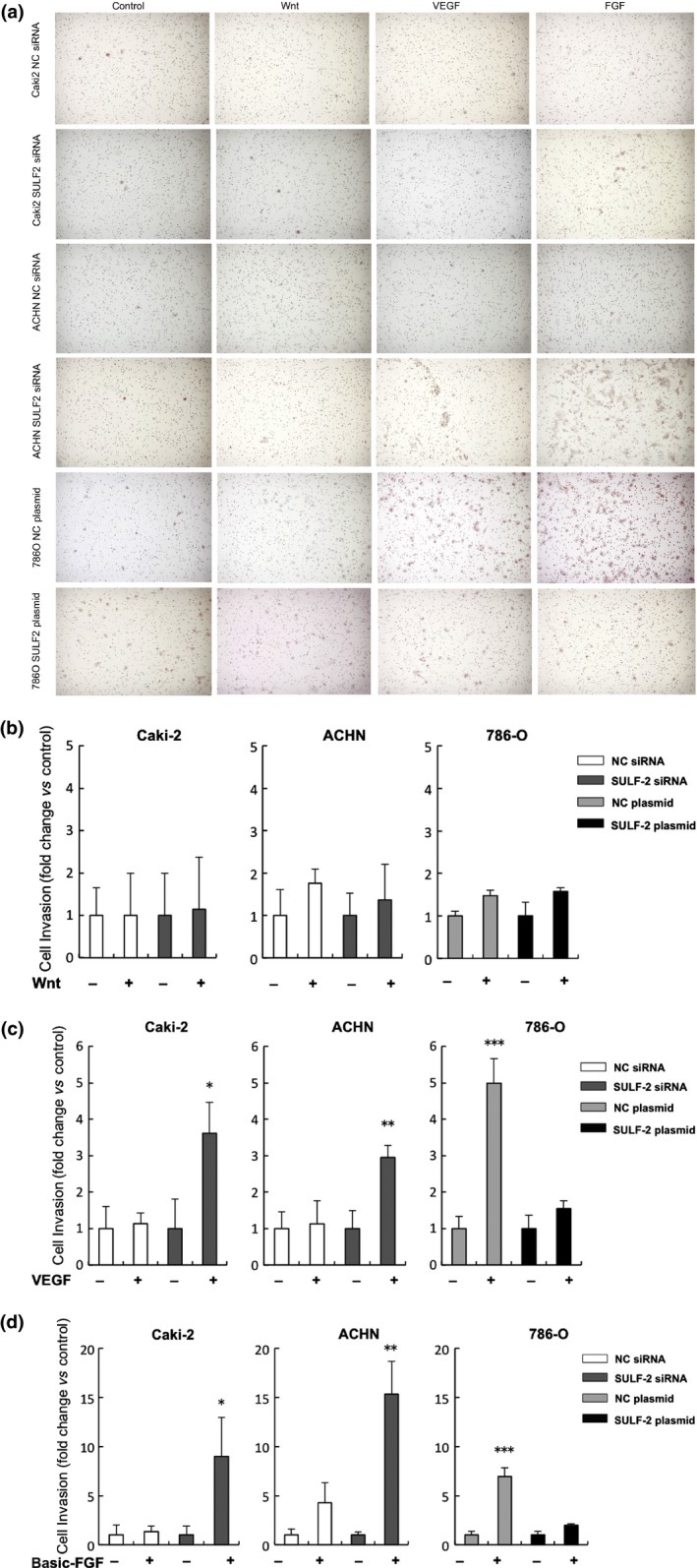
Analysis of the effect of cellular SULF‐2 levels on Wnt, VEGF or FGF regulation of cell invasion. Cells invading the BD Matrigel Basement Membrane Matrix are shown (a). The effect of cellular SULF‐2 level on Wnt (b), VEGF (c) or basic FGF (d) regulation of cell invasion was determined using a cell invasion assay. Fold change in cell invasion compared to non‐treated control is shown. For (C), *P < 0.001, **P = 0.01 (compared with NC siRNA‐transfected cells with VEGF stimulation), ***P = 0.001 (compared with SULF‐2 plasmid‐transfected cells with VEGF stimulation) and for (d), *P = 0.02, **P = 0.008 (compared with NC siRNA‐transfected cells with FGF stimulation), ***P < 0.001(compared with SULF‐2 plasmid‐transfected cells with FGF stimulation).
Discussion
We have demonstrated that among clinical RCC specimens, some showed higher levels of SULF‐2 expression and others lower levels of SULF‐2 expression compared to the adjacent normal kidney tissue. From our results, high SULF‐2‐expressing RCC were generally associated with favorable prognoses, an indication of nonaggressive cancers. In contrast, low SULF‐2‐expressing RCC were associated with worse prognoses. It was also demonstrated that there were similarly high SULF‐2‐expressing cells and low SULF‐2‐expressing cells among the RCC cell lines tested. Caki‐2 and ACHN cells showed higher levels of SULF‐2 expression and 786‐O cells showed a lower level of SULF‐2 expression. The manner of progression as well as the prognosis of RCC might differ with differences in the expression level of SULF‐2 as SULF‐2 increases the binding affinity of Wnt to its specific receptor and decreases the binding affinity of VEGF/FGF to their specific receptors via its desulfation of HSPG, which acts as a co‐receptor for these ligands.7, 8, 9, 10 Indeed, in our study, NC siRNA‐transfected Caki‐2 and ACHN cells did not show any increase in p‐ERK on stimulation by VEGF/FGF, although 786‐O cells transfected with NC plasmid showed an increase in p‐ERK. In contrast, enhancement of the Wnt pathway was observed in NC siRNA‐transfected Caki‐2 and ACHN cells but not in NC plasmid‐transfected 786‐O cells. The signal transduction of these ligands could be affected by the baseline expression level of SULF‐2 in each cell line.
Our results showed that SULF‐2 might positively regulate Wnt signaling and negatively regulate VEGF/FGF signaling in RCC cells, as reported in other cancers.12, 13, 14 The enhancement of Wnt signaling associated with high levels of SULF‐2 expression increased cell proliferation but did not affect cell invasion in the in vitro assay employed in the present study. In contrast, the enhancement of VEGF/FGF pathways associated with low levels of SULF‐2 expression increased not only cell proliferation but also cell invasion.
It has been reported that Wnt plays an oncogenic role in RCC.21 Higher levels of SULF‐2 expression might be correlated with enhanced carcinogenesis of RCC, as reported in several other cancers.12, 13, 22 Approximately 85% of high SULF‐2‐expressing RCC were diagnosed as T1 tumors or clinically early stage cancers without pathologically invasive properties in our study. If high levels of SULF‐2 expression are involved in the carcinogenesis of RCC, it seems reasonable that early stage RCC would generally display high SULF‐2 expression.
Renal cell carcinomas generally have receptors for VEGF23, 24, 25 and FGF,26, 27 and the expression status of VEGF in RCC cell lines varies. The expression level of VEGF is 4–5‐fold higher in 786‐O cells than in Caki‐2 and ACHN cells.28 RCC cells with low SULF‐2 expression, such as 786‐O cells, might have acquired their marked invasiveness via the enhanced VEGF pathway. Indeed, as we observed in our current study, approximately 80% of low SULF‐2‐expressing RCC were clinically advanced cancers with venous invasion. One plausible explanation is that a reduction in SULF‐2 expression might contribute to an increase in the potential for metastasis and tumor growth in RCC via VEGF/FGF signaling. The results of experiments with the cell lines appear to corroborate the clinical data.
SULF‐2 mRNA expression in normal kidney tissues in clinical 3–4 stage cases or cases with positive venous invasion seems higher than that in stage 1–2 cases and cases with negative venous invasion. One explanation for this is that a new carcinoma might be developing in the neighboring normal renal tubules in these cases with advanced or aggressive cancers, if high SULF‐2 expression is involved in carcinogenesis. Another possibility is that SULF‐2 mRNA in normal kidney tissues may be overexpressed as a biological reaction against cancers in advanced cases. For example, in normal kidney tissues, reactions such as inflammation directly affected by the growing cancer might be involved in the high level of SULF‐2 expression. Indeed, SULF‐2 overexpression is seen in osteoarthritic cartilage, perhaps to maintain cartilage homeostasis.29
The precise mechanisms resulting in the upregulation and downregulation of SULF‐2 have not been elucidated in the present study. Several possible mechanisms of SULF‐2 regulation can be provided. For example, transforming growth factor‐β1 (TGF‐β1) was reported to be an inducer of SULF‐2,30 and promoter hypermethylation was suggested as one of the causes of SULF‐2 silencing.31 VHL mutation and hypoxia could also negatively control the level of SULF‐2 expression.19 Therefore, VHL mutation and hypoxia might be important factors. Although VHL is well known as a tumor suppressor gene in RCC, VHL mutation is, in fact, found in only 29–51% of RCC patients.32 VHL positively regulates SULF‐2 and its mutation or hypoxia could lead to the suppression of SULF‐2 via the hypoxia‐HIF‐pathway.19 Furthermore, the status of the three RCC cell lines employed in our study differs in terms of VHL mutation. Caki‐2 and ACHN cells have wild type VHL, whereas 786‐O cells have a mutant VHL encoding a deficient VHL protein.28, 33 Our study demonstrated that SULF‐2 expression level was significantly higher in Caki‐2 and ACHN cells than in 786‐O cells. It seems reasonable to consider that differences in the expression levels of SULF‐2 might be caused by VHL mutation in RCC.
This study suggests that RCC may exhibit distinct behaviors dependent on differences in SULF‐2 expression levels. When the SULF‐2 expression level is low, even if the tumor is resected at an early stage, the cancer may have already metastasized and show a greater tendency to relapse near the local lesion or at remote sites in the future. A lack of SULF‐2 might be a good indicator of aggressiveness in RCC. Moreover, it has been reported that tyrosine kinases might be involved not only in VEGF or FGF signal transduction but also in Wnt signal transduction.34, 35 It would be interesting to evaluate the outcome of treatments for the RCC patients targeting tyrosine kinases in relation to differences in the SULF‐2 expression level as the intracellular signal transduction pathways of Wnt, VEGF and FGF may share some common tyrosine‐kinases.
In conclusion, this study demonstrates that RCC with higher SULF‐2 expression appear to have lower invasiveness whereas RCC with lower SULF‐2 expression show a higher potential for cell invasion and proliferation, leading to a poorer prognosis for the RCC patients.
Disclosure Statement
The authors have no conflict of interest to declare.
Supporting information
Fig. S1. SULF‐2 expression levels in renal cell carcinomas (RCC) cell lines.
Fig. S2. Suppression or overexpression of SULF‐2 by siRNA or plasmid in renal cell carcinomas (RCC) cell lines.
Acknowledgments
We thank Masaki Yanagishita for helpful discussion about HSPG and SULF as well as for his critical reading of the manuscript.
Cancer Sci 107 (2016) 1632–1641
Funding Information
No sources of funding were declared in this study.
References
- 1. Morimoto‐Tomita M, Uchimura K, Werb Z, Hemmerich S, Rosen SD. Cloning and characterization of two extracellular heparin‐degrading endosulfatases in mice and humans. J Biol Chem 2002; 277: 49175–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sarrazin S, Lamanna WC, Esko JD. Heparan sulfate proteoglycans. Cold Spring Harb Perspect Biol 2011; 3: doi: 10.1101/cshperspect.a004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Perrimon N, Bernfield M. Specificities of heparan sulphate proteoglycans in developmental processes. Nature 2000; 404: 725–8. [DOI] [PubMed] [Google Scholar]
- 4. Lindahl U, Kusche‐Gullberg M, Kjellen L. Regulated diversity of heparan sulfate. J Biol Chem 1998; 273: 24979–82. [DOI] [PubMed] [Google Scholar]
- 5. Lamanna WC, Kalus I, Padva M, Baldwin RJ, Merry CL, Dierks T. The heparanome–the enigma of encoding and decoding heparan sulfate sulfation. J Biotechnol 2007; 129: 290–307. [DOI] [PubMed] [Google Scholar]
- 6. Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine‐tune mammalian physiology. Nature 2007; 446: 1030–7. [DOI] [PubMed] [Google Scholar]
- 7. Dhoot GK, Gustafsson MK, Ai X, Sun W, Standiford DM, Emerson CP Jr. Regulation of Wnt signaling and embryo patterning by an extracellular sulfatase. Science 2001; 293: 1663–6. [DOI] [PubMed] [Google Scholar]
- 8. Mitsi M, Hong Z, Costello CE, Nugent MA. Heparin‐mediated conformational changes in fibronectin expose vascular endothelial growth factor binding sites. Biochemistry 2006; 45: 10319–28. [DOI] [PubMed] [Google Scholar]
- 9. Uchimura K, Morimoto‐Tomita M, Bistrup A et al HSulf‐2, an extracellular endoglucosamine‐6‐sulfatase, selectively mobilizes heparin‐bound growth factors and chemokines: effects on VEGF, FGF‐1, and SDF‐1. BMC Biochem 2006; 7: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lai J, Chien J, Staub J et al Loss of HSulf‐1 up‐regulates heparin‐binding growth factor signaling in cancer. J Biol Chem 2003; 278: 23107–17. [DOI] [PubMed] [Google Scholar]
- 11. Bret C, Moreaux J, Schved JF, Hose D, Klein B. SULFs in human neoplasia: implication as progression and prognosis factors. J Transl Med 2011; 9: 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lemjabbar‐Alaoui H, van Zante A, Singer MS et al Sulf‐2, a heparan sulfate endosulfatase, promotes human lung carcinogenesis. Oncogene 2010; 29: 635–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nawroth R, van Zante A, Cervantes S, McManus M, Hebrok M, Rosen SD. Extracellular sulfatases, elements of the Wnt signaling pathway, positively regulate growth and tumorigenicity of human pancreatic cancer cells. PLoS ONE 2007; 2: e392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Peterson SM, Iskenderian A, Cook L et al Human Sulfatase 2 inhibits in vivo tumor growth of MDA‐MB‐231 human breast cancer xenografts. BMC Cancer 2010; 10: 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dai Y, Yang Y, MacLeod V et al HSulf‐1 and HSulf‐2 are potent inhibitors of myeloma tumor growth in vivo. J Biol Chem 2005; 280: 40066–73. [DOI] [PubMed] [Google Scholar]
- 16. Yang JD, Sun Z, Hu C et al Sulfatase 1 and sulfatase 2 in hepatocellular carcinoma: associated signaling pathways, tumor phenotypes, and survival. Genes Chromosom Cancer 2011; 50: 122–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014; 64: 9–29. [DOI] [PubMed] [Google Scholar]
- 18. Cohen HT, McGovern FJ. Renal‐cell carcinoma. N Engl J Med 2005; 353: 2477–90. [DOI] [PubMed] [Google Scholar]
- 19. Khurana A, Tun HW, Marlow L, Copland JA, Dredge K, Shridhar V. Hypoxia negatively regulates heparan sulfatase 2 expression in renal cancer cell lines. Mol Carcinog 2012; 51: 565–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Morimoto‐Tomita M, Uchimura K, Bistrup A et al Sulf‐2, a proangiogenic heparan sulfate endosulfatase, is upregulated in breast cancer. Neoplasia 2005; 7: 1001–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hsu RJ, Ho JY, Cha TL et al WNT10A plays an oncogenic role in renal cell carcinoma by activating WNT/beta‐catenin pathway. PLoS ONE 2012; 7: e47649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Najdi R, Holcombe RF, Waterman ML. Wnt signaling and colon carcinogenesis: beyond APC. J Carcinog 2011; 10: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tsuchiya N, Sato K, Akao T et al Quantitative analysis of gene expressions of vascular endothelial growth factor‐related factors and their receptors in renal cell carcinoma. Tohoku J Exp Med 2001; 195: 101–13. [DOI] [PubMed] [Google Scholar]
- 24. Ljungberg BJ, Jacobsen J, Rudolfsson SH, Lindh G, Grankvist K, Rasmuson T. Different vascular endothelial growth factor (VEGF), VEGF‐receptor 1 and ‐2 mRNA expression profiles between clear cell and papillary renal cell carcinoma. BJU Int 2006; 98: 661–7. [DOI] [PubMed] [Google Scholar]
- 25. Kluger HM, Siddiqui SF, Angeletti C et al Classification of renal cell carcinoma based on expression of VEGF and VEGF receptors in both tumor cells and endothelial cells. Lab Invest 2008; 88: 962–72. [DOI] [PubMed] [Google Scholar]
- 26. Tsimafeyeu I, Demidov L, Stepanova E, Wynn N, Ta H. Overexpression of fibroblast growth factor receptors FGFR1 and FGFR2 in renal cell carcinoma. Scand J Urol Nephrol 2011; 45: 190–5. [DOI] [PubMed] [Google Scholar]
- 27. Cirovic S, Vjestica J, Mueller CA et al NCAM and FGFR1 coexpression and colocalization in renal tumors. Int J Clin Exp Pathol 2014; 7: 1402–14. [PMC free article] [PubMed] [Google Scholar]
- 28. Shinojima T, Oya M, Takayanagi A, Mizuno R, Shimizu N, Murai M. Renal cancer cells lacking hypoxia inducible factor (HIF)‐1alpha expression maintain vascular endothelial growth factor expression through HIF‐2alpha. Carcinogenesis 2007; 28: 529–36. [DOI] [PubMed] [Google Scholar]
- 29. Otsuki S, Hanson SR, Miyaki S et al Extracellular sulfatases support cartilage homeostasis by regulating BMP and FGF signaling pathways. Proc Natl Acad Sci USA 2010; 107: 10202–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yue X, Lu J, Auduong L, Sides MD, Lasky JA. Overexpression of Sulf2 in idiopathic pulmonary fibrosis. Glycobiology 2013; 23: 709–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang L, Xie L, Wang J, Shen J, Liu B. Correlation between the methylation of SULF2 and WRN promoter and the irinotecan chemosensitivity in gastric cancer. BMC Gastroenterol 2013; 13: 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cowey CL, Rathmell WK. VHL gene mutations in renal cell carcinoma: role as a biomarker of disease outcome and drug efficacy. Curr Oncol Rep 2009; 11: 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jiang Y, Zhang W, Kondo K et al Gene expression profiling in a renal cell carcinoma cell line: dissecting VHL and hypoxia‐dependent pathways. Mol Cancer Res 2003; 1: 453–62. [PubMed] [Google Scholar]
- 34. Lu W, Yamamoto V, Ortega B, Baltimore D. Mammalian Ryk is a Wnt coreceptor required for stimulation of neurite outgrowth. Cell 2004; 119: 97–108. [DOI] [PubMed] [Google Scholar]
- 35. Krejci P, Aklian A, Kaucka M et al Receptor tyrosine kinases activate canonical WNT/beta‐catenin signaling via MAP kinase/LRP6 pathway and direct beta‐catenin phosphorylation. PLoS ONE 2012; 7: e35826. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. SULF‐2 expression levels in renal cell carcinomas (RCC) cell lines.
Fig. S2. Suppression or overexpression of SULF‐2 by siRNA or plasmid in renal cell carcinomas (RCC) cell lines.