

Abstract
In this article, we review how mitochondrial Ca2+ transport (mitochondrial Ca2+ uptake and Na+/Ca2+ exchange) is involved in T cell biology, including activation and differentiation through shaping cellular Ca2+ signals. Based on recent observations, we propose that the Ca2+ crosstalk between mitochondria, endoplasmic reticulum and cytoplasm may form a proportional–integral–derivative (PID) controller. This PID mechanism (which is well known in engineering) could be responsible for computing cellular decisions. In addition, we point out the importance of analogue and digital signal processing in T cell life and implication of mitochondrial Ca2+ transport in this process.
Keywords: mitochondria, Ca2+ signalling, PID controller, digital and analogue signals, T lymphocytes, systems biology
1. Introduction
Activation of T lymphocytes is a complex process associated with remodelling of signalling network and metabolism. Initiated by the triggering of T cell receptor (TCR) at the plasma membrane, activation propagates into the nucleus, resulting in transcriptional changes. Obviously, the amplitude and duration of T cell activation should be tightly controlled owing to undesirable outcomes of under- or overactivation, such as immunodeficiency or autoimmunity, respectively. Furthermore, T lymphocytes should respond adequately to the type of pathogen (i.e. viral, bacterial, tumour) and to its dose. Finally, T cell memory serves to fight the repeated pathogen encounters and needs remodelling of metabolic processes to meet re-activation requirements.
Studies of the last 10–15 years shed light on metabolic reprogramming in T cells. Currently, it is well accepted that resting T cells mostly use oxidative phosphorylation and beta-oxidation to retrieve energy from nutrients. However, upon activation, T cells switch from respiration to more active glucose and glutamine uptake, as well as aerobic glycolysis [1,2]. The latter serves for active production of monomers required for nucleic acid synthesis and is in turn necessary for cell proliferation and expansion of specific T cell clones [3,4]. This effect is reminiscent of the well-known Warburg effect [2].
Recent data in mitochondrial biology allow us to consider mitochondria simply as cellular energetic stations, but also as important signalling hubs. Indeed, first results in this field came from findings that mitochondria produce reactive oxygen species (ROS) and communicate with the nucleus via oxidative modification of proteins including transcription factors [5–8]. Another important step forward was made after the identification of the molecular nature of the mitochondrial Ca2+ uptake/uniport (mtCU). Although the existence of a process of Ca2+ inward transport (from cytosol into mitochondrial matrix) was known for at least 50 years, the pore-forming and the Ca2+ sensor regulatory subunits of the mtCU, mitochondrial Ca2+ uniporter (MCUa) and mitochondrial Ca2+ uptake 1 (MICU1), respectively, have been described for the first time only a few years ago [9–12]. Currently, the role of several more proteins (such as MICU2–3, MCUb, EMRE, MCUR1, UCP2) involved in the control of mtCU has been described [13–17]. Finally, NCLX1 (Na+/Ca2+ exchanger 1) was identified as a protein molecule responsible for Na+/Ca2+ exchange, a process that balances mtCU-induced Ca2+ accumulation and prevents matrix Ca2+ overload, which could potentially lead to apoptosis [18,19]. Although the MCU and NCLX1 are the prominent and best identified pathways for Ca2+ influx and efflux in and out of mitochondria, other pathways have been reported to play a role in these exchange processes [20].
Further studies in mitochondrial biology progressed towards the understanding of interorganelle interactions, in particular Ca2+ transfer between the endoplasmic reticulum (ER) and mitochondria that is mediated by molecular bridges (figure 1). Major players that form ER/mitochondria tethers are Grp75/HSPA9, inositol trisphosphate receptor (IP3R), mitofusin 2 (MFN2) from the outside surface of the ER and the voltage-dependent anion channel 1 (VDAC1), and mitofusins MFN1 or MFN2 from outside mitochondrial membrane (OMM) [21]. The presence of physical contacts between the ER and mitochondria explains the paradox of mitochondrial Ca2+ accumulation: mtCU has an intrinsic low affinity (i.e. mitochondria import Ca2+ only after significant Ca2+ elevations around 3–10 µM), whereas most of global cellular Ca2+ spikes do not reach this level [15,22–24]. This apparent discrepancy was resolved by showing that protein complexes tether ER to mitochondria, thus close positioning these organelles and creating local Ca2+ domains with ion concentration of 10 µm and above [25–27]. Thus, the ER–mitochondria interface represents a local signalling platform where cytosolic Ca2+ elevations are transmitted to mitochondria [28,29] (figure 1).
Figure 1.
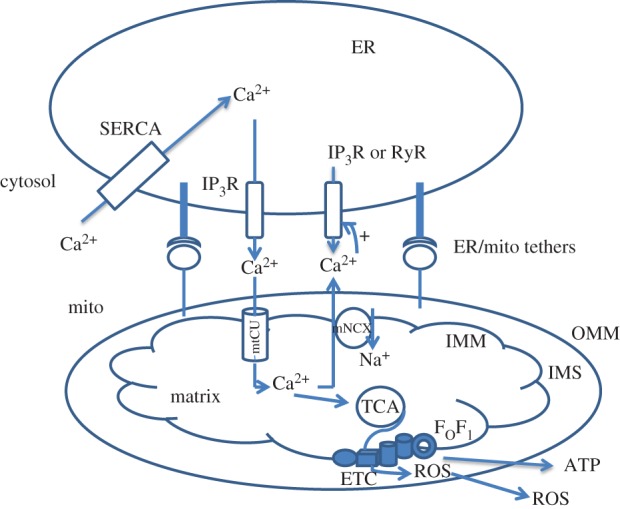
General scheme of Ca2+ exchange between the endoplasmic reticulum and mitochondria. Multiple proteins form tethers controlling the ER/mitochondria junction and maintaining ER/mitochondria Ca2+ crosstalk. Ca2+ release induced by the opening of RyRs or IP3Rs leads to elevation of Ca2+ at the ER/mitochondria interface. When Ca2+ reaches a concentration of 3–10 µM (threshold for mtCU), the MCU opens and allows Ca2+ ions to be imported into the matrix where they stimulate the tricarboxylic cycle (TCA). As a consequence, the electron transport chain (ETC) starts producing ROS, which are utilized by antioxidant systems. At the same time, Ca2+ ions are directed from mitochondria into the cytosol by the mitochondrial Na+/Ca2+ exchanger (mNCX) at the expense of Na+ import. This Ca2+ activates IP3Rs and RyRs in the ER/mitochondria space, thereby leading to Ca2+ release from the ER. OMM, outer mitochondrial membrane; IMM, inner mitochondrial membrane; IMS, intermembrane space.
Mitochondria are able to accumulate large amounts of Ca2+ ions, but the increase in free Ca2+ induced by cytosolic Ca2+ transients is limited by buffering by anions present in mitochondrial matrix, such as phosphate (Pi), acetate, bicarbonate, etc. [30,31]. Mitochondrial Pi buffers sink extra amounts of Ca2+ until reaching the buffer capacity; after this point, Ca2+ phosphates trigger mPTP opening [31,32]. mtCU and Pi buffer work cooperatively to respond to cytosolic Ca2+ increases. If rapid and submicromolar concentrations of cytosolic Ca2+ elevations induce fast accumulation of free matrix Ca2+ (mCU1 mode), which upregulates electron-transport chain (ETC; see below), then sustained mtCU activation (mCU2) leads to the expansion of the compartment of matrix Ca2+ sequestered by phosphate buffer [31].
How might all these molecular mechanisms be related to T cell functions, activation and fate? The most obvious hypothesis is that mitochondria participate in the supply of ATP in activated T cells, and in turn in ATP-dependent transport processes in immune synapse, where the majority of signalling events are triggered [33,34]. Second, mitochondria may be involved through the opening of the mPTP in its high-conductance mode. Mitochondria are indeed involved in apoptosis induction during activation-induced cell death caused by TCR stimulation and aimed at restricting further clonal expansion to prevent autoimmune disorders [35,36]. Finally, mitochondria produce ROS, which accompany upregulation of nuclear factor kappa B (NFκB) and nuclear factor of activated T cells (NFAT), two master regulators of TCR-stimulated transcription changes [37–41]. These three processes are stimulated by an increase in mitochondrial Ca2+, as this ion stimulates the tricarbon acid (TCA) cycle in the mitochondrial matrix, leading to an increase in proton pumping, ATP synthesis and ROS production [42,43] (figure 1). It is noteworthy that the mtCU inhibitor, Ru360, prevents ROS formation in activated T lymphocytes, thus showing that the mtCU is the mechanistic link between TCR-induced Ca2+ elevation and ROS production for mitochondria–nucleus communication [40]. In this review, we propose that mitochondrial Ca2+ transport and mitochondria could also play a key role in the central regulation of T cell fate. In addition, we try to understand why activated T lymphocytes use mitochondria for cell decisions, in particular from the perspective of Ca2+ transport.
2. The importance of mitochondrial Ca2+ for T cell life and fate
The importance of the mtCU in physiology and pathology of T cells is demonstrated by several different examples. Components of Ca2+ signalling machinery in T cells, ryanodine receptors (RyR), IP3R and Orai1 channel subunits mediating store-operated calcium entry (SOCE), are coordinately associated with MCU expression; downregulation of these proteins in activated T cells leads to a compromised upregulation of the MCU mediated by the CREB transcriptional factor [44]. The significance of mitochondria in the control of cytosolic Ca2+ turnover has been shown for the Jurkat T cell line: dissipation of mitochondrial membrane potential (ΔΨm), the driving force for mtCU, causes early Ca2+ release-activated channel (CRAC) inactivation (discussed below), Ca2+ signal reduction and decreased nuclear import of NFAT [45]. Apparently, changes in SOCE activation via elevated mtCU mechanism may explain the fact that T lymphocytes from systemic lupus erythematosus display sustained increased Ca2+ response after their activation, which is proposed to be triggered by upregulated nitric oxide-dependent mitochondrial biogenesis [46,47]. Protein p13 of human T cell leukaemia virus type 1 (HTLV-1) modulates ΔΨm, thus limiting Ca2+ uptake and affecting T cell activation and death [48]. Further, we showed that in CD4+ T cells lacking mitochondrial tumour suppressor Fus1/Tusc2 with a potential Ca2+-binding domain (also discussed below), upregulation of NFAT- and NFκB-driven genes after activation was diminished [49]. An interesting role of the mtCU-mediated ROS production in negative feedback regulatory loop has been described in T cells: mitoROS produced by elevations in cytosolic Ca2+ stimulate protein kinase D, which in turn activates DRAK2, thus negatively regulating the influx of external Ca2+ [50]. Considering the ER/mitochondria Ca2+ crosstalk, new linkers with profound effect on T cell responses have been identified. In T cells, transglutaminase 2 (TG2) binds RAP1, the guanine nucleotides exchange factor of small GTPases, which enhances ER Ca2+ release and induces mitochondrial Ca2+ accumulation [51]. The absence of TG2 led to a diminished mouse CD8+ T cell activation and memory cell formation in vivo [52]; in Jurkat cells, TG2 overexpression leads to apoptosis in vitro [51]. Another protein, Tespa1, is localized closed to ER/mitochondria contacts, and directly interacts with Grp75 and IP3R [53]. Mitochondrial GTPase of the immune-associated nucleotide-binding protein 5 (GIMAP5) links mitochondria with microtubule cytoskeleton, helping them to localize at sites with high Ca2+ concentration and to maintain membrane Ca2+ currents via SOCE, thus favouring T cell survival [54]. Finally, mitochondrial Ca2+ accumulation triggers ATP synthesis and secretion via panx1 transporter in the activated Jurkat and human CD4+ T cells, whereas secreted ATP activates P2 purinergic receptors involved in the maintenance of intracellular Ca2+ elevation [55]. These examples are convincing about the diversity of roles of mitochondrial Ca2+ in T cell life and fate.
However, the significance of mitochondria in T cell activation and homeostasis will not be fully appreciated without quoting mitochondrial dynamics, a process of mitochondrial remodelling via fusion, fission, autophagy and movement [56]. These processes are tightly controlled by GTPases: mitofusins 1 and 2 and OPA1 for fusion, DRP1 and its receptor Fis1 for fission, and finally Miro 1 and 2 for movement [57–59]. Ca2+/calmodulin (CaM) kinase I alpha phosphorylates DRP1, leading to its binding with Fis1 and mitochondrial fission [60]. Other Ca2+-dependent proteins, Miro 1 and 2 possessing Ca2+-binding domains called EF-hands, promote mitochondrial fragmentation after interaction with Ca2+ ions [61]. In T cells, TCR triggering is associated with translocation of mitochondria to the immune synapse controlled by DRP1, which positions fragmented mitochondria in close proximity to the peripheral supramolecular activation cluster (pSMAC) [62]. Upon stimulation and supplying the immune synapse with ATP, CD3 molecules providing TCR proximal signalling move from pSMAC towards central SMAC and become internalized. In the absence of DRP1, CD3 molecules remain in the immune synapse and continue sending signals inside cells, thus increasing TCR response strength (e.g. IL-2 synthesis) [33]. It is noteworthy that intensity of TCR-triggered signalling defines Th polarization with mostly Th1 differentiation upon strong stimulation [63]. Obviously, the ability of mitochondria to take up Ca2+ should affect the processes of mitochondrial dynamics similar to other Ca2+-dependent processes such as activation of PKB/Akt [64] and NADPH oxidase [65].
Interesting connections between mitochondrial dynamics, respiration and metabolic reprograming have been recently described for T cells facing the choice between effector (Te) and memory (Tm) phenotypes. In particular, it was demonstrated that downregulation of DRP1 function leads to mitochondrial fusion-favouring activation of respiration over aerobic glycolysis in Tm cells, whereas the opposite effect was observed in Te lymphocytes [66]. It is noteworthy that DRP1 is activated by phosphorylation while its inhibition is associated with dephosphorylation by Ca2+-dependent phosphatase calcineurin (CaN) [67]. In this respect, modulation of Ca2+ signals including mtCU would coordinate mitochondrial dynamics with metabolic demands and execute metabolic reprogramming.
Th polarization is another process that correlates with Ca2+ dynamics in T cells after their activation. Upon identical stimulation, Th1 lymphocytes display higher mtCU activity compared with Th2 [68]. Accordingly, Th2 cells possess more efficient mechanisms for cytosolic Ca2+ clearance [69]. Finally, differences between Th1, Th2 and Th17 in terms of Ca2+ dynamics have been found. Th1 cells display high-amplitude elevations and multiple oscillations after TCR activation, whereas Th2 cells exhibit only a few post-stimulation oscillations and fast recovery to baseline, albeit a significant initial rise in Ca2+ level; Th17 lymphocytes show an intermediate pattern with Ca2+ response amplitude higher than Th2 but lower than Th1 and Th1-type oscillations [70]. During IL-6-driven Th2 differentiation, increased mitochondrial Ca2+ and NCX are required to sustain late NFAT accumulation during activation of CD4+ T cells (see below for more details) [71].
3. T cell Ca2+ dynamics
Obviously, the physiological and pathological function of mitochondrial Ca2+ transport should be tightly associated with its biochemical and biophysical properties. As mentioned above, the mtCU has a rather low affinity for Ca2+ ions, thus setting a high threshold for Ca2+ signals in mitochondria [15,22,24,36]. In this way, subthreshold Ca2+ signals are not transmitted to—nor affected by—mitochondria [72] (figure 2a). From a molecular perspective, this threshold is related to the expression of the MICU1 protein as in its absence mitochondria accumulate Ca2+ already at submicromolar content [73–75]. Further, MICU1 amplifies and accelerates Ca2+ uptake at higher Ca2+ concentration by preventing MCU self-inhibition [74] (figure 2b). De la Fuente et al. [74] conclude that the ability of MICU1 to lag Ca2+ response in mitochondria in time-consuming mode may result in their unresponsiveness to small amplitude and fast Ca2+ changes; downregulation of MICU1 would ‘give a green light’ for Ca2+ signals with these characteristics to be transmitted to mitochondria [11,74]. Because of this regulation of mtCU by MICU1, Ca2+ dependence of mtCU is biphasic: Ca2+ release from ER via IP3R triggers mtCU activation, whereas sustained cytosolic Ca2+ response suppresses mtCU [23]. Activation occurs on a timescale of approximately 10 s, whereas inactivation takes several minutes to establish [74]. Another player setting the mtCU threshold was identified in our study: a mitochondrial resident tumour suppressor Fus1/Tusc2 possessing Ca2+-binding EF-hand motif similar to MICU1 protein [49,76]. In the absence of Fus1, lymphocytes and immortalized epithelial cells demonstrated 1.5 to 2 times greater levels of mitochondrial Ca2+ levels than wild-type cells [49] (figure 2b).
Figure 2.

Mitochondrial decoding of cytosolic Ca2+ signals. (a) Elevations of cytosolic Ca2+ with low amplitude and short duration will not produce a significant rise in mitochondrial Ca2+, whereas Ca2+ signals with sufficient amplitude and duration will increase the Ca2+ content of mitochondria. (b) Binding of calcium ions with the Ca2+-binding protein MICU1 (and possibly with the tumour suppressor Fus1) promotes the opening of the Ca2+-permeable channel MCU (pore component of mtCU mechanism) leading to the influx of Ca2+ ions into mitochondria (right). At steady state, MICU1 (and probably Fus1) functions as the MCU gatekeeper to prevent basal accumulation of Ca2+ (left). (c) After Ca2+ loading, mitochondrial Ca2+ uptake can self-inhibit. The activation of the MCU by MICU1 allows for a regulatory loop preventing MCU self-inhibition, thus prolonging the open state of the channel.
To better understand how mitochondria shape intracellular Ca2+ signals and patterns, we need to review the mechanisms of Ca2+ oscillations in non-excitable cells such as T lymphocytes. Non-excitable cells use so-called SOCE and Ca2+-induced Ca2+ release (CICR) mechanisms to orchestrate Ca2+ oscillations [77–79]. Ligation of membrane receptors (e.g. TCR) triggers the formation of the second messenger IP3, which binds its receptor on ER stores leading to the opening of IP3R channels and massive Ca2+ release [78] (figure 3). ER possesses transmembrane Ca2+ sensor called stromal interaction molecule 1 (STIM1) containing EF-hand domain facing intraluminal space. The Ca2+ drop in the ER causes conformational alterations in STIM1 and its oligomerization, resulting in its interaction with the plasmalemmal Orai1 protein, the pore-forming subunit of CRAC, followed by the opening of Orai1 and Ca2+ influx into the cell [78] (figure 3). Another mechanism, CICR, maintains Ca2+ release from ER store: after reaching the threshold level, locally increased Ca2+ sensitizes IP3R and RyR to opening and propagation of Ca2+ waves along the ER [77,80,81]. It is noteworthy that both channels, CRAC and IP3R, create local Ca2+ domains with high Ca2+ concentration, which can autoinhibit channels [82,83]. This self-suppression is prevented by mtCU restricting local Ca2+ concentration [79,84] (figure 3). Finally, plasma membrane (PMCA) and ER store (SERCA) Ca2+ ATPases contribute to decay phase of oscillations and terminate Ca2+ signal; lower affinity of Ca2+ ATPases to Ca2+ and slower rates, compared with IP3R and RyR, allow them to enter the process of Ca2+ signal propagation later than Ca2+ channels, thus allowing Ca2+ response to develop [79].
Figure 3.

General scheme of store-operated Ca2+ entry (SOCE) signalling in non-excitable cells such as T lymphocytes. Activation of plasma membrane receptors (e.g. T cell receptors) leads to the production of second messengers (IP3, cADPR, etc.) involved in Ca2+ release from the ER. Ca2+ that is released from the ER accumulates in the mitochondria via mitochondrial Ca2+ uptake (mtCU) mechanisms. In Ca2+-depleted ER, Ca2+-binding protein STIM1 changes conformation and oligomerizes; it induces its interaction with the plasma membrane channel ORAI1, the pore-forming subunit of Ca2+ release-activated channel (CRAC) and allows the entry of Ca2+ ions from extracellular space. ORAI1, IP3Rs and RyRs have the ability to self-inhibit after significant Ca2+ rise, which prevents cell from Ca2+ overloading. By taking up extra amounts of Ca2+ close to Ca2+ release channels, mitochondria extend the open state of these three channels. Furthermore, the mNCX provides a positive feedback to ER Ca2+ release via activation of IP3Rs and RyRs through local elevations of Ca2+ ions at the ER/mitochondria interface.
A mathematical model shows that the optimal distance between the ER and mitochondria should be 30–85 nm; when it is less, mitochondria prevent positive feedback of low Ca2+ on IP3R, whereas above 30–85 nm, mitochondria take up Ca2+ and preserve IP3R from Ca2+-induced inhibition [85]. Moreover, the ability of mitochondria to accumulate Ca2+ released from the ER store indirectly causes STIM1 oligomerization and activation of the CRAC current [86,87]. Finally, accumulated Ca2+ is released from mitochondria by the mNCX and owing to the proximity of the ER, mitochondria create local Ca2+ domains, which sensitize IP3R and RyR to Ca2+ release, thus triggering CICR [88,89]. It is important that both the mtCU and the mNCX have an influence on the shape and the dynamics of regenerative Ca2+ oscillations [20,89,90]. Finally, the NCLX is a key for refuelling the ER with Ca2+ ions in B cells, whereas NCLX-mediated Ca2+ mitochondria/ER recycling is central in antigen stimulation of B cells [91]. Collectively, these data indicate that mitochondria provide cellular Ca2+ influx mechanisms with positive feedback loops, which are important for generation of digital signals (discussed below).
Oscillations in cytosolic Ca2+ translate into transcriptional changes based on frequency, amplitude and spatial decoding of Ca2+ signals [79,92,93]. Classical examples are NFAT and NFκB transcriptional factors dependent on the frequency of Ca2+ oscillations: NFAT requires more frequent oscillations (approx. every 6 min) to maintain a transcriptionally active state, whereas NFκB needs less frequent fluctuations (every 30 min) [94]. This is explained by the mechanism of activation and cytosol/nuclear shuttling of NFAT and NFκB. Ca2+ activates the CaM-dependent phosphatase CaN, leading to dephosphorylation of inactive NFAT followed by its translocation into the nucleus, whereas re-phosphorylation of NFAT promotes its return into the cytosolic compartment. Thus, rapid periodic oscillations are required for NFAT activation state via maintenance of its dephosphorylated state [95]. At the same time, the active state of NFκB is defined by Ca2+-induced proteolysis of its inhibitor, IkB, and relocation of NFκB from the nucleus into the cytosol, which requires binding of NFκB with IkB. Re-synthesis of IkB is counterbalanced by its Ca2+-triggered proteolysis; thereby, Ca2+ fluctuations with a period of 30 min are able to maintain NFκB in the activated state [95].
Coming back to the initial question of why T cells use mitochondria to make decisions involving Ca2+ signalling, we assume a simple concept formulated in the next way: if mitochondria are used for this process, it means that they meet requirements/criteria for the computational tasks of signal processing they are provided with. However, how do T cells decode the information transferred from the APC to T cells' nuclei, and help in computing cell decisions? To respond to this question, we have to take into consideration specificity of T cell activation (i.e. how T lymphocytes distinguish specific antigenic peptides from non-specific at the levels of information processing and decision-making). Altan-Bonnet & Germain [96] showed that both types of peptides, specific and non-specific, bind TCR and induce tyrosine kinase cascade together with the negative feedback loop, including tyrosine phosphatase SHP-1 dephosphorylating TCR targets and overall signal dampening. At the same time, only agonistic peptides stimulate an MAPK module via a positive feedback loop originating from TCR triggering. This effect is based on the longer lifetime of TCR/MHC/peptide complexes characteristic for specific interaction [96]. Negative (SHP-1) and positive (MAPK) feedback loops are activated consequently: SHP-1 becomes activated after short TCR/MHC interaction, whereas MAPK activation needs a longer time for TCR engagement by pMHC. That means that T cells discriminate specific recognition from a non-specific one by a time lag in signalling: if the pMHC complex does not match the target TCR, then downstream signalling will not activate MAPK, because SHP-1 will barely suppress TCR downstream signals; on the contrary, a longer interaction of pMHC with TCR will result in the activation of MAPK and the consequent maintenance of the T cell activation programme [96]. This important role of time lags in T cells reading Ca2+ signals has been demonstrated in another work focused on the signal discrimination by autocorrelation of detrended time series [97]. Autocorrelation (serial correlation) is a mathematical function allowing the discovery of patterns in signal dynamics, based on the quantification of the level of self-similarity of a signal observed at different time intervals. Detrending of time series consists of removing a time-delayed tendency of time series, resulting in signal differencing/rationing. Translating to cellular processes, this work [97] proposed that oscillation frequency-dependent shuttling dynamics of NFAT and NFκB provide cells with detrending procedure/computing similarly to described above. Further, activation of NFAT- and NFκB-dependent transcription starts after overcoming threshold in average intracellular Ca2+ concentration leading to filtering out non-functional noise from functional data [94,97].
Thus, cellular systems involved in signal reading should demonstrate specific features such as filtering properties (thresholding) and memory in signal dynamics to compute difference between two time points (series). The importance of signal memory has been demonstrated in vivo for tumour rejection: prolonged activation of TCR leads to sustained nuclear NFAT accumulation and transcription of IFNg gene, whereas short-term transitory stimulation is accompanied by only transitory NFAT nuclear translocation and Egr2 gene upregulation inducing CD8+ T cell tolerance [98].
At this point, we can already draw some conclusions about the involvement of mitochondria in Ca2+ signalling from the perspective of information processing. Mitochondria have (i) a threshold for Ca2+ signals (MICU1, Fus1) [49,73–76], and (ii) a ‘lagging’ mechanism between onsets of cytosolic and mitochondrial Ca2+ elevations, which allow to process the incoming information (input) by analysing both the amplitude and the duration of Ca2+ responses (MICU1-mediated block) [74,99]. This delay is due to the overall slower Ca2+ dynamics in mitochondria induced in large part by the large Ca2+ buffering capacity of this organelle. Also, (iii) MICU1 removes self-inhibition (negative feedback loop) of mtCU [74], and (iv) Ca2+ removal from mitochondria through the mNCX triggers Ca2+ oscillations, after the opening of mtCU and accumulation of mitochondrial Ca2+ (positive feedback loop) [89,90]. The importance of the coordination between mtCU and mNCX mechanisms in the general scheme of cellular Ca2+ signalling is supported by the fact that MICU1 and Fus1 are both involved in the control of mNCX besides their effect on mtCU [49,73,76]. Thus, mitochondria ‘send’ a triggering signal to ER store, but it is possible after filling the mitochondrial compartment with Ca2+ [100]. In turn, the input signal should have enough strength and duration, otherwise, it will be filtered out by mitochondria [72]. Such a complex character of the transport mechanism of mitochondrial Ca2+ might be resolved if one considers that mitochondrial Ca2+ transport is a part of a molecular proportional−integral−derivative (PID) controller often used in engineering applications. In the following, we analyse mitochondria from this prospective.
4. Mitochondria as a module of cellular proportional–integral–derivative controller
PID controllers are sensitive to the magnitude of input signals and correct it in the course of time according to a controller function u(t) defined as
where u(t) is the controller output, Kpe(t) is the proportional function, 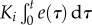 is the integral term and
is the integral term and 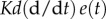 is the differential component of the PID controller [101]. Error e(t) represents the difference (e
= SP − PV) between a well-defined set point (SP) and the current value of a given variable (process variable, PV; cytosolic Ca2+ concentration in this example). The PID controller computes the output signal based on the sum of the proportional (P), integral (I) and derivative (D) terms (figure 4a). The proportional term helps to adapt the process variable proportionally to the error. The derivative term responds to the error depending on how fast the error is approaching zero. Finally, the integral term operates based on the accumulated error and thus set PV proportionally to past errors (figure 4a). If the PID controller overshoots the SP, the output signal would oscillate around the SP with increasing, decreasing or constant amplitude, thereby reflecting the system stability [101].
is the differential component of the PID controller [101]. Error e(t) represents the difference (e
= SP − PV) between a well-defined set point (SP) and the current value of a given variable (process variable, PV; cytosolic Ca2+ concentration in this example). The PID controller computes the output signal based on the sum of the proportional (P), integral (I) and derivative (D) terms (figure 4a). The proportional term helps to adapt the process variable proportionally to the error. The derivative term responds to the error depending on how fast the error is approaching zero. Finally, the integral term operates based on the accumulated error and thus set PV proportionally to past errors (figure 4a). If the PID controller overshoots the SP, the output signal would oscillate around the SP with increasing, decreasing or constant amplitude, thereby reflecting the system stability [101].
Figure 4.

Mitochondrial Ca2+ transport is analogous to a proportional–integral–derivative (PID) controller. (a) Block diagram of a PID controller, an engineering application that uses feedbacks to continuously generate an output signal in a dynamic system until it reaches a well-defined reference value. An error e(t) is generated continuously by comparing the SP with the current value of the parameter controlled by the system. Three elements within the PID controller produce three modes of output: proportional (P), integrative (I) and derivative (D) functions of e(t). The outcome of the three terms is then summed (∑) to give a system's output. (b) Mitochondrial Ca2+ transport as the PID controller for Ca2+ oscillations. Error signal (e(t)) represents the difference between mtCU threshold (SP) and current Ca2+ concentration (PV). Mitochondrial Ca2+ transport and associated metabolic processes display the same modes as those of the PID controller elements. Mitochondrial Ca2+ uptake (mtCU) and export (mNCX) mechanisms are proportional (P) to the Ca2+ loading of mitochondria, whereas mtCU, Ca2+-dependent NADP(H) response and the PTP in its low-conductance mode represent the I element. Finally, mitochondrial ROS production corresponds to the D element.
PID controllers are widely used in industry for regulation of temperature, speed, pressure, flow, etc. [101]. The erythropoietin system represents an example of molecular PID controller in living systems, where an optimal O2 concentration acts as the reference SP, whereas the signal error (e(t)) is erythropoietin (Epo) secretion induced via HIF1/HIF2 activation by low oxygen content [102]. Early erythroblasts form a PID controller based on the findings that EPO (i) proportionally downregulates Bim and Fas, which promote apoptosis of these cells (proportional function), and (ii) increases in a digital mode the expression of Bcl-xL, a pro-survival Bcl-2 family member (derivative function). It results in erythroblast expansion during erythropoietic stress in vivo. However, what is an integrative component is still not defined although it might be a self-renewal process [102]. The resulting output will be a change in red cell mass and amount of tissue O2. The difference between current O2 concentration and optimal (set) O2 content defines the amount of Epo secretion [102]. On a molecular level, the PI controller model has been recently applied for mammalian central carbon metabolic pathway in normal and cancer cells [103]. At the organism level, the PI controller was proposed for homeostatic mechanisms responsible for inflammation and glucose level control [104]. Undoubtedly, in the future, other biological PI(D) controllers will be described.
Although speculative, it seems that mitochondria may act as part of a PID controller for T cell activation through the regulation of cellular Ca2+. The PV would be a given value of cytosolic Ca2+, which is continuously adjusted to the SP by the appropriate Ca2+ fluxes between the cytosol and the mitochondria. Which are the processes that might be considered as proportional, integral and derivative terms? Most probably, transmitochondrial Ca2+ transport (mtCU + mNCX) represents the proportional element of PID controller. Mitochondria proportionally increase Ca2+ uptake after an increase in cell Ca2+ load [105]. The ability of mtCU to respond proportionally to a cytosolic Ca2+ increase is apparently based on the mtCU cooperativity determined by the presence of two Ca2+-binding sites on the MICU1 protein, the regulatory subunit of MCU [73], and on the presence of Ca2+ buffers (e.g. Pi) in mitochondrial matrix [31,106]. The Hill coefficient (defining the degree of cooperativity) for mtCU has a value of 2.4 [105]; in the classical example of cooperative binding of oxygen with haemoglobin, the Hill coefficient is approximately 2.3–3 [107]. Pi buffer helps to maintain ΔΨm polarized after initial Ca2+ uptake, thereby helping mitochondria to take up Ca2+ ions. It is important to note that the rate of dynamic change in ΔΨm is proportional to the rate of gain in Ca2+ uptake [106].
There is another, more indirect way by which the mtCU produces an output value that is proportional to the current error value (i.e. the current cytosolic Ca2+ concentration). The cytosolic Ca2+ response including Ca2+ oscillations indeed relies on the process of STIM1-dependent oligomerization and activation of the SOCE Ca2+ pathway. In turn, STIM1 oligomerization is proportional to the depletion of the endoplasmic Ca2+ store, which is indirectly controlled by the mtCU through both the MCU and UCP2 [86]. Moreover, the magnitudes of the mitochondrial and cytosolic Ca2+ responses directly correlate [86], thus demonstrating that the gains in cytosolic and mitochondrial Ca2+ are proportional. This effect depends on mtCU activity. Finally, gradual dissipation of ΔΨm, the driving force for Ca2+ exchanges between the cytosol and mitochondria, proportionally diminishes Ca2+ oscillations in mast cell line RBL-1 [90]. Besides the mtCU, the mNCX might also contribute to shaping the P-term of PID controller (figure 4b). Indeed, the significance of mNCX for cytosolic Ca2+ oscillations is well documented and based on the ability of the mNCX to fuel the ER store with the amount of Ca2+ ions necessary to trigger CICR and the accompanying Ca2+ oscillations [88,89].
Another term of the mitochondrial PID controller, integrative, might involve some components responsible for Ca2+ transport and accumulation that demonstrate an ability to summarize incoming signals. In particular, mtCU cooperatively with Pi buffers integrates repetitive cytosolic Ca2+ signals resulting in a frequency-dependent net accumulation of Ca2+ ions in mitochondria [105,108] (figure 4b). As mentioned above, Ca2+ activates the TCA cycle and production of NAD(P)H [43]. The redox response usually recovers to basal levels much later than the Ca2+ signal triggering the changes in NAD(P)H production [72], although the mNCX significantly accelerates the return of the redox state to its steady values [109–111]. Furthermore, an increase in the frequency of Ca2+ signals leads to a larger NAD(P) reduction even if the decline in NADP(H) from a previous signal still propagates, thereby summarizing responses from few inputs [72]. Thereby, the metabolic status of mitochondria might play the role of integrator in the proposed mitochondrial PID controller. Finally, Ca2+ can leave mitochondria via the mPTP in its low-conductance mode, a process that also shows the characteristics of an integrative element in physiological conditions. Fast rates of repeated Ca2+ stimulation favour low-conductance flickering of the mPTP (permeability < 300 Da) and consequent Ca2+ release [112]. MPTP opening is dependent on pH and ΔΨm [112,113], two processes that depend on mitochondrial Ca2+ accumulation after repeated cytosolic Ca2+ stimuli.
The derivative element of the PID controller anticipates the system behaviour based on the calculation of the slope of the error with respect to time. We propose here that mitochondrial ROS production might represent the D term of the mitochondrial PID controller. Treatment of cells with mitochondria-targeted antioxidants abolishes cytosolic Ca2+ oscillations induced by physiological stimulation [114–116]. This effect is based on the ability of ROS to oxidize protein thiols and inhibit PMCA and SERCA while sensitizing RyR and IP3R to opening and Ca2+ release [114,115]. It is important that the effect of ROS on ER Ca2+ release channels is dual: RyR opens at moderate concentrations of ROS but becomes inhibited at high ROS concentration [117]. ROS accumulation in mitochondria is balanced by its production in the ETC and utilization by antioxidant systems (SOD, catalase, peroxidase) [118]. Production of ROS is stimulated by elevation in matrix Ca2+ leading to an increase in the electron flux of the ETC [119]. It has been shown that mitochondria-derived ROS regulate cytosolic Ca2+ sparks in a time/history-dependent manner. During the first phase, ROS-sensitized ER channels release Ca2+ (figure 5a). Emptying Ca2+ ER store is accompanied by expansion of mitochondrial ROS production. In the second phase, high concentrations of ROS significantly depress Ca2+ sparks [116] (figure 5b). Thus, the level of Ca2+ is dynamically regulated by the rate of the mitochondrial ROS changes. Besides, the buffering of ROS in mitochondria is tightly connected to the mitochondrial NADH pool as alterations of only one parameter in the antioxidant system (e.g. superoxide dismutase concentration) can significantly change the amplitude and the frequency of mitochondrial oscillations (e.g. ROS outcome of mitochondrial oscillator) [118,120,121] (figure 4b).
Figure 5.

Dual regulation of Ca2+ signalling by ROS. At low concentration (a), ROS, stimulated by Ca2+ accumulation in mitochondria and stimulation of TCA and ETC, activate Ca2+ release from the ER and inhibit ER ATPases thereby promoting a Ca2+ response in the cell. At high concentration (b), ROS inhibit Ca2+ channels, thus dampening further expansion of the Ca2+ response.
ROS also play a role in the dynamic and concentration-dependent regulation of glucose metabolism that couples glycolysis and ETC fuelling with reducing equivalents. Indeed, short exposure of cells to ROS triggers glucose uptake, its conversion into pyruvate and hyperpolarization of the inner mitochondrial membrane. By contrast, prolonged presentation of ROS leads to downregulation of glucose transport in order to prevent excessive feeding of ETC with electron-donor molecules [122]. Thus, ROS potentially couples Ca2+ signalling, ETC activation and metabolic reprograming (in particular, alterations in glucose transport) in a manner that depends on the temporal changes in ROS production associated with T cell development.
PID controllers have an important limitation: they have a low capability to minimize noise. In engineering, to remove noise in the system using PID-based control system, low-pass filters (e.g. Kalman filter) for feedback response are used [101]. In the case of mitochondria, such a role might be ascribed to the MICU1 protein, which has two important characteristics as a low-pass filter: (i) MICU1 filters out signals with low amplitude [72]; (ii) the protein removes MCU block in a time-dependent mode proportionally to the strength of cytosolic Ca2+ signal [74].
After performing the analogue computational procedures, the PID controller combines outcomes from all three terms (proportional, integrative and derivative) and sends a final (summarized) decision to elements of the driving process [101]. How does the mitochondrial PID controller compute the resulting outcome based on its P, I and D terms? If we imagine that individual cells would experience sustained Ca2+ signals with high amplitude, then we could expect that mitochondria will be proportionally accumulating Ca2+ with equal emptying of the ER store as the mNCX would provide the IP3R and RyR with enough Ca2+ ions to trigger their opening. That would favour CRAC channels in an active state. Concurrently, Ca2+ response will be tuned out by the amount of ROS and availability of antioxidant buffers. Thus, the resulting response will be directed by the summation of PID controller terms. If the value of any parameter (beginning with Ca2+ signal length/amplitude/frequency and ending with NAD(P)H concentration) is changed, then the PID controller outcome will be adjusted according to these modifications. In our case, the resulting process might involve Ca2+ signalling and Ca2+-dependent signal transduction systems such as NFAT cytosol/nuclear shuttling. For example, during Ca2+ responses, the treatment of cells with pro-oxidant (analogue of excessive mitoROS production) leads to the CaN inhibition and the dampening of Ca2+-induced NFAT nuclear translocation, whereas the prevention of oxidative stress (analogue of high NAD(P)H production) suppresses this effect [123].
Before further discussion, we return to the properties of the mtCU and remind that (i) the mtCU acts as a low-pass filter, which has a high threshold [15,22–24,99]; (ii) the dissipation of ΔΨm proportionally decreases Ca2+ oscillations [90]. We propose that the Ca2+ threshold for mtCU activation might serve as an SP for Ca2+ oscillations. Indeed, Ca2+ oscillations would be maintained until the error would reach zero; in other words, when SP = PV and e = SP − PV = 0. However, while PV is greater than SP, the Ca2+ system will be oscillating owing to the difference between the cytosolic Ca2+ concentration and the threshold for mtCU opening. It is noteworthy that these two parameters are coupled via the mtCU-dependence of STIM1 oligomerization and further interaction with CRAC1, the pore unit of SOCE currents [86]. Mitochondria indeed take up Ca2+ from the ER store, thus emptying it and allowing the ER Ca2+ level to drop below the Kd for Ca2+ binding to the EF-hand of STIM1, thereby leading to its oligomerization [84,124].
As mentioned above, if the PID controller overshoots the SP, the output signal will oscillate around the SP [101]. In the framework of the dynamics of nonlinear systems, oscillations (cellular Ca2+ oscillations, mitochondrial oscillations, etc.) arise at special points called Hopf bifurcations [120,125,126]. The bifurcation point represents a point in the parameter space where the system changes behaviour in a qualitative mode (e.g. from a steady state to self-sustained oscillations) [126]. In this regard, e = SP − PV = 0 would be analogous to the bifurcation point between these two states.
The SP, PV and e are flexible as cells can use different strategies to shape cellular Ca2+ signals through post-translation or transcriptional regulation of proteins involved in ER/mitochondria Ca2+ crosstalk:
(1) reduction of Ca2+ currents by depolarization of cellular membrane potential, downregulation of IP3R or RyR, modified efflux of Ca2+ by Ca2+ ATPase (PMCA or SERCA);
(2) changes in sensitivity of the mtCU to Ca2+ (MICU1, Fus1), mitochondrial Ca2+ export or expression of its components (e.g. NCLX1), ΔΨm increase or decrease; and
(3) alterations in the distance between the ER and mitochondria (e.g. up/downregulation of mitofusins, Grp75/HSPA9).
How can T cells use these strategies in order to regulate their fate? IP3R-mediated Ca2+ release is important for the initial production of cytokines [127] but not for late expression of cytokines by activated CD4+ T cells [128] where mitochondria are the most plausible source for cellular Ca2+ exchange [71]. It is well documented that T cells use two major types of K+ channels: voltage-gated Kv1.3 and Ca2+/calmodulin(CaM)-regulated KCa3.1. The Kv1.3 channel has a voltage sensor located in the fourth transmembrane domain containing four arginine residues, whereas the C-terminus of KCa3.1 channel bears intracellular CaM-binding motif and senses Ca2+ elevations [129]. Naive T cells express mostly Kv1.3 channels, whereas T cells upregulate KCa3.1 channels after TCR-triggered activation [129]. There is also a difference in Kv1.3 and KCa3.1 usage described for central-memory T(CM) and effector-memory T(EM) cells: upon activation, T(CM) lymphocytes display dependence on KCa3.1 channels, whereas T(EM) rely on Kv1.3. It is believed that in resting T cells, the opening of Kv1.3 channels repolarizes membrane potential after transient depolarization, whereas blocking a Kv1.3 channel leads to diminished Ca2+ currents owing to a decrease in the driving force for Ca2+ [130]. An increase in the intensity and the density of Ca2+ currents in activated T cells is accompanied by an upregulation of KCa3.1 channels, which hyperpolarizes the membrane potential and maintains CRAC influx [131–134]. The Th1 subset demonstrates higher KCa2+ currents compared with Th2, which is translated into stronger and sustained Ca2+ signals in Th1 cells [69]. In contrast, Th2 have an elevated level of Trpm4, a Ca2+-dependent Na+ channel; opening of Trpm4 leads to depolarization of the membrane potential and reduced Ca2+ oscillation patterns [135]. Thus, changes in expression of K+, Ca2+ or Na+ channels pursue the goal of fine-tuning Ca2+ responses according to a programme dictated by different steps in T cell life. The role of mitochondria in these processes might be to set the parameters responsible for the spatio-temporal Ca2+ patterns such as oscillations.
5. Analogue and digital signalling in T lymphocytes
Another important aspect of T lymphocyte life is the transduction of stimuli with different intensity into analogue (gradual) or digital (binary) outcomes in order to adequately respond to environmental signals. An early study demonstrated that in T cells, parameters of Ca2+ mobilization (magnitude, increase rate, frequency of oscillations, etc.) after specific stimulation display incremental characteristics when increasing the amount of MHC/peptide molecules [136]. However, a recent investigation showed that a single peptide/MHC complex can initiate the response of CD4+ T cell and that additional pMHC molecules do not increase the secretion of effector cytokines (such as IL-2 or TNFa), demonstrating an ‘all or none’ type of response [137]. The follow-up of the secretion of Th2 cytokine, IL-4, strictly requires the presence of NFAT in the nucleus and analogue differences in TCR triggering are converted into digital outcome of NFAT nuclear translocation [138]. It is noteworthy that activation of NFAT itself is a digital process because it requires the complete—highly cooperative—dephosphorylation by CaN of 13 serine residues, which hide the nuclear localization signal segment inside the protein [139]. Moreover, CRAC-driven activation of NFAT is also described as an ‘all or nothing’ response: opening of CRAC channels creates local Ca2+ domains, which cause CaN activation if they reach the threshold Ca2+ concentration; CaN-dependent dephosphorylation of NFAT overcomes another threshold set by the highly phosphorylated state of inactive NFAT [140]. As described above, TCR specificity is dependent on a digital ERK activation based on the high amplification of the MAPK module [96]. At the same time, MAPK responds gradually to stimulation of Jurkat cells with SDF-1, reflecting plasticity in the reaction of T cells to chemokine gradients [141]. In addition, the activation of NFκB showed analogue [138] and digital [142] modes after gradual T cell stimulation.
The importance of digital and gradual signals has been demonstrated for the MAPK module activation in PC-12 cells. Endothelial (EGF) and neuronal (NGF) growth factors both activate Erk1/2 MAPK, but with different modalities: gradual for EGF and digital for NGF [143]. Functionally, EGF stimulates proliferation, whereas NGF directs cells toward differentiation. The mechanism of rewiring of the MAPK pathway from analogue to digital mode relies on a positive feedback loop: NGF stimulates PKC, which in turn phosphorylates and inhibits Raf kinase inhibitory protein (RKIP), thus keeping the MAPK phosphorylation cascade turned on (figure 6a, stimulus I). In the case of EGF, this mechanism is lacking as only a negative feedback loop is present, which implies that the strength of signal transduction is determined exclusively by the amount of ligand (input signal; figure 6a, stimulus II) [143]. Another example that represents a high-fidelity analogue–digital–analogue converter is based on Ras nanoclusters [144]. Ras is a small GTPase protein anchored in cellular membranes, which participates in signal transduction from membrane receptors to the MAPK module. After ligand–receptor binding, Ras coordinately aggregates into nanoclusters with about seven molecules per group and a lifetime of about 0.5 s. Then, Ras clusters send a downstream signal towards ERK1/2 via interaction with the Raf kinase (figure 6b, top). Ras nanocluster formation is digital in its nature, because GTPase molecules start clustering after reaching a given threshold. Below this threshold, Ras signalling does not provide any significant output. The amount of clusters, however, depends on the ligand concentration and displays analogue characteristics when Ras is above the threshold. The signal output then rises proportionally to the input. This process is called signal integration and is necessary for high-fidelity signal transduction when the output (ERK activation) should precisely reflect the input (ligand concentration) signal (figure 6b, bottom graphs) [144,145].
Figure 6.

Digital and analogue activation of the MAPK cascade. (a) Top: stimulus I leads to activation of MAPK cascade together with PKC, which keeps MAPK stimulated over a long time as RKIP, a suppressor of MAPK signalling, is inhibited. This creates a positive feedback forward loop (digital mode). Bottom: stimulus II does not activate PKC, which results in the decrease of MAPK cascade activation with time; the strength of such a response will depend on the amount of stimulus II (analogue mode). (b) Top: upon EGF stimulation, Ras molecules form nanoclusters, which stimulate MAPK cascade. Bottom graphs: each nanocluster gives 1 digital unit of ERK activation while combining responses induced by distinct nanoclusters leads to a higher overall response. The level of response is determined by the concentration of EGF capable to stimulate the formation of more Ras nanoclusters, via binding to the receptor (analogue output).
For T lymphocytes, fine-tuning of NFAT dynamics requires high-fidelity signal transduction owing to the extreme importance of NFAT-mediated transcription in cell decisions. Disturbances in this process result in severe pathology [146–148]. As an example, the length of the NFAT half-life in the nucleus defines Th differentiation: after similar strength of stimulus, NFAT is present in the nucleus much longer in Th1 and Th17 when compared with Th2 cells [70,149,150]. NFAT nuclear accumulation and transcriptional regulation demonstrate signs of hysteresis or molecular memory when the system continues producing output signals even when the input trigger is no longer active [98,151]. Indeed, NFAT upregulates its targets even when receptor activation is gone. This process is based on the binding of CaN with NFAT in the nucleus; propagation of Ca2+ waves into the nucleus (most probably from perinuclear mitochondria) leads to strong association of both proteins, preventing NFAT from re-phosphorylation and further Crm1-mediated nuclear export [152]. At the same time, sustained dynamics of NFAT nuclear accumulation provides evidences of the presence of a positive feedback loop similar to the MAPK module described above. Moreover, NFAT nuclear retention reflects the longevity of the Ca2+ response, thereby exhibiting graded mode. This inter-relationship is determined by the ability of mitochondria to slowly release Ca2+ via the mNCX after its accumulation during cytosolic elevations in sensory neurons [123] and CD4+ T cells [71]. It is known that the close proximity of mitochondria with ER store allows the mNCX to activate IP3R and RyR owing to the local increase of Ca2+ concentration [88,89]. Although the mechanism of NFAT activation and translocation has a digital nature [139,153], transcription factor retention in the nucleus has more analogue characteristics although the curve of NFAT/Ca2+ response dependence is very steep and close to a switch-like response [123]. We propose that the ability of NFAT to titrate the duration of the Ca2+ responses is very important for its biological effects, because NFAT can trigger different transcription programmes depending on the nuclear lifetime (e.g. activation versus tolerance) [148,154]. The process of mNCX-controlled nuclear NFAT retention reminds us of the signal integration for Ras clusters described above. Indeed, IP3Rs also form dynamic clusters, which are responsible for elementary Ca2+ events [81,155] (figure 7). IP3Rs form single quantum (binary) circuits and summation of Ca2+ release quanta gives graded response depending on agonist concentration at submaximal doses [156–159]. We assume that mitochondria in this case could be considered as integrating detectors owing to their ability to accumulate Ca2+ and release it during the plateau phase following the peak (figure 7), thereby maintaining NFAT in an ‘on’ state in the nucleus. Furthermore, mitochondria are involved in CRAC-driven NFAT activation, which has a digital nature as described above [140]. Taken together, mitochondria might represent a domain involved in analogue–digital and digital–analogue conversions. As mentioned above, cells use this type of system when high fidelity of signal transduction is necessary [144,145].
Figure 7.

The possible role of mitochondria in gradual transformation of Ca2+ signals. (a) After an IP3-induced Ca2+ release of low magnitude (frequency, duration or amplitude), Ca2+ recycling maintained by mitochondria will lead to a low activation of IP3Rs and RyRs (1 relative quantum of signal). (b) Higher amplitude of Ca2+ release from the ER will result in larger Ca2+ released from mitochondria and in a proportional gain of the overall Ca2+ response (three relative quanta). Thus, the integration by mitochondria of discrete Ca2+ pulses from individual clusters of channels in the ER might be considered as a digital–analogue conversion. As the output of each ER storage channel (IP3Rs, RyRs) is dumped into ER/mitochondria cleft, the digital pulses are summed to give a final analogue output expressed by the activity of transcription factors (i.e. NFAT, NFκB). The existence of such biological circuits is necessary for the generation of high-fidelity signals.
We now briefly review possible mechanisms related to the involvement of mitochondria in the rewiring of the signalling pathways, in order to switch between the various T cell programmes. To achieve this aim, we need to imagine a hypothetical stimulus S, which in a simple assumption activates two signalling pathways Y1 and Y2 via competing sensors X1 and X2, respectively. Both sensors are specific to the mediator Z, but possess different activation parameters (affinity, kinetics, etc.). If the level of Z, reaches the threshold parameters for X2, then the latter will buffer mediator Z and occlude it from interaction with X1, thus keeping pathway Y1 inactive (figure 8a). At the same time, if the level of Z is below threshold parameters for X2, then stimulus S will favour activation of sensor X1 via mediator Z, resulting in activation of pathway Y1 (figure 8a). Applying this assumption to mitochondria, mtCU opening and Ca2+ uptake can limit availability of Ca2+ ions to other Ca2+-dependent targets. Thus, Akt/PKB-mediated survival requires the release of mitochondrial Ca2+, and in the absence of mNCX, the apoptotic programme is triggered [64]. Short-term translocation of NFAT in the nucleus induces a tolerance programme, whereas prolonged retention upregulates the activation programme [98]. Sustained residence of NFAT in the nucleus is strictly necessary for IL-4 synthesis [138]. As described above, EGFR and NGFR can rewire the MAPK module depending on activation of PKC involving a positive feedback loop [143]. It is known that in non-polarized conditions (PMA/ionomycin, CD3/CD28), T cells mostly favour Th1 phenotype with prevalent secretion of IFNg [160–162]. Common features of these responses are high-amplitude and long-term activation of Ca2+ responses, which correlate with the requirements for Th1 differentiation [69,70]. At the same time, suppression of transmitochondrial Ca2+ transport would lead to different patterns of Ca2+ signalling, including lack of regenerative Ca2+ oscillations [89,90]. Indeed, blocking the mtCU in human lymphoblasts upregulates the NADPH oxidase, which normally requires Ca2+-dependent PKC activation [65]. In turn, NADPH oxidase 5 (NOX5)-deficient T cells show under-regulated phospho-Stat5 and diminished Th2 differentiation [163] (figure 8b). Thus, the threshold for mtCU opening might serve as a switch for rewiring between signalling pathways responsible for different T cell programmes.
Figure 8.

Rewiring of the Ca2+-signalling pathways triggering different cellular programmes. (a) General scheme of rewiring dependent on transmitochondrial Ca2+ transport. If the mtCU and the mNCX are active, elevated Ca2+ ions undergo periodic fluctuations and stimulate Ca2+ oscillation-dependent proteins (pattern I), whereas cytosolic Ca2+ retention will keep these targets inactive (pattern II; left side). Deficiency in mitochondrial transport will favour stimulation of pattern II at the expense of pattern I (right side). (b) Proposed scheme of rewiring in Th differentiation. Intense transmitochondrial Ca2+ transport leads to strong NFAT activation and bias towards Th1 lymphocytes and suppression of Th2 stemming (left side), whereas diminished mtCU in T cells will be accompanied by a PKC-dependent stimulation of NADPH oxidase, which requires accumulation of intracellular Ca2+ leading to Th2 differentiation (right side).
6. Concluding remarks
In this article, we have attempted to characterize mitochondrial Ca2+ transport as an engineering application with capability to perform computational tasks based on characteristics of the input signal. Thanks to the mitochondrial regulations, the outcome signal possesses well-defined characteristics able to trigger specific programmes. At the same time, the adjustment of the parameters of the proposed mitochondrial PID controller (e.g. Ca2+ threshold) would change the cellular output. Translating this hypothesis to T cells, we expect multiple remodelling processes in the Ca2+ signalling network during activation, differentiation, apoptosis, etc. Multiple cytokines are crucial players in the regulation of the T cell states and we believe that their role is to provide cells with correct instructions, including how to remodel Ca2+ signalling patterns, in order to respond adequately to modifications of the environment. This view is confirmed by observations that T cell activation is associated with coordinated expression of RyR, IP3R, Orai1 together with MCU expression [131], while Ca2+ signalling in T cells is subjected to remodelling in terms of Ca2+ sources depending on the stage of activation [71].
The present model focuses on the roles of Ca2+ fluxes as key mediators of mitochondrial control of T cell activation. It does not provide a complete description of the molecular pathways responsible for T cell activation, which involve many other important factors such as nitric oxide, mitochondrial mass, mTOR signalling, etc. The model could be applied to other cell types owing to similarity in the fundamental mechanisms and principles of signalling regulation among different cell types. As an example, decoding of Ca2+ oscillations by cardiomyocytes and hypertrophic response are executed by the Ca2+-sensitive NFAT/CaN system, which can act as an integrator [164] in the same way as in T cells [94]. Further, recent observations indicate that an increase in Ca2+ concentration results in primary salivary gland epithelial cells acquiring an acinar-like phenotype. This process is associated with the remodelling of the Ca2+ signalling machinery: upregulation of Orai1, STIM1, STIM2 and NFAT1, accompanied by an elevation in SOCE [165]. According to our hypothesis, this remodelling might be considered necessary to fine-tune the Ca2+ response and the parameters of output of the PID controller that result in the realization of specific programmes. Finally, these conclusions might have not only fundamental interest, but also therapeutic implications, because they would allow us to predict which spots in cellular Ca2+ signalling have the most crucial effect in different pathological states.
Acknowledgements
We are grateful to Prof. John Wikswo (Vanderbilt Institute for Integrative Biosystems Research and Education, Vanderbilt University, Nashville, TN) for his careful proofreading and comments, which essentially helped to improve the article.
Competing interests
The authors declare that no conflict of interest exists.
Funding
A.S. is supported by U54CA091408 (NCI), 5 U54 RR026140-03 (NCRR), 8 U54 MD007593-0yy3 (NIMHD), 5P50CA 090949 (NCI), R01CA175370 (NCI) and SCI CA182843 (NCI). G.D. is a research director at the Belgian FNRS and is supported by the FNRS Research Credit ‘Auto-organization in cell signaling’.
References
- 1.Byersdorfer CA. 2014. The role of fatty acid oxidation in the metabolic reprograming of activated T-cells. Front. Immunol. 5, 641 (doi:10.3389/fimmu.2014.00641) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang R, Green DR. 2012. Metabolic reprogramming and metabolic dependency in T cells. Immunol. Rev. 249, 14–26. (doi:10.1111/j.1600-065X.2012.01155.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maciolek JA, Pasternak JA, Wilson HL. 2014. Metabolism of activated T lymphocytes. Curr. Opin. Immunol. 27, 60–74. (doi:10.1016/j.coi.2014.01.006) [DOI] [PubMed] [Google Scholar]
- 4.Pearce EL, Poffenberger MC, Chang CH, Jones RG. 2013. Fueling immunity: insights into metabolism and lymphocyte function. Science 342, 1242454 (doi:10.1126/science.1242454) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galganska H, Karachitos A, Wojtkowska M, Stobienia O, Budzinska M, Kmita H. 2010. Communication between mitochondria and nucleus: putative role for VDAC in reduction/oxidation mechanism. Biochim. Biophys. Acta 1797, 1276–1280. (doi:10.1016/j.bbabio.2010.02.004) [DOI] [PubMed] [Google Scholar]
- 6.Holley AK, Dhar SK, St Clair DK. 2010. Manganese superoxide dismutase vs. p53, regulation of mitochondrial ROS. Mitochondrion 10, 649–661. (doi:10.1016/j.mito.2010.06.003) [DOI] [PubMed] [Google Scholar]
- 7.Kotiadis VN, Duchen MR, Osellame LD. 2014. Mitochondrial quality control and communications with the nucleus are important in maintaining mitochondrial function and cell health. Biochim. Biophys. Acta 1840, 1254–1265. (doi:10.1016/j.bbagen.2013.10.041) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pesaresi P, Schneider A, Kleine T, Leister D. 2007. Interorganellar communication. Curr. Opin. Plant Biol. 10, 600–606. (doi:10.1016/j.pbi.2007.07.007) [DOI] [PubMed] [Google Scholar]
- 9.Baughman JM, et al. 2011. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. (doi:10.1038/nature10234) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. 2011. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340. (doi:10.1038/nature10230) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK. 2010. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 467, 291–296. (doi:10.1038/nature09358) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Csordas G, Varnai P, Golenar T, Sheu SS, Hajnoczky G. 2012. Calcium transport across the inner mitochondrial membrane: molecular mechanisms and pharmacology. Mol. Cell Endocrinol. 353, 109–113. (doi:10.1016/j.mce.2011.11.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Stefani D, Patron M, Rizzuto R. 2015. Structure and function of the mitochondrial calcium uniporter complex. Biochim. Biophys. Acta 1853, 2006–2011. (doi:10.1016/j.bbamcr.2015.04.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hajnoczky G, et al. 2014. Reliance of ER-mitochondrial calcium signaling on mitochondrial EF-hand Ca2+ binding proteins: Miros, MICUs, LETM1 and solute carriers. Curr. Opin. Cell Biol. 29, 133–141. (doi:10.1016/j.ceb.2014.06.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marchi S, Pinton P. 2014. The mitochondrial calcium uniporter complex: molecular components, structure and physiopathological implications. J. Physiol. 592, 829–839. (doi:10.1113/jphysiol.2013.268235) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bondarenko AI, Parichatikanond W, Madreiter CT, Rost R, Waldeck-Weiermair M, Malli R, Graier WF. 2015. UCP2 modulates single-channel properties of a MCU-dependent Ca2+ inward current in mitochondria. Pflugers Arch. 467, 2509–2518. (doi:10.1007/s00424-015-1727-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pendin D, Greotti E, Pozzan T. 2014. The elusive importance of being a mitochondrial Ca2+ uniporter. Cell Calcium 55, 139–145. (doi:10.1016/j.ceca.2014.02.008) [DOI] [PubMed] [Google Scholar]
- 18.Palty R, et al. 2010. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl Acad. Sci. USA 107, 436–441. (doi:10.1073/pnas.0908099107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palty R, Sekler I. 2012. The mitochondrial Na+/Ca2+ exchanger. Cell Calcium 52, 9–15. (doi:10.1016/j.ceca.2012.02.010) [DOI] [PubMed] [Google Scholar]
- 20.Wacquier B, Combettes L, Van Nhieu GT, Dupont G. 2016. Interplay between intracellular Ca2+ Oscillations and Ca2+-stimulated mitochondrial metabolism. Sci. Rep. 6, 19316 (doi:10.1038/srep19316) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Vliet AR, Verfaillie T, Agostinis P. 2014. New functions of mitochondria associated membranes in cellular signaling. Biochim. Biophys. Acta 1843, 2253–2262. (doi:10.1016/j.bbamcr.2014.03.009) [DOI] [PubMed] [Google Scholar]
- 22.Bragadin M, Pozzan T, Azzone GF. 1979. Kinetics of Ca2+ carrier in rat liver mitochondria. Biochemistry 18, 5972–5978. (doi:10.1021/bi00593a033) [DOI] [PubMed] [Google Scholar]
- 23.Moreau B, Nelson C, Parekh AB. 2006. Biphasic regulation of mitochondrial Ca2+ uptake by cytosolic Ca2+ concentration. Curr. Biol. 16, 1672–1677. (doi:10.1016/j.cub.2006.06.059) [DOI] [PubMed] [Google Scholar]
- 24.Olson ML, Chalmers S, McCarron JG. 2012. Mitochondrial organization and Ca2+ uptake. Biochem. Soc. Trans. 40, 158–167. (doi:10.1042/BST20110705) [DOI] [PubMed] [Google Scholar]
- 25.Csordas G, Varnai P, Golenar T, Roy S, Purkins G, Schneider TG, Balla T, Hajnoczky G. 2010. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell 39, 121–132. (doi:10.1016/j.molcel.2010.06.029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giacomello M, Drago I, Bortolozzi M, Scorzeto M, Gianelle A, Pizzo P, Pozzan T. 2010. Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store-operated Ca2+ channels. Mol. Cell 38, 280–290. (doi:10.1016/j.molcel.2010.04.003) [DOI] [PubMed] [Google Scholar]
- 27.Rizzuto R, Brini M, Murgia M, Pozzan T. 1993. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 262, 744–747. (doi:10.1126/science.8235595) [DOI] [PubMed] [Google Scholar]
- 28.Bononi A, et al. 2012. Mitochondria-associated membranes (MAMs) as hotspot Ca2+ signaling units. Adv. Exp. Med. Biol. 740, 411–437. (doi:10.1007/978-94-007-2888-2_17) [DOI] [PubMed] [Google Scholar]
- 29.Rizzuto R, et al. 2009. Ca2+ transfer from the ER to mitochondria: when, how and why. Biochim. Biophys. Acta 1787, 1342–1351. (doi:10.1016/j.bbabio.2009.03.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seifert EL, Ligeti E, Mayr JA, Sondheimer N, Hajnoczky G. 2015. The mitochondrial phosphate carrier: role in oxidative metabolism, calcium handling and mitochondrial disease. Biochem. Biophys. Res. Commun. 464, 369–375. (doi:10.1016/j.bbrc.2015.06.031) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei AC, Liu T, Winslow RL, O'Rourke B. 2012. Dynamics of matrix-free Ca2+ in cardiac mitochondria: two components of Ca2+ uptake and role of phosphate buffering. J. Gen. Physiol. 139, 465–478. (doi:10.1085/jgp.201210784) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chalmers S, Nicholls DG. 2003. The relationship between free and total calcium concentrations in the matrix of liver and brain mitochondria. J. Biol. Chem. 278, 19 062–19 070. (doi:10.1074/jbc.M212661200) [DOI] [PubMed] [Google Scholar]
- 33.Junker C, Hoth M. 2011. Immune synapses: mitochondrial morphology matters. EMBO J. 30, 1187–1189. (doi:10.1038/emboj.2011.72) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quintana A, et al. 2011. Calcium microdomains at the immunological synapse: how ORAI channels, mitochondria and calcium pumps generate local calcium signals for efficient T-cell activation. EMBO J. 30, 3895–3912. (doi:10.1038/emboj.2011.289) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arnold R, Brenner D, Becker M, Frey CR, Krammer PH. 2006. How T lymphocytes switch between life and death. Eur. J. Immunol. 36, 1654–1658. (doi:10.1002/eji.200636197) [DOI] [PubMed] [Google Scholar]
- 36.Roth D, Krammer PH, Gulow K. 2014. Dynamin related protein 1-dependent mitochondrial fission regulates oxidative signalling in T cells. FEBS Lett. 588, 1749–1754. (doi:10.1016/j.febslet.2014.03.029) [DOI] [PubMed] [Google Scholar]
- 37.Kaminski MM, Roth D, Krammer PH, Gulow K. 2013. Mitochondria as oxidative signaling organelles in T-cell activation: physiological role and pathological implications. Arch. Immunol. Ther. Exp. (Warsz) 61, 367–384. (doi:10.1007/s00005-013-0235-0) [DOI] [PubMed] [Google Scholar]
- 38.Kaminski MM, Roth D, Sass S, Sauer SW, Krammer PH, Gulow K. 2012. Manganese superoxide dismutase: a regulator of T cell activation-induced oxidative signaling and cell death. Biochim. Biophys. Acta 1823, 1041–1052. (doi:10.1016/j.bbamcr.2012.03.003) [DOI] [PubMed] [Google Scholar]
- 39.Kaminski MM, et al. 2012. T cell activation is driven by an ADP-dependent glucokinase linking enhanced glycolysis with mitochondrial reactive oxygen species generation. Cell Rep 2, 1300–1315. (doi:10.1016/j.celrep.2012.10.009) [DOI] [PubMed] [Google Scholar]
- 40.Sena LA, et al. 2013. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38, 225–236. (doi:10.1016/j.immuni.2012.10.020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weinberg SE, Sena LA, Chandel NS. 2015. Mitochondria in the regulation of innate and adaptive immunity. Immunity 42, 406–417. (doi:10.1016/j.immuni.2015.02.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. 2004. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 287, C817–C833. (doi:10.1152/ajpcell.00139.2004) [DOI] [PubMed] [Google Scholar]
- 43.Feissner RF, Skalska J, Gaum WE, Sheu SS. 2009. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front. Biosci. (Landmark Ed) 14, 1197–1218. (doi:10.2741/3303) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shanmughapriya S, et al. 2015. Ca2+ signals regulate mitochondrial metabolism by stimulating CREB-mediated expression of the mitochondrial Ca2+ uniporter gene MCU. Sci. Signal. 8, ra23. (doi:10.1126/scisignal.2005673) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoth M, Button DC, Lewis RS. 2000. Mitochondrial control of calcium-channel gating: a mechanism for sustained signaling and transcriptional activation in T lymphocytes. Proc. Natl Acad. Sci. USA 97, 10 607–10 612. (doi:10.1073/pnas.180143997) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nagy G, Koncz A, Perl A. 2003T. cell activation-induced mitochondrial hyperpolarization is mediated by Ca2+- and redox-dependent production of nitric oxide. J. Immunol. 171, 5188–5197. (doi:10.4049/jimmunol.171.10.5188) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nagy G, Barcza M, Gonchoroff N, Phillips PE, Perl A. 2004. Nitric oxide-dependent mitochondrial biogenesis generates Ca2+ signaling profile of lupus T cells. J. Immunol. 173, 3676–3683. (doi:10.4049/jimmunol.173.6.3676) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Biasiotto R, Aguiari P, Rizzuto R, Pinton P, D'Agostino DM, Ciminale V. 2010. The p13 protein of human T cell leukemia virus type 1 (HTLV-1) modulates mitochondrial membrane potential and calcium uptake. Biochim. Biophys. Acta 1797, 945–951. (doi:10.1016/j.bbabio.2010.02.023) [DOI] [PubMed] [Google Scholar]
- 49.Uzhachenko R, Ivanov SV, Yarbrough WG, Shanker A, Medzhitov R, Ivanova AV. 2014. Fus1/Tusc2 is a novel regulator of mitochondrial calcium handling, Ca2+-coupled mitochondrial processes, and Ca2+-dependent NFAT and NF-kappaB pathways in CD4+ T cells. Antioxid. Redox Signal. 20, 1533–1547. (doi:10.1089/ars.2013.5437) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Newton RH, Leverrier S, Srikanth S, Gwack Y, Cahalan MD, Walsh CM. 2011. Protein kinase D orchestrates the activation of DRAK2 in response to TCR-induced Ca2+ influx and mitochondrial reactive oxygen generation. J. Immunol. 186, 940–950. (doi:10.4049/jimmunol.1000942) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hsieh YF, et al. 2013. Transglutaminase 2 contributes to apoptosis induction in Jurkat T cells by modulating Ca2+ homeostasis via cross-linking RAP1GDS1. PLoS ONE 8, e81516 (doi:10.1371/journal.pone.0081516) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim JH, Hong JM, Jeong EM, Lee WJ, Kim HR, Kang JS, Kim IG, Hwang YI. 2014. Lack of transglutaminase 2 diminished T-cell responses in mice. Immunology 142, 506–516. (doi:10.1111/imm.12282) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matsuzaki H, Fujimoto T, Tanaka M, Shirasawa S. 2013. Tespa1 is a novel component of mitochondria-associated endoplasmic reticulum membranes and affects mitochondrial calcium flux. Biochem. Biophys. Res. Commun. 433, 322–326. (doi:10.1016/j.bbrc.2013.02.099) [DOI] [PubMed] [Google Scholar]
- 54.Chen XL, Serrano D, Mayhue M, Wieden HJ, Stankova J, Boulay G, Ilangumaran S, Ramanathan S. 2013. GTPase of the immune-associated nucleotide-binding protein 5 (GIMAP5) regulates calcium influx in T-lymphocytes by promoting mitochondrial calcium accumulation. Biochem. J. 449, 353–364. (doi:10.1042/BJ20120516) [DOI] [PubMed] [Google Scholar]
- 55.Ledderose C, Bao Y, Lidicky M, Zipperle J, Li L, Strasser K, Shapiro NI, Junger WG. 2014. Mitochondria are gate-keepers of T cell function by producing the ATP that drives purinergic signaling. J. Biol. Chem. 289, 25 936–25 945. (doi:10.1074/jbc.M114.575308) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mishra P, Chan DC. 2014. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 15, 634–646. (doi:10.1038/nrm3877) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mishra P, Chan DC. 2016. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 212, 379–387. (doi:10.1083/jcb.201511036) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Quintana A, Hoth M. 2012. Mitochondrial dynamics and their impact on T cell function. Cell Calcium 52, 57–63. (doi:10.1016/j.ceca.2012.02.005) [DOI] [PubMed] [Google Scholar]
- 59.Hom J, Sheu SS. 2009. Morphological dynamics of mitochondria--a special emphasis on cardiac muscle cells. J. Mol. Cell Cardiol. 46, 811–820. (doi:10.1016/j.yjmcc.2009.02.023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Han XJ, et al. 2008. CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J. Cell Biol. 182, 573–585. (doi:10.1083/jcb.200802164) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, Hajnoczky G. 2008. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc. Natl Acad. Sci. USA 105, 20 728–20 733. (doi:10.1073/pnas.0808953105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baixauli F, Martin-Cofreces NB, Morlino G, Carrasco YR, Calabia-Linares C, Veiga E, Serrador JM, Sanchez-Madrid F. 2011. The mitochondrial fission factor dynamin-related protein 1 modulates T-cell receptor signalling at the immune synapse. EMBO J. 30, 1238–1250. (doi:10.1038/emboj.2011.25) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van Panhuys N, Klauschen F, Germain RN. 2014. T-cell-receptor-dependent signal intensity dominantly controls CD4+ T cell polarization in vivo. Immunity 41, 63–74. (doi:10.1016/j.immuni.2014.06.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Feldman B, Fedida-Metula S, Nita J, Sekler I, Fishman D. 2010. Coupling of mitochondria to store-operated Ca2+-signaling sustains constitutive activation of protein kinase B/Akt and augments survival of malignant melanoma cells. Cell Calcium 47, 525–537. (doi:10.1016/j.ceca.2010.05.002) [DOI] [PubMed] [Google Scholar]
- 65.Dikalov SI, Li W, Doughan AK, Blanco RR, Zafari AM. 2012. Mitochondrial reactive oxygen species and calcium uptake regulate activation of phagocytic NADPH oxidase. Am. J. Physiol. Regul. Integr. Comp. Physiol. 302, R1134–R1142. (doi:10.1152/ajpregu.00842.2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Buck MD, et al. 2016. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 166, 63–76. (doi:10.1016/j.cell.2016.05.035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cribbs JT, Strack S. 2007. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 8, 939–944. (doi:10.1038/sj.embor.7401062) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Toldi G, Kaposi A, Zsembery A, Treszl A, Tulassay T, Vasarhelyi B. 2012. Human Th1 and Th2 lymphocytes are distinguished by calcium flux regulation during the first 10 min of lymphocyte activation. Immunobiology 217, 37–43. (doi:10.1016/j.imbio.2011.08.007) [DOI] [PubMed] [Google Scholar]
- 69.Fanger CM, Neben AL, Cahalan MD. 2000. Differential Ca2+ influx, KCa channel activity, and Ca2+ clearance distinguish Th1 and Th2 lymphocytes. J. Immunol. 164, 1153–1160. (doi:10.4049/jimmunol.164.3.1153) [DOI] [PubMed] [Google Scholar]
- 70.Weber KS, Miller MJ, Allen PM. 2008. Th17 cells exhibit a distinct calcium profile from Th1 and Th2 cells and have Th1-like motility and NF-AT nuclear localization. J. Immunol. 180, 1442–1450. (doi:10.4049/jimmunol.180.3.1442) [DOI] [PubMed] [Google Scholar]
- 71.Yang R, et al. 2015. Mitochondrial Ca2+ and membrane potential, an alternative pathway for Interleukin 6 to regulate CD4 cell effector function. Elife 4, e06376 (doi:10.7554/eLife.06376) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. 1995. Decoding of cytosolic calcium oscillations in the mitochondria. Cell 82, 415–424. (doi:10.1016/0092-8674(95)90430-1) [DOI] [PubMed] [Google Scholar]
- 73.Csordas G, et al. 2013. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metab. 17, 976–987. (doi:10.1016/j.cmet.2013.04.020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.de la Fuente S, Matesanz-Isabel J, Fonteriz RI, Montero M, Alvarez J. 2014. Dynamics of mitochondrial Ca2+ uptake in MICU1-knockdown cells. Biochem. J. 458, 33–40. (doi:10.1042/BJ20131025) [DOI] [PubMed] [Google Scholar]
- 75.Mallilankaraman K, et al. 2012. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell 151, 630–644. (doi:10.1016/j.cell.2012.10.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Uzhachenko R, Shanker A, Yarbrough WG, Ivanova AV. 2015. Mitochondria, calcium, and tumor suppressor Fus1, At the crossroad of cancer, inflammation, and autoimmunity. Oncotarget 6, 20 754–20 772. (doi:10.18632/oncotarget.4537) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dupont G, Combettes L, Bird GS, Putney JW. 2011. Calcium oscillations. Cold Spring Harb. Perspect. Biol. 3, ppii. a004226 (doi:10.1101/cshperspect.a004226) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hogan PG, Lewis RS, Rao A. 2010. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu. Rev. Immunol. 28, 491–533. (doi:10.1146/annurev.immunol.021908.132550) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Parekh AB. 2011. Decoding cytosolic Ca2+ oscillations. Trends Biochem. Sci. 36, 78–87. (doi:10.1016/j.tibs.2010.07.013) [DOI] [PubMed] [Google Scholar]
- 80.Dupont G, Goldbeter A. 1994. Properties of intracellular Ca2+ waves generated by a model based on Ca2+-induced Ca2+ release. Biophys. J. 67, 2191–2204. (doi:10.1016/S0006-3495(94)80705-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rahman T. 2012. Dynamic clustering of IP3 receptors by IP3. Biochem. Soc. Trans. 40, 325–330. (doi:10.1042/BST20110772) [DOI] [PubMed] [Google Scholar]
- 82.Bezprozvanny I, Watras J, Ehrlich BE. 1991. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature 351, 751–754. (doi:10.1038/351751a0) [DOI] [PubMed] [Google Scholar]
- 83.Mullins FM, Lewis RS. 2016. The inactivation domain of STIM1 is functionally coupled with the Orai1 pore to enable Ca2+-dependent inactivation. J. Gen. Physiol. 147, 153–164. (doi:10.1085/jgp.201511438) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Watson R, Parekh AB. 2012. Mitochondrial regulation of CRAC channel-driven cellular responses. Cell Calcium 52, 52–56. (doi:10.1016/j.ceca.2012.02.003) [DOI] [PubMed] [Google Scholar]
- 85.Qi H, Li L, Shuai J. 2015. Optimal microdomain crosstalk between endoplasmic reticulum and mitochondria for Ca2+ oscillations. Sci. Rep. 5, 7984 (doi:10.1038/srep07984) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Deak AT, Blass S, Khan MJ, Groschner LN, Waldeck-Weiermair M, Hallstrom S, Graier WF, Malli R. 2014. IP3-mediated STIM1 oligomerization requires intact mitochondrial Ca2+ uptake. J. Cell Sci. 127, 2944–2955. (doi:10.1242/jcs.149807) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zeng B, Chen GL, Xu SZ. 2012. Store-independent pathways for cytosolic STIM1 clustering in the regulation of store-operated Ca2+ influx. Biochem. Pharmacol. 84, 1024–1035. (doi:10.1016/j.bcp.2012.07.013) [DOI] [PubMed] [Google Scholar]
- 88.Hernandez-SanMiguel E, Vay L, Santo-Domingo J, Lobaton CD, Moreno A, Montero M, Alvarez J. 2006. The mitochondrial Na+/Ca2+ exchanger plays a key role in the control of cytosolic Ca2+ oscillations. Cell Calcium 40, 53–61. (doi:10.1016/j.ceca.2006.03.009) [DOI] [PubMed] [Google Scholar]
- 89.Ishii K, Hirose K, Iino M. 2006. Ca2+ shuttling between endoplasmic reticulum and mitochondria underlying Ca2+ oscillations. EMBO Rep. 7, 390–396. (doi:10.1038/sj.embor.7400620) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Samanta K, Douglas S, Parekh AB. 2014. Mitochondrial calcium uniporter MCU supports cytoplasmic Ca2+ oscillations, store-operated Ca2+ entry and Ca2+-dependent gene expression in response to receptor stimulation. PLoS ONE 9, e101188 (doi:10.1371/journal.pone.0101188) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kim B, Takeuchi A, Koga O, Hikida M, Matsuoka S. 2012. Pivotal role of mitochondrial Na+ − Ca2+ exchange in antigen receptor mediated Ca2+ signalling in DT40 and A20 B lymphocytes. J. Physiol. 590, 459–474. (doi:10.1113/jphysiol.2011.222927) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Di Capite J, Ng SW, Parekh AB. 2009. Decoding of cytoplasmic Ca2+ oscillations through the spatial signature drives gene expression. Curr. Biol. 19, 853–858. (doi:10.1016/j.cub.2009.03.063) [DOI] [PubMed] [Google Scholar]
- 93.Dupont G. 2014. Modeling the intracellular organization of calcium signaling. Wiley Interdiscip. Rev. Syst. Biol. Med. 6, 227–237. (doi:10.1002/wsbm.1261) [DOI] [PubMed] [Google Scholar]
- 94.Dolmetsch RE, Xu K, Lewis RS. 1998. Calcium oscillations increase the efficiency and specificity of gene expression. Nature 392, 933–936. (doi:10.1038/31960) [DOI] [PubMed] [Google Scholar]
- 95.Lewis RS. 2003. Calcium oscillations in T-cells: mechanisms and consequences for gene expression. Biochem. Soc. Trans. 31, 925–929. (doi:10.1042/bst0310925) [DOI] [PubMed] [Google Scholar]
- 96.Altan-Bonnet G, Germain RN. 2005. Modeling T cell antigen discrimination based on feedback control of digital ERK responses. PLoS Biol. 3, e356 (doi:10.1371/journal.pbio.0030356) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Utzny C, Faroudi M, Valitutti S. 2005. Frequency encoding of T-cell receptor engagement dynamics in calcium time series. Biophys. J. 88, 1–14. (doi:10.1529/biophysj.103.038216) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Marangoni F, Murooka TT, Manzo T, Kim EY, Carrizosa E, Elpek NM, Mempel TR. 2013. The transcription factor NFAT exhibits signal memory during serial T cell interactions with antigen-presenting cells. Immunity 38, 237–249. (doi:10.1016/j.immuni.2012.09.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Waldeck-Weiermair M, Alam MR, Khan MJ, Deak AT, Vishnu N, Karsten F, Imamura H, Graier WF, Malli R. 2012. Spatiotemporal correlations between cytosolic and mitochondrial Ca2+ signals using a novel red-shifted mitochondrial targeted cameleon. PLoS ONE 7, e45917 (doi:10.1371/journal.pone.0045917) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Takeuchi A, Kim B, Matsuoka S. 2015. The destiny of Ca2+ released by mitochondria. J. Physiol. Sci. 65, 11–24. (doi:10.1007/s12576-014-0326-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Aström KJ, Murray RM. 2010. Feedback systems: an introduction for scientists and engineers. Princeton, NJ: Princeton University Press. [Google Scholar]
- 102.Koulnis M, Porpiglia E, Hidalgo D, Socolovsky M. 2014. Erythropoiesis: from molecular pathways to system properties. Adv. Exp. Med. Biol. 844, 37–58. (doi:10.1007/978-1-4939-2095-2_3) [DOI] [PubMed] [Google Scholar]
- 103.Paul D, Dasgupta A, De RK. 2015. Exploring the altered dynamics of mammalian central carbon metabolic pathway in cancer cells: a classical control theoretic approach. PLoS ONE 10, e0137728 (doi:10.1371/journal.pone.0137728) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kotas ME, Medzhitov R. 2015. Homeostasis, inflammation, and disease susceptibility. Cell 160, 816–827. (doi:10.1016/j.cell.2015.02.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sedova M, Dedkova EN, Blatter LA. 2006. Integration of rapid cytosolic Ca2+ signals by mitochondria in cat ventricular myocytes. Am. J. Physiol. Cell Physiol. 291, C840–C850. (doi:10.1152/ajpcell.00619.2005) [DOI] [PubMed] [Google Scholar]
- 106.Wei AC, Liu T, O'Rourke B. 2015. Dual effect of phosphate transport on mitochondrial Ca2+ dynamics. J. Biol. Chem. 290, 16 088–16 098. (doi:10.1074/jbc.M114.628446) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Edelstein SJ. 1971. Extensions of the allosteric model for haemoglobin. Nature 230, 224–227. (doi:10.1038/230224a0) [DOI] [PubMed] [Google Scholar]
- 108.Huser J, Blatter LA, Sheu SS. 2000. Mitochondrial calcium in heart cells: beat-to-beat oscillations or slow integration of cytosolic transients? J. Bioenerg. Biomembr. 32, 27–33. (doi:10.1023/A:1005556227425) [DOI] [PubMed] [Google Scholar]
- 109.De Marchi U, Santo-Domingo J, Castelbou C, Sekler I, Wiederkehr A, Demaurex N. 2014. NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced NAD(P)H production and modulating matrix redox state. J. Biol. Chem. 289, 20 377–20 385. (doi:10.1074/jbc.M113.540898) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kohlhaas M, Liu T, Knopp A, Zeller T, Ong MF, Bohm M, O'Rourke B, Maack C. 2010. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation 121, 1606–1613. (doi:10.1161/CIRCULATIONAHA.109.914911) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kohlhaas M, Maack C. 2010. Adverse bioenergetic consequences of Na+-Ca2+ exchanger-mediated Ca2+ influx in cardiac myocytes. Circulation 122, 2273–2280. (doi:10.1161/CIRCULATIONAHA.110.968057) [DOI] [PubMed] [Google Scholar]
- 112.Ichas F, Mazat JP. 1998. From calcium signaling to cell death: two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochim. Biophys. Acta 1366, 33–50. (doi:10.1016/S0005-2728(98)00119-4) [DOI] [PubMed] [Google Scholar]
- 113.Halestrap AP. 2009. What is the mitochondrial permeability transition pore? J. Mol. Cell Cardiol. 46, 821–831. (doi:10.1016/j.yjmcc.2009.02.021) [DOI] [PubMed] [Google Scholar]
- 114.Camello-Almaraz C, Gomez-Pinilla PJ, Pozo MJ, Camello PJ. 2006a. Mitochondrial reactive oxygen species and Ca2+ signaling. Am. J. Physiol. Cell Physiol. 291, C1082–C1088. (doi:10.1152/ajpcell.00217.2006) [DOI] [PubMed] [Google Scholar]
- 115.Camello-Almaraz MC, Pozo MJ, Murphy MP, Camello PJ. 2006b. Mitochondrial production of oxidants is necessary for physiological calcium oscillations. J. Cell Physiol. 206, 487–494. (doi:10.1002/jcp.20498) [DOI] [PubMed] [Google Scholar]
- 116.Yan Y, Liu J, Wei C, Li K, Xie W, Wang Y, Cheng H. 2008. Bidirectional regulation of Ca2+ sparks by mitochondria-derived reactive oxygen species in cardiac myocytes. Cardiovasc. Res. 77, 432–441. (doi:10.1093/cvr/cvm047) [DOI] [PubMed] [Google Scholar]
- 117.Favero TG, Zable AC, Abramson JJ. 1995. Hydrogen peroxide stimulates the Ca2+ release channel from skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. 270, 25 557–25 563. (doi:10.1074/jbc.270.43.25557) [DOI] [PubMed] [Google Scholar]
- 118.Aon MA, Cortassa S, Maack C, O'Rourke B. 2007. Sequential opening of mitochondrial ion channels as a function of glutathione redox thiol status. J. Biol. Chem. 282, 21 889–21 900. (doi:10.1074/jbc.M702841200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Adam-Vizi V, Starkov AA. 2010. Calcium and mitochondrial reactive oxygen species generation: how to read the facts. J. Alzheimers Dis. 20(Suppl 2), S413–S426. (doi:10.3233/JAD-2010-100465) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Aon MA, Cortassa S, O'Rourke B. 2008. Mitochondrial oscillations in physiology and pathophysiology. Adv. Exp. Med. Biol. 641, 98–117. (doi:10.1007/978-0-387-09794-7_8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Slodzinski MK, Aon MA, O'Rourke B. 2008. Glutathione oxidation as a trigger of mitochondrial depolarization and oscillation in intact hearts. J. Mol. Cell Cardiol. 45, 650–660. (doi:10.1016/j.yjmcc.2008.07.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Liemburg-Apers DC, Willems PH, Koopman WJ, Grefte S. 2015. Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch. Toxicol. 89, 1209–1226. (doi:10.1007/s00204-015-1520-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kim MS, Usachev YM. 2009. Mitochondrial Ca2+ cycling facilitates activation of the transcription factor NFAT in sensory neurons. J. Neurosci. 29, 12 101–12 114. (doi:10.1523/JNEUROSCI.3384-09.2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Parekh AB. 2008. Mitochondrial regulation of store-operated CRAC channels. Cell Calcium 44, 6–13. (doi:10.1016/j.ceca.2007.12.006) [DOI] [PubMed] [Google Scholar]
- 125.Goldbeter A, Gonze D, Houart G, Leloup JC, Halloy J, Dupont G. 2001. From simple to complex oscillatory behavior in metabolic and genetic control networks. Chaos 11, 247–260. (doi:10.1063/1.1345727) [DOI] [PubMed] [Google Scholar]
- 126.Schuster S, Marhl M, Hofer T. 2002. Modelling of simple and complex calcium oscillations. From single-cell responses to intercellular signalling. Eur. J. Biochem. 269, 1333–1355. (doi:10.1046/j.0014-2956.2001.02720.x) [DOI] [PubMed] [Google Scholar]
- 127.Feske S. 2007. Calcium signalling in lymphocyte activation and disease. Nat. Rev. Immunol. 7, 690–702. (doi:10.1038/nri2152) [DOI] [PubMed] [Google Scholar]
- 128.Nagaleekar VK, et al. 2008. IP3 receptor-mediated Ca2+ release in naive CD4T cells dictates their cytokine program. J. Immunol. 181, 8315–8322. (doi:10.4049/jimmunol.181.12.8315) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Feske S, Skolnik EY, Prakriya M. 2012. Ion channels and transporters in lymphocyte function and immunity. Nat. Rev. Immunol. 12, 532–547. (doi:10.1038/nri3233) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hou P, Zhang R, Liu Y, Feng J, Wang W, Wu Y, Ding J. 2014. Physiological role of Kv1.3 channel in T lymphocyte cell investigated quantitatively by kinetic modeling. PLoS ONE 9, e89975 (doi:10.1371/journal.pone.0089975) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Fanger CM, Rauer H, Neben AL, Miller MJ, Wulff H, Rosa JC, Ganellin CR, Chandy KG, Cahalan MD. 2001. Calcium-activated potassium channels sustain calcium signaling in T lymphocytes. Selective blockers and manipulated channel expression levels. J. Biol. Chem. 276, 12 249–12 256. (doi:10.1074/jbc.M011342200) [DOI] [PubMed] [Google Scholar]
- 132.Fomina AF, Fanger CM, Kozak JA, Cahalan MD. 2000. Single channel properties and regulated expression of Ca2+ release-activated Ca2+ (CRAC) channels in human T cells. J. Cell Biol. 150, 1435–1444. (doi:10.1083/jcb.150.6.1435) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Grissmer S, Nguyen AN, Cahalan MD. 1993. Calcium-activated potassium channels in resting and activated human T lymphocytes: expression levels, calcium dependence, ion selectivity, and pharmacology. J. Gen. Physiol. 102, 601–630. (doi:10.1085/jgp.102.4.601) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hess SD, Oortgiesen M, Cahalan MD. 1993. Calcium oscillations in human T and natural killer cells depend upon membrane potential and calcium influx. J. Immunol. 150, 2620–2633. [PubMed] [Google Scholar]
- 135.Weber KS, Hildner K, Murphy KM, Allen PM. 2010. Trpm4 differentially regulates Th1 and Th2 function by altering calcium signaling and NFAT localization. J. Immunol. 185, 2836–2846. (doi:10.4049/jimmunol.1000880) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rotnes JS, Bogen B. 1994. Ca2+ mobilization in physiologically stimulated single T cells gradually increases with peptide concentration (analog signaling). Eur. J. Immunol. 24, 851–858. (doi:10.1002/eji.1830240412) [DOI] [PubMed] [Google Scholar]
- 137.Huang J, Brameshuber M, Zeng X, Xie J, Li QJ, Chien YH, Valitutti S, Davis MM. 2013. A single peptide-major histocompatibility complex ligand triggers digital cytokine secretion in CD4+ T cells. Immunity 39, 846–857. (doi:10.1016/j.immuni.2013.08.036) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kock J, et al. 2014. Nuclear factor of activated T cells regulates the expression of interleukin-4 in Th2 cells in an all-or-none fashion. J. Biol. Chem. 289, 26 752–26 761. (doi:10.1074/jbc.M114.587865) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Podtschaske M, Benary U, Zwinger S, Hofer T, Radbruch A, Baumgrass R. 2007. Digital NFATc2 activation per cell transforms graded T cell receptor activation into an all-or-none IL-2 expression. PLoS ONE 2, e935 (doi:10.1371/journal.pone.0000935) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kar P, Nelson C, Parekh AB. 2012. CRAC channels drive digital activation and provide analog control and synergy to Ca2+-dependent gene regulation. Curr. Biol. 22, 242–247. (doi:10.1016/j.cub.2011.12.025) [DOI] [PubMed] [Google Scholar]
- 141.Lin J, Harding A, Giurisato E, Shaw AS. 2009. KSR1 modulates the sensitivity of mitogen-activated protein kinase pathway activation in T cells without altering fundamental system outputs. Mol. Cell Biol. 29, 2082–2091. (doi:10.1128/MCB.01634-08) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kingeter LM, Paul S, Maynard SK, Cartwright NG, Schaefer BC. 2010. Cutting edge: TCR ligation triggers digital activation of NF-kappaB. J. Immunol. 185, 4520–4524. (doi:10.4049/jimmunol.1001051) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Santos SD, Verveer PJ, Bastiaens PI. 2007. Growth factor-induced MAPK network topology shapes Erk response determining PC-12 cell fate. Nat. Cell Biol. 9, 324–330. (doi:10.1038/ncb1543) [DOI] [PubMed] [Google Scholar]
- 144.Tian T, Harding A, Inder K, Plowman S, Parton RG, Hancock JF. 2007. Plasma membrane nanoswitches generate high-fidelity Ras signal transduction. Nat. Cell Biol. 9, 905–914. (doi:10.1038/ncb1615) [DOI] [PubMed] [Google Scholar]
- 145.Harding A, Hancock JF. 2008. Ras nanoclusters: combining digital and analog signaling. Cell Cycle 7, 127–134. (doi:10.4161/cc.7.2.5237) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hermann-Kleiter N, Baier G. 2010. NFAT pulls the strings during CD4+ T helper cell effector functions. Blood 115, 2989–2997. (doi:10.1182/blood-2009-10-233585) [DOI] [PubMed] [Google Scholar]
- 147.Srikanth S, Gwack Y. 2013. Orai1-NFAT signalling pathway triggered by T cell receptor stimulation. Mol. Cells 35, 182–194. (doi:10.1007/s10059-013-0073-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Valdor R, Macian F. 2013. Induction and stability of the anergic phenotype in T cells. Semin. Immunol. 25, 313–320. (doi:10.1016/j.smim.2013.10.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Izsepi E, Himer L, Szilagyi O, Hajdu P, Panyi G, Laszlo G, Matko J. 2013. Membrane microdomain organization, calcium signal, and NFAT activation as an important axis in polarized Th cell function. Cytometry A 83, 185–196. (doi:10.1002/cyto.a.22234) [DOI] [PubMed] [Google Scholar]
- 150.Porter CM, Clipstone NA. 2002. Sustained NFAT signaling promotes a Th1-like pattern of gene expression in primary murine CD4+ T cells. J. Immunol. 168, 4936–4945. (doi:10.4049/jimmunol.168.10.4936) [DOI] [PubMed] [Google Scholar]
- 151.Tomida T, Hirose K, Takizawa A, Shibasaki F, Iino M. 2003. NFAT functions as a working memory of Ca2+ signals in decoding Ca2+ oscillation. EMBO J. 22, 3825–3832. (doi:10.1093/emboj/cdg381) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhu J, McKeon F. 1999. NF-AT activation requires suppression of Crm1-dependent export by calcineurin. Nature 398, 256–260. (doi:10.1038/18473) [DOI] [PubMed] [Google Scholar]
- 153.Okamura H, et al. 2000. Concerted dephosphorylation of the transcription factor NFAT1 induces a conformational switch that regulates transcriptional activity. Mol. Cell 6, 539–550. (doi:10.1016/S1097-2765(00)00053-8) [DOI] [PubMed] [Google Scholar]
- 154.Baine I, Abe BT, Macian F. 2009. Regulation of T-cell tolerance by calcium/NFAT signaling. Immunol. Rev. 231, 225–240. (doi:10.1111/j.1600-065X.2009.00817.x) [DOI] [PubMed] [Google Scholar]
- 155.Taylor CW, Tovey SC, Rossi AM, Lopez Sanjurjo CI, Prole DL, Rahman T. 2014. Structural organization of signalling to and from IP3 receptors. Biochem. Soc. Trans. 42, 63–70. (doi:10.1042/BST20130205) [DOI] [PubMed] [Google Scholar]
- 156.Bootman M. 1994. Quantal Ca2+ release from InsP3-sensitive intracellular Ca2+ stores. Mol. Cell. Endocrinol. 98, 157–166. (doi:10.1016/0303-7207(94)90134-1) [DOI] [PubMed] [Google Scholar]
- 157.Irvine R. 1990. ‘Quantal’ Ca2+ release and the control of Ca2+ entry by inositol phosphates—a possible mechanism. FEBS Lett. 263, 5–9. (doi:10.1016/0014-5793(90)80692-C) [DOI] [PubMed] [Google Scholar]
- 158.Parys J, Missiaen L, Smedt H, Sienaert I, Casteels R. 1996. Mechanisms responsible for quantal Ca2+ release from inositol trisphosphate-sensitive calcium stores. Pflugers Arch. 423, 359–367. (doi:10.1007/s004240050145) [DOI] [PubMed] [Google Scholar]
- 159.Dupont G, Swillens S, Clair C, Tordjmann T, Combettes L. 2000. Hierarchical organization of calcium signals in hepatocytes: from experiments to models. Biochim. Biophys. Acta 1498, 134–152. (doi:10.1016/S0167-4889(00)00090-2) [DOI] [PubMed] [Google Scholar]
- 160.Baran J, Kowalczyk D, Ozog M, Zembala M. 2001. Three-color flow cytometry detection of intracellular cytokines in peripheral blood mononuclear cells: comparative analysis of phorbol myristate acetate-ionomycin and phytohemagglutinin stimulation. Clin. Diagn. Lab Immunol. 8, 303–313. (doi:10.1128/CDLI.8.2.303-313.2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Madden J, Howarth PP, Godfrey RR, Frew AA. 1999. The kinetics and stimulant dependence of cytokine production by blood and bronchoalveolar lavage T cells evaluated at the single cell level. Cytokine 11, 510–517. (doi:10.1006/cyto.1998.0456) [DOI] [PubMed] [Google Scholar]
- 162.Rostaing L, Tkaczuk J, Durand M, Peres C, Durand D, de Preval C, Ohayon E, Abbal M. 1999. Kinetics of intracytoplasmic Th1 and Th2 cytokine production assessed by flow cytometry following in vitro activation of peripheral blood mononuclear cells. Cytometry 35, 318–328. (doi:10.1002/(SICI)1097-0320(19990401)35:4<318::AID-CYTO4>3.0.CO;2-4) [DOI] [PubMed] [Google Scholar]
- 163.Shatynski KE, Chen H, Kwon J, Williams MS. 2012. Decreased STAT5 phosphorylation and GATA-3 expression in NOX2-deficient T cells: role in T helper development. Eur. J. Immunol. 42, 3202–3211. (doi:10.1002/eji.201242659) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Colella M, Grisan F, Robert V, Turner JD, Thomas AP, Pozzan T. 2008. Ca2+ oscillation frequency decoding in cardiac cell hypertrophy: role of calcineurin/NFAT as Ca2+ signal integrators. Proc. Natl Acad. Sci. USA 105, 2859–2864. (doi:10.1073/pnas.0712316105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Jang SI, Ong HL, Liu X, Alevizos I, Ambudkar IS. 2016. Up-regulation of store-operated Ca2+ entry and nuclear factor of activated T cells promote the acinar phenotype of the primary human salivary gland cells. J. Biol. Chem. 291, 8709–8720. (doi:10.1074/jbc.M115.701607) [DOI] [PMC free article] [PubMed] [Google Scholar]