

A comprehensive study of 100 Arabidopsis accessions at the phenotype and transcriptome level elucidated extraordinary plasticity of growth responses to mild drought.
Abstract
Plant growth and crop yield are negatively affected by a reduction in water availability. However, a clear understanding of how growth is regulated under nonlethal drought conditions is lacking. Recent advances in genomics, phenomics, and transcriptomics allow in-depth analysis of natural variation. In this study, we conducted a detailed screening of leaf growth responses to mild drought in a worldwide collection of Arabidopsis thaliana accessions. The genetic architecture of the growth responses upon mild drought was investigated by subjecting the different leaf growth phenotypes to genome-wide association mapping and by characterizing the transcriptome of young developing leaves. Although no major effect locus was found to be associated with growth in mild drought, the transcriptome analysis delivered further insight into the natural variation of transcriptional responses to mild drought in a specific tissue. Coexpression analysis indicated the presence of gene clusters that co-vary over different genetic backgrounds, among others a cluster of genes with important regulatory functions in the growth response to osmotic stress. It was found that the occurrence of a mild drought stress response in leaves can be inferred with high accuracy across accessions based on the expression profile of 283 genes. A genome-wide association study on the expression data revealed that trans regulation seems to be more important than cis regulation in the transcriptional response to environmental perturbations.
INTRODUCTION
Due to their sessile lifestyle, plants constantly need to adapt to the environment and find a balance between growth and survival (Claeys and Inzé, 2013). One of the main environmental factors impacting plants is the availability of water. Until now, the focus of many studies has primarily been on the mechanisms regulating plant survival during severe drought stress, but also mild, nonlethal drought conditions significantly alter growth as a result of reduced cell proliferation and/or expansion (Aguirrezabal et al., 2006; Pereyra-Irujo et al., 2008; Clauw et al., 2015). The regulatory mechanisms behind this growth reduction are distinct from those during severe drought and are poorly understood (Claeys and Inzé, 2013).
Arabidopsis thaliana grows in a wide variety of habitats throughout the Eurasian continent and beyond (Hoffmann, 2002). Different accessions (naturally occurring inbred lines) have been found to be genetically adapted to their specific environments (Fournier-Level et al., 2011). Extensive phenotypic variation is observed for traits such as flowering time, water use efficiency, nitrate uptake and nitrate use efficiency, heat and drought stress responses, salt tolerance, and growth responses upon moderate drought stress (Kenney et al., 2014; Bouchabke et al., 2008; Chardon et al., 2010; Vile et al., 2012; Katori et al., 2010; Clauw et al., 2015).
Recent sequencing efforts have made available high-resolution genotypes for over 1000 Arabidopsis accessions (The 1001 Genomes Consortium, 2016). At the same time, automated phenotyping platforms have been developed (Granier et al., 2006; Skirycz et al., 2011b; Dhondt et al., 2013; Tisné et al., 2013; Wuyts et al., 2015; Flood et al., 2016), allowing for precise and high-throughput measurements of plant growth. These developments enhance our ability to map causal genetic polymorphisms through genome-wide association studies (GWAS), an approach that has been applied successfully to many different traits, ranging from genetically simple traits, such as biotic stress resistance (Huard-Chauveau et al., 2013), to more complex features, such as root system architecture and rosette growth (Rosas et al., 2013; Meijón et al., 2014; Bac-Molenaar et al., 2015).
Any causal polymorphism should either affect gene function or gene expression. Variability in gene expression between different accessions can be substantial and is thought to be an important cause of phenotypic variation. For example, gene expression variability of the auxin-responsive genes is more important than protein-encoding sequence polymorphisms for the variation in the phenotypic response to auxin treatment (Delker et al., 2010). Similarly, exposure to salicylic acid or mild drought stress elicits substantially different genome-wide expression responses in different natural accessions (van Leeuwen et al., 2007; Des Marais et al., 2012; Clauw et al., 2015). However, part of this variation can be considered neutral or close to neutral and is buffered at the phenotypic level (Fu et al., 2009).
In this study, we characterized growth-related phenotypes and responses to mild drought stress of 98 accessions. To gather more insight into the underlying genetic architecture, the genetic loci associated with the differential growth observed upon mild drought were mapped using a multitrait mixed model GWAS (Korte et al., 2012). In addition, genome-wide transcriptome profiling was conducted on young, developing leaves (at the final day of the cell proliferation phase) in control and mild drought conditions for 89 accessions. The transcriptional response was characterized using a clustering approach that highlighted an important gene regulatory network for the growth response to mild drought stress. A classification model, based on the expression profiles of 283 genes exhibiting a significant treatment effect across accessions, showed that the occurrence of mild drought stress can be predicted efficiently from leaf transcriptomics data. Finally, genome-wide association mapping was performed on the expression data in order to identify regulators of the transcriptional response to mild drought, revealing a major regulatory role for trans-acting loci.
The genetic background of quantitative traits, like growth, is expected to involve a large number of loci with small effects, which require extensive statistical power to map. By analyzing, in actively growing leaves, the transcriptional responses to a growth-reducing environmental perturbation, like mild drought, we could identify genes and gene clusters that are plausible components of the complex network that shapes leaf growth.
RESULTS
Natural Variation in Leaf Growth Responses to Mild Drought
To study the diversity of mild drought stress responses in Arabidopsis, we selected a set of 98 accessions that reflects the large genetic and phenotypic variation in this species (Supplemental Data Set 1). The accessions originate from diverse regions ranging from the Cape Verde Islands in the south to Scandinavia in the north and spanning the entire Eurasian continent including Japan. Leaf growth responses were analyzed in plants grown in well-watered soil or in nonlethal, mild drought conditions as described by Clauw et al. (2015). The mild drought treatment was initiated 4 d after stratification (DAS), 1 d before the initiation of the third leaf from the shoot apical meristem (Skirycz et al., 2010). The overall growth response was determined by measuring the projected rosette area (PRA), using the automated phenotyping platform WIWAMxy (Skirycz et al., 2011b). A more detailed view on growth responses was obtained by specifically quantifying different growth-related phenotypes of the third emerging leaf (Figure 1).
Figure 1.

Distribution of Leaf Growth-Related Phenotypes in 98 Arabidopsis Accessions.
Histograms of the distribution of the different phenotypes measured in 98 accessions under control (blue) and mild drought (pink) conditions. The distribution of percent reduction is given in the inset histograms colored in green.
(B) Third leaf size at maturity (23 DAS).
(C) Third leaf size at the proliferation stage.
(D) Pavement cell area of the third leaf at maturity.
(E) Pavement cell number of the third leaf at maturity.
(F) Stomatal index of the third leaf at maturity. The Ler-1 accession was not included in the histogram for pavement cell number due to its extremely high number of cells (117,165 pavement cells).
At maturity, under control conditions, both rosette and third leaf areas showed an almost 4-fold difference between the smallest and the largest accession (Figures 1A and 1B). Also, the reduction in leaf and rosette areas caused by mild drought stress was highly variable between the accessions (Figures 1A and 1B). Rosette and third leaf areas at maturity correlated well in both control (r = 0.62; P < 0.0001) and mild drought conditions (r = 0.76; P < 0.0001; Supplemental Figures 1A and 1B). On average, rosette areas of plants grown under mild drought were 62% smaller than those grown under control conditions; the average reduction of the third leaf area was similar (57%; Table 1). The correlation of the relative reduction under mild drought stress with the respective sizes under control conditions was nonsignificant for the rosette area (r = 0.10, Pearson correlation coefficient; P = 0.33, t test; Supplemental Figure 1C) and significant, although not very highly, for the area of the third leaf (r = 0.39; P < 0.0001; Supplemental Figure 1D). This indicates that large plants are not necessarily more sensitive to mild drought stress. In general, accessions that were large under control conditions remained large under mild drought stress. This is shown by the significant positive correlation between the rosette areas (r = 0.73; P < 0.001; Supplemental Figure 1E) and between the third leaf areas (r = 0.59; P < 0.001; Supplemental Figure 1F) at maturity in control and mild drought stress conditions.
Table 1. Overview of the Growth-Related Parameters.
| Phenotype | Control | Mild Drought Stress | Percentage of Reduction |
|---|---|---|---|
| Rosette area (mm2) | 677 ± 7 | 257 ± 7 | 62% (P < 0.0001) |
| Third leaf area M (mm2) | 121 ± 2 | 51 ± 2 | 57% (P < 0.0001) |
| Third leaf area P (mm2) | 0.027 ± 0.100 | 0.023 ± 0.153 | 18% (P = 0.1782) |
| Pavement cell area (µm2) | 3,565 ± 17 | 2,000 ± 17 | 42% (P < 0.0001) |
| Pavement cell number | 33,789 ± 64 | 22,773 ± 64 | 30% (P < 0.0001) |
| Stomatal index (%) | 45 ± 2 | 41 ± 2 | 6% (P < 0.0001) |
Averages of the phenotypes of the 98 accessions and their percentage of reductions under mild drought stress compared with control conditions. Values represent averages ± se except for the third leaf area at proliferation, for which the median was calculated because of the non-normal distribution of the values. P values are the significance of the difference between control and mild drought treatment over all accessions (Mann-Whitney U test). M, at maturity; P, at proliferation.
We identified the most tolerant or most sensitive accessions, based on the responses of both rosette and leaf areas at maturity to mild drought. NFA-10, Pna-10, and ICE-163 were the three accessions showing the lowest size reduction, while ICE-61, Can-0, and Rubezhnoe-1 showed the highest reduction upon exposure to mild drought stress (Supplemental Figure 2).
In conclusion, imposing mild drought stress early during leaf development did impair leaf and rosette areas for all accessions, but the response differed strongly between accessions. We also observed that tolerance or sensitivity to mild drought was not, or only weakly, related to the plant size in control conditions.
Cellular Parameters Defining Leaf Area
To study how cell proliferation and cell expansion, the two main drivers of leaf growth (Andriankaja et al., 2012), are affected by mild drought, pavement cell number, pavement cell area, and stomatal index were measured for all accessions in the third leaf at maturity.
For both pavement cell area and pavement cell number, an up to 4-fold difference was observed between the accessions under control conditions (Figures 1D and 1E). This range reflects what was found for rosette and third leaf areas. For further analysis of pavement cell numbers, we excluded Ler-1 because its number of pavement cells (117,165 ± 4174 cells) is doubled compared with the second-ranked accession (Bl-1; 58,531 ± 4139 cells), a known consequence of the ERECTA mutation in the Ler-1 accession (Tisné et al., 2011).
To quantify the relationship between the cellular parameters and the final leaf size in the 98 accessions, Pearson correlations were calculated. Both pavement cell area and number separately showed moderate but significant (P < 0.0001) correlations with final leaf area (r = 0.41 and r = 0.54, respectively; Supplemental Figures 1I and 1J). Taking both cellular traits together, however, explains 72% (multiple R2; P < 0.0001) of the variation in final leaf area. There was a negative correlation between pavement cell area and pavement cell number, which was moderate but significant (r = −0.46; P < 0.0001; Figure 2). When analyzing the relation between cell number, cell area, and final leaf area, we found that none of the accessions with the 10% largest pavement cells was present in the category having the 10% highest pavement cell numbers (Figure 2). From the 20 accessions within the category of either 10% largest or most pavement cells, 16 accessions had larger than average leaf sizes. Other accessions with larger leaves had both an intermediate pavement cell number and area. This shows that the accessions use all three possible strategies to produce large leaves: either many cells or large cells, or an intermediate number of cells with intermediate sizes.
Figure 2.

Pavement Cell Area and Pavement Cell Number in Relation to the Third Leaf Area at Maturity.
Dots are color-coded according to third leaf size under control conditions, from small (green) to large (red). Dashed lines represent the 90% quantile, where 10% of the accessions have a higher value for cell number or area. Accessions that are above the 90% quantiles of pavement cell area or pavement cell number are labeled.
Upon exposure to mild drought stress, the accessions showed on average a 42% reduction in pavement cell area, whereas the pavement cell number was on average 30% reduced (Table 1). Interestingly, although three accessions (Pna-10, Br-0, and Ga-0) showed an increase in pavement cell number of 20, 9, and 4%, respectively, under mild drought stress, the leaf area was reduced due to a proportionally larger reduction in pavement cell area (36, 59, and 63%, respectively).
Under mild drought stress, the reduction in the area of the third leaf correlated significantly with both the reduction in pavement cell area (r = 0.53; P < 0.0001; Supplemental Figure 1G) and the reduction in pavement cell number (r = 0.70; P < 0.0001; Supplemental Figure 1H).
The stomatal index, i.e., the percentage of stomata on total cell number, was significantly affected by mild drought (P < 0.0001; Table 1), but the effect size of the mild drought was smaller compared with the other phenotypes studied. On average, stomatal indices were 6% reduced, with changes ranging from a reduction of 26% (Ler-1) to an increase of 13% (Wt-5). Under control conditions, an average stomatal index of 0.45 was recorded (Table 1, Figure 1F).
The cellular analysis demonstrates that both cell division and expansion were negatively affected by the mild drought stress and that the response differed between accessions.
Effect of Mild Drought Stress on the Third Leaf during Proliferation
Because the mild drought treatment was already initiated at 4 DAS, the effect on early leaf development could be studied. To this end, the third leaf was harvested at the last day at which all leaf cells are still dividing, as determined for each accession by microscopy analysis (see Methods). Depending on the accession, this cell proliferation-to-cell expansion transition occurred at 8 to 10 DAS. At these time points, the third leaf areas of plants grown in well-watered conditions ranged from 0.009 to 0.149 mm2 (Figure 1C). There was no significant correlation between leaf size at the last day of full proliferation and the leaf size at maturity (r = −0.04; P = 0.7) or the pavement cell number at maturity (r = −0.15; P = 0.2). On average, the mild drought stress caused a reduction of 18% in the third leaf area at this early developmental time point (Table 1), but we could not find a significant correlation (r = −0.02; P = 0.85) between the leaf size reductions under mild drought at this proliferation stage and at maturity.
Genome-Wide Association Mapping
Next, a multitrait mixed model (Korte et al., 2012) was used to scan the genome for polymorphisms associated with the phenotypes discussed above. The multitrait model enables the specific detection of genetic loci that are associated with a differential response to mild drought by treating the phenotype as a function of genotype (G), environment (E), and the interaction of genotype and environment (G×E). The multitrait model has proven to deliver more power in comparison to separate single-trait GWAS, which delivered little to no significant results for our phenotypes (Supplemental Figure 3 and Supplemental Table 1).
Although all traits were highly heritable (Table 2), marginal single nucleotide polymorphism (SNP) associations reached genome-wide significance only for the differential response to mild drought of the third leaf at the proliferation stage. For this trait, a highly significant association on chromosome 4 was found (Supplemental Figure 4A). The 10-kb window covering the association contains five genes: NODULE-INCEPTION-LIKE PROTEIN7, a pre-tRNA encoding gene (AT4G24025), two genes encoding unknown proteins (AT4G24026 and AT4G24030), and TREHALASE1 (TRE1).
Table 2. Heritability Estimates.
| Phenotype | Heritability-Control | Heritability-Mild Drought |
|---|---|---|
| Rosette area | 0.64 ± 0.13 | 0.91 ± 0.13 |
| Third leaf area M | 0.87 ± 0.13 | 0.61 ± 0.18 |
| Third leaf area P | 0.22 ± 0.29 | 0.7 ± 0.31 |
| Pavement cell area | 0.45 ± 0.21 | 0.99 ± 0.01 |
| Pavement cell number | 0.99 ± 0.05 | 0.44 ± 0.31 |
| Stomatal index | 0.69 ± 0.16 | 0.84 ± 0.13 |
Overview of the heritability estimates from the multitrait mixed model ± se for the different phenotypes in control and mild drought conditions. M, at maturity; P, at proliferation. Heritability estimates form the single-trait mixed model can be found in Supplemental Table 1.
Although the SNP associations were not highly significant, the estimated effect sizes of the most significant associations were around 20% (Supplemental Table 2). However, previous simulations (Korte and Farlow, 2013) have shown that with the limited sample size of this study only a quarter of SNPs with this effect size are expected to be significant at the Bonferroni threshold. In addition, we can expect that the majority of the variation will be explained by loci with small phenotypic effects. This notion is supported by the presence of obvious subthreshold peaks associated with the differential response to mild drought stress (Supplemental Figures 3 and 4). For third leaf size at maturity, there were several highly suggestive association peaks on chromosomes 1, 3, and 5 (Supplemental Figure 4B), covering 30 candidate genes (Supplemental Table 3). One of these genes encodes miR171c, which is involved in controlling cell differentiation at the periphery of the shoot apical meristem (Schulze et al., 2010) and regulating chlorophyll biosynthesis and leaf growth in the light through gibberellic acid (GA)-DELLA signaling (Ma et al., 2014). Promising peaks of association were also identified for rosette area, pavement cell area, pavement cell number, and stomatal index (Supplemental Figure 5 and Supplemental Data Set 2).
Natural Variation in Gene Expression upon Mild Drought
The lack of significant associations with leaf growth-related phenotypes reflects the high complexity of the underlying genetic architecture. To further investigate the genetic architecture of these complex quantitative traits and their response to mild drought, we analyzed the natural variation of gene expression. Plants were grown under well-watered or mild drought conditions, and the third leaf was harvested for RNA-seq analysis at the last day of proliferation, as determined through microscopy analysis for each accession (see Methods). The transcriptional responses to drought were highly variable between accessions; therefore, we used CAST (cluster affinity search technique) clustering (Ben-Dor et al., 1999) to identify clusters of genes that covaried over the different accessions in their transcriptional response to drought.
The obtained clusters showed variable expression profiles, with up- or downregulation of the clustered genes upon drought being highly accession dependent (Supplemental Figure 6). The clusters described here in more detail were selected on size (more than 10 genes), enrichment for drought- and growth-related Gene Ontology (GO) categories, and presence of genes likely involved in the drought response used in this particular experimental setup. This selection resulted in eight clusters that were enriched for genes involved in drought/osmotic stress responses, cell wall modifications, or cell cycle (Supplemental Data Set 3 and Supplemental Figure 6). Remarkably, the largest of these clusters (cluster A; Supplemental Data Set 4), with 145 genes, contains the main components of a gene regulatory network that was previously proposed to regulate the osmotic stress response of leaf growth (Dubois et al., 2013, 2015), with genes such as ACC-SYNTHASE6 (ACS6), ETHYLENE RESPONSE FACTOR6 (ERF6), ERF11, MAP KINASE3 (MPK3), WRKY DNA BINDING PROTEIN 33 (WRKY33), and SALT TOLERANCE ZINC FINGER (STZ). Furthermore, the cluster contains 53 jasmonic acid (JA)-related genes (Supplemental Data Set 4), including the main JA biosynthesis and primary response genes such as JASMONATE INSENSITIVE1 (JAI1/JIN1/MYC2) and multiple JASMONATE ZIM DOMAIN (JAZ) protein-encoding genes (Dombrecht et al., 2007; Pauwels and Goossens, 2011). In addition, the cluster contains well-known drought-responsive genes such as CALCINEURIN B-LIKE PROTEIN1, DEHYDRATION-RESPONSIVE ELEMENT BINDING PROTEIN 1B (DREB1B), DREB1D, EARLY-RESPONSIVE TO DEHYDRATION7 (ERD7), ERD10, and ERD15.
Cluster C contains 41 genes and was enriched for response to water deprivation, osmotic stress, and abscisic acid (ABA). Among these are typical drought-related genes such as nine late embryogenesis abundant proteins (Hundertmark and Hincha, 2008) and DESSICATION RESPONSIVE PROTEIN 29A (Supplemental Data Set 5).
Of the eight selected clusters, cluster D was found to be enriched for genes involved in the cell cycle. Three genes are involved in spindle formation (END BINDING PROTEIN 1B, AUGMIN SUBUNIT2, and BUB1-RELATED) (Green et al., 2005; Hotta et al., 2012; Paganelli et al., 2015), one gene encodes a cyclin-dependent kinase (CDT1A), and one encodes a cyclin (CYCLIN H;1). Interestingly, CYCH1-1 is also involved in regulating drought responses (Zhou et al., 2013).
Predicting the Occurrence of Mild Drought Conditions from Gene Expression
We also investigated if the transcriptomic profile could be used to predict whether a plant has been subjected to mild drought stress. To reduce the dimensionality of the problem, we only considered genes for which treatment effects (control versus mild drought) explained a significant portion of the genes’ expression variation observed across the data set. One-way ANOVA analysis identified 283 genes with a significant treatment effect (false discovery rate [FDR]-adjusted P < 0.05; Supplemental Data Set 6). Based on the expression profiles of the 283 selected genes, a support vector machine (SVM) classification model was built to distinguish between the samples in the two treatment groups. The trained model was highly successful in separating control from mild drought-treated samples: the model’s F-score of 0.859 indicated high sensitivity and precision, whereas the area under the curve (AUC) of 0.926 implied a good separation of both treatment groups.
GO enrichment analysis on the set of 283 so-called “stress predictor” genes revealed that the set was highly enriched for genes responding to water deprivation, osmotic stress, and ABA (Table 3) and also for the common drought genes identified by Clauw et al. (2015) (P < 0.001; 17-fold enrichment). Eleven of the 283 genes exhibited the same direction of expression change upon mild drought for at least 80 out of 89 accessions analyzed (Figure 3, Table 4). Moreover, these 11 genes (except CHLOROPLAST VESICULATION) were previously found to be among a conserved transcription response (in six accessions) to mild drought (Clauw et al., 2015).
Table 3. Twenty of the Most Significantly Enriched GO Categories in the 283 Stress Predictors.
| Description | GO Term | Log2 Enrichment | Q-Value |
|---|---|---|---|
| Response to water deprivation | GO:0009414 | 2.43 | 3.52E-15 |
| Response to water | GO:0009415 | 2.42 | 5.10E-15 |
| Response to superoxide | GO:0000303 | 4.43 | 7.80E-11 |
| Response to inorganic substance | GO:0010035 | 1.44 | 1.29E-10 |
| Response to abscisic acid | GO:0009737 | 1.8 | 1.30E-09 |
| Hyperosmotic salinity response | GO:0042538 | 3.03 | 1.53E-09 |
| Response to oxygen-containing compound | GO:1901700 | 1.07 | 2.93E-09 |
| Response to lipid | GO:0033993 | 1.61 | 3.73E-09 |
| Single-organism cellular process | GO:0044763 | 0.51 | 4.41E-09 |
| Response to alcohol | GO:0097305 | 1.64 | 1.08E-08 |
| Cellular response to ABA stimulus | GO:0071215 | 2.23 | 1.52E-08 |
| Response to chemicals | GO:0042221 | 0.82 | 2.88E-08 |
| Single-organism process | GO:0044699 | 0.4 | 3.03E-08 |
| Hyperosmotic response | GO:0006972 | 2.55 | 4.80E-08 |
| Cellular response to lipid | GO:0071396 | 1.91 | 1.50E-07 |
| Cellular response to oxygen-containing compound | GO:1901701 | 1.42 | 1.53E-07 |
| Cellular response to alcohol | GO:0097306 | 2.02 | 1.61E-07 |
| ABA-activated signaling pathway | GO:0009738 | 2.15 | 1.76E-07 |
| Response to stimulus | GO:0050896 | 0.55 | 3.27E-07 |
| Response to stress | GO:0006950 | 0.72 | 3.54E-07 |
The Q-values are Bonferroni corrected P values (family-wise error rate threshold set at 0.05).
Figure 3.

Differential Expression of Eleven “Signature” Genes in 89 Accessions.
The genes displayed show similar fold changes upon mild drought stress in at least 80 accessions. Fold changes are arcsinh transformed (0.7 equals a 2-fold change).
Table 4. Genes with Similar Transcription Responses to Mild Drought Stress in 90% of the Accessions.
| Gene | Name | Process | Reference |
|---|---|---|---|
| AT1G69260 | AFP1 | ABA signaling | Garcia et al. (2008) |
| AT3G11410 | PP2CA/AHG3 | ABA signaling | Rodrigues et al. (2013) |
| AT2G46680 | HB7 | ABA signaling/leaf growth in drought conditions | (Olsson et al. (2004), Ré et al. (2014), Valdés et al. (2012) |
| AT2G39800 | P5CS1 | Proline biosynthesis | Strizhov et al. (1997) |
| AT3G57520 | SIP2 | Raffinose-specific α-galactoside hydrolase | Peters et al. (2010) |
| AT5G53390 | FOP1 | Wax ester biosynthesis | Takeda et al. (2013) |
| AT2G03090 | EXPA15 | Cell wall modification | Lee et al. (2001) |
| AT5G64570 | XYL4 | Cell wall modification | Minic et al. (2004) |
| AT1G08650 | PPCK1 | Rosette growth and development | Meimoun et al. (2009) |
| AT2G25625 | CV | Chloroplast disruption | Wang and Blumwald (2014) |
| AT1G60960 | IRT3 | Zn/Fe transporter | Lin et al. (2009) |
In total, 160 of the 283 stress predictor genes were in common with genes that are differentially expressed in mature leaves exposed to severe drought (Matsui et al., 2008; Huang et al., 2008; Harb et al., 2010) or mild drought (Baerenfaller et al., 2012; Des Marais et al., 2012; Harb et al., 2010) and as such they can be considered as a highly robust series of drought stress-responsive genes independent of whether the affected tissue is still growing or has reached maturity (Supplemental Figure 7 and Supplemental Data Set 4). Not unexpectedly, these 160 genes were enriched for genes that respond to water deprivation, salt stress, and ABA (FDR corrected P values < 0.001). On the other hand, 42 of the 283 stress predictor genes were previously not reported to be differentially expressed by mild or severe drought stress in mature leaves and did not reveal an enrichment of any abiotic stress-related GO category (Supplemental Data Set 4). These genes might have a specific role in drought stress responses in proliferating leaves.
Expression GWAS
To investigate the genetic basis of expression variation both between accessions and between treatments, a multitrait mixed model was applied to the transcriptomics data. To get a high-confidence set for further analysis, genes for which obvious model inflation was detected were removed (see Methods). This stringent statistical selection resulted in a set of 509 genes for which SNPs were found to associate with treatment-independent gene expression differences across accessions (Figure 4A) and a set of 158 genes for which SNPs associated with differential expression upon mild drought (Figure 4B). The multiple testing problems arising through testing association with gene expression of tens of thousands of genes were ignored. Instead, an amenable significance threshold of 10-7 for each gene was used, as we focused on the comparison of specific patterns and peaks of associations for each gene.
Figure 4.

Overview of eGWAS Results.
The scatterplots show for each gene (positions on y axes) the SNPs that associate with treatment-independent expression (common test; [A]) and SNPs that associate with treatment-dependent expression (trait-specific test; [B]). SNPs were considered to be in trans at a threshold of 5 kb from the transcription start and stop site.
Regulators of Treatment-Independent Gene Expression
In total, 509 genes had one or more SNPs that associated with the variation in treatment-independent gene expression (control and mild drought taken together). A clear diagonal band is visible (Figure 4A), containing SNPs located in the proximity of the gene whose expression they are associated with, indicating cis-regulatory SNPs. From the spatial distribution of the SNPs associated with treatment-independent expression, we observed that the proportion of SNPs clearly increased in the promoter region 2 kb upstream of the transcription start site. The largest density of associated SNPs was located within the 1-kb upstream region, after which the proportion of SNPs rapidly declined (Figure 5; Supplemental Figure 8). Associated SNPs were also located in the transcribed regions. These SNPs could be in linkage disequilibrium with upstream causative markers or could be involved in intron-mediated regulation of gene expression (Rose, 2008). Alternatively, changes in intronic sequences may cause differences in alternative splicing (Kesari et al., 2012). Downstream of the transcribed region, the number of SNPs that regulate expression rapidly declined.
Figure 5.

Histogram of Locations of SNPs Associated with Treatment-Independent Expression.
The transcription start site (TSS) and stop site (Terminus) are the untranslated region boundaries as determined by the Arabidopsis genome annotation (TAIR10; www.arabidopsis.org). Indicated in red are the associated SNPs located upstream of the TSS; the 1 kb, 2 kb, and SNPs further upstream are colored in different shades of red. In blue are the associated SNPs downstream of the transcription stop site; different shades of blue indicate the different distances downstream of the transcription stop site (1 kb, 2 kb, and further). In green are the associated SNPs located in the gene itself, mapped relative to the gene length (distribution of absolute SNP positions in the gene are shown in Supplemental Figure 8). Numbers of SNPs are raw SNP counts, and bin size is 100 bp.
Regulators of Differential Gene Expression upon Mild Drought
SNPs associated with differential expression upon mild drought were found for 158 genes. These genes were not enriched for known stress response genes. In total, 869 SNPs were found to be associated with differential expression of these 158 genes, which is more than expected by chance (443 SNPs). In contrast to the SNPs associated with treatment-independent gene expression, none of the associated SNPs was located in cis. In the used multitrait mixed model, there are no differences in statistical power between treatment-independent or -dependent expression, nor between cis- and trans-located loci (Korte et al., 2012).
Of the 869 SNPs, 187 were located in genes with various functions but without enrichment for the two GO categories transcriptional regulation or transcription factors. Only two genes (HOMEODOMAIN GLABROUS10 and U12 SMALL NUCLEOLAR RNA) overlapped with genes identified in the phenotypic GWAS, as described in Supplemental Table 3 and Supplemental Data Set 2. It is noteworthy that 172 of the 869 associated SNPs were located in transposable elements (Supplemental Data Set 7).
By conducting an additional variance component analysis, we could identify that most of the variation of gene expression could be explained by variation in trans and trans × environment (Supplemental Figure 9). Together with the expression GWAS (eGWAS) results, this shows that variation in trans seems to explain much more variation of the transcriptional response to mild drought compared with cis-located loci. It is important to note that these trans regulators are not necessarily having a direct regulatory effect but may interact with other regulators or may affect gene expression indirectly through induced physiological changes. Illustrative for this is the finding that among the potential trans regulators we were unable to observe a major effect on variation between accessions in environmental regulation of gene expression by classical regulators like transcription factors.
DISCUSSION
Phenotypic Variation among Accessions
The phenotypic responses of accessions to mild drought were found to differ up to 4-fold. The lack of a strong correlation between size reduction caused by mild drought stress and the size under well-watered conditions for the third leaf and rosette area indicates that larger accessions are not per se more sensitive to mild drought stress, nor do smaller accessions tolerate the stress better in terms of size reduction. The size independency of the mild drought response has recently also been observed when the water deficit was applied later during development (Bac-Molenaar et al., 2016). This observation fits within the current ideas that mild drought/osmotic stress actively reduces growth, rather than being a secondary effect of insufficient resources (water, and indirectly, due to stomatal closure, also carbon) (Skirycz et al., 2011a, 2011b). With insufficient resources, larger plants will indeed be at a disadvantage because they need more resources for maintenance. However, in this setup, water is added daily to the plants, which compensates for water spoilage of plants with a lower water use efficiency. The growth reduction that is observed is hence purely an effect of the plant’s active mechanism to optimize/reduce growth corresponding to the prevailing conditions.
Different Cellular Mechanisms Determine Final Leaf Size in Natural Variants
The two processes that determine leaf size are cell division and cell expansion, and the negative correlation between pavement cell number and cell area shows the existence of a trade-off between both. Our data indicate the existence of three different strategies to produce large leaves. A first strategy is through the production of a limited number of cells that are larger than average. The smaller number of cells can be the result of either a limited number of cells recruited from the shoot apical meristem to the leaf primordium, a low cell proliferation rate or a short, developmentally determined cell proliferation phase (Gonzalez et al., 2012). The cells may increase in size through more extensive cell expansion or an extended cell expansion phase. The phenomenon by which cell expansion makes up for a reduced cell proliferation has previously been referred to as compensation (Tsukaya, 2002; Beemster et al., 2003). In the second strategy, a large leaf size is reached mainly by investing in an increased cell number, whereas cells are not larger than average. Many genes known to affect cell number mainly delay the developmentally determined transition between cell division and cell expansion (Gonzalez et al., 2012). In a third strategy, the mature leaf consists of an intermediate number of cells with intermediate sizes. Interestingly, another potential strategy that would combine a high number of cells with large cell sizes is not found, suggesting that there is a trade-off between both mechanisms. The accession Wa-1 has the largest number of cells with the largest size, and noteworthy, this is the only known tetraploid in the accessions investigated in this study (Henry et al., 2005). Polyploidy has been related to an increase in organ sizes; however, the relationship is elusive.
The trade-off between epidermal cell area and cell number has also been observed in a Ler × An-1 recombinant inbred line (RIL) population, but only for RILs that lacked the erecta mutation (Tisné et al., 2008). The erecta mutation has a strong effect on cell division, resulting in a positive correlation between both cellular traits for RILs with the mutation and an extremely high number of pavement cells in the Ler accession in our study.
The absence of plants having the combination of more and larger cells argues for an organ-wide control mechanism that optimizes leaf size. This optimal leaf size would be the result of integrating intrinsic developmental programs and signals, as well as a plethora of environmental factors (such as light, temperature, nutrients, and water availability), which together determine the leaf’s energy economics (Wright et al., 2004). However, some of the restrictions and trade-offs can be overcome through engineering, as is shown by the observed synergistic effects of combining cell division- and cell expansion-stimulating mutations (Vanhaeren et al., 2014, 2016). Nevertheless, the question remains why certain accessions evolved toward investing more energy in cell division, while others more in cell expansion. Is there an accession-dependent fitness advantage of doing one over the other, or is this selectively neutral so that only general leaf size constraints are important? Or, is it rather an effect of the unnatural experimental growth conditions, which is abolished in the field?
Drought Affects Both Cell Division and Cell Expansion
The experimental setup used in this study subjected the third leaf to mild drought stress during its development from 4 to 23 DAS. The effect of the induced mild drought stress was already visible 4 d after stress onset, at the last day of full proliferation of the third leaf. The reduction in the third leaf area at maturity was caused by alterations in pavement cell number and area, as was previously described for different Arabidopsis leaves (Aguirrezabal et al., 2006; Pereyra-Irujo et al., 2008; Baerenfaller et al., 2012; Clauw et al., 2015). Aguirrezabal et al. (2006) showed that cell division and cell expansion were affected independently from each other in response to different drought scenarios in the An-1 and Cvi-0 accessions. Both processes were therefore suggested to be partly uncoupled. Also in our study, in a large number of different genetic backgrounds, there was no correlation between the reduction in cell number and the reduction in cell size. However, the trade-off between cell proliferation and cell expansion in well-watered conditions either suggests crosstalk between both processes or an overarching regulatory mechanism. It is therefore reasonable to assume that regulation of cell division and cell expansion are also linked in mild drought conditions. Variations in the regulatory interactions and coupling of both processes between accessions may explain the lack of correlation between the reduction in cell size and cell number.
Genetic Architecture of Mild Drought Responses in Leaves
The performed GWAS specifically aimed at retrieving genetic associations with the differential response to mild drought stress. While all analyzed traits showed a high heritability, significant associations with SNPs that are linked to TRE1, encoding the Arabidopsis trehalase, were only found for the differential response upon mild drought stress of the third leaf area at the proliferation stage. TRE1 is the only enzyme known in this species to specifically hydrolyze trehalose into glucose (Müller et al., 2001; Lunn et al., 2006). Loss of endogenous trehalase activity by mutation results in increased trehalose levels (Van Houtte et al., 2013). Therefore, natural variation in TRE1 functionality or expression level likely results in varying levels of trehalose, which is a known osmoprotectant (Elbein et al., 2003), and consequently can contribute to the variability observed in the mild drought response. Moreover, as a metabolic signal, trehalose 6-phosphate is involved in regulating starch content in the leaf and has been implicated in leaf growth (Lunn et al., 2014). For a complex trait such as growth, many loci with small phenotypic effects are expected to be involved. Raising the statistical power, through increasing the sample number (Korte and Farlow, 2013), may allow for detecting these small effect loci at the resolution offered by whole-genome sequencing data. Independent of the effect size of the loci, increasing the sample number will also decrease the amount of rare alleles. Successful associations with traits of similar complexity have been found using collections of over 200 accessions (Meijón et al., 2014; Bac-Molenaar et al., 2015, 2016; Li et al., 2010). Another option is to split up the complex trait in its cellular subtraits, as proposed by Meijón et al. (2014). However, in this study, GWAS for the response of pavement cell area and number to mild drought did not deliver more significant associations. The high heritabilities that were observed for these traits suggest a strong genetic component determining the observed phenotypic variability. Allelic heterogeneity, epistasis, and the involvement of many loci with small effects can explain why, with these high heritabilities, the statistical significance of the associations remained low. However, it is worth noting that heritabilities estimated from mixed models might be overestimated (Heckerman et al., 2016).
To further elucidate the genetic architecture of the growing leaf’s responses to mild drought, the natural variation in transcriptome responses was investigated. Measuring transcriptional differences between Arabidopsis accessions has indicated a substantial variation in gene expression in different conditions (van Leeuwen et al., 2007; Delker et al., 2010; Des Marais et al., 2012; Clauw et al., 2015). Also in our specific experimental setup, considerable expression variation was observed between the different accessions. Variation in gene expression may be important for the adaptation to mild drought stress in different accessions. For some traits, the expression variation of the regulatory network can be more important than polymorphisms that affect gene function. This was shown for the regulatory network of auxin responses (Delker et al., 2010). Adaptation by gene expression variation may be especially important in quantitative traits for which characteristics of the function, such as the rate, size, or duration, differ.
Although a large variability in differential expression was detected between the accessions, genes did not vary randomly in expression between accessions. Coexpression analysis identified clusters of genes with similar responses to mild drought between the accessions, suggesting functional relatedness of these genes (Allocco et al., 2004). Based on GO enrichment analyses and the presence of genes whose expression can distinguish stress-treated samples from control samples, we selected eight interesting clusters. The suggested cofunctionality of the genes in the respective clusters may be interesting for further characterization in mild drought responses. Moreover, the GO enrichment clearly suggests cofunctionality of significant parts of the clusters. One of the clusters almost completely contained a gene regulatory network that was previously shown to regulate growth in osmotic stress (Dubois et al., 2013, 2015). Central in this network are ERF5, ERF6, and ERF11, which are proposed to regulate both a stress defense response and a growth response when activated by ethylene, through the action of MPK3/MPK6. The stress defense response was found to involve WRKY33, STZ/ZAT10, and MYB51, while the growth response is regulated by a decrease of GA levels and subsequent DELLA protein stabilization (Dubois et al., 2013, 2015). Six major components of this network (ACS6, MPK3, ERF6, ERF11, WRKY33, and STZ) were part of this cluster. Although no genes directly involved in GA biosynthesis or degradation were detected in the coexpression cluster, two AP2 transcription factors (DDF1 and DDF2) that control GA levels through regulating GA2OX7 (Magome et al., 2004, 2008) were present. Moreover, DDF1 is known to be involved in heat, cold, and drought responses (Kang et al., 2011). DDF1 and the closely related DDF2 are therefore interesting candidates for transcriptionally regulating GA biosynthesis in the growth regulatory network proposed by Dubois et al. (2013, 2015). Besides ERF6, other members of the ERF/AP2 transcription factor family were present in this coexpression cluster (ABR1, ERF1, ERF4, ERF13, ERF98/TDR1, ERF109/RRTF1, and RAP2.6). The ERFs are proposed to be important hubs for hormone signaling in various abiotic stresses (Sewelam et al., 2013). Interestingly, cluster A also contains most genes involved in the biosynthesis of JA (DEFECTIVE ANTHER DEHISCENCE1, LIPOXYGENASE3 [LOX3], LOX4, ALLENE OXIDE CYCLASE3, ALLENE OXIDE SYNTHASE, OXOPHYTODIENOATE-REDUCTASE3, OPC-8:0 COA LGASE1) and JA-Ile catabolism (CYTOCHROME P450 FAMILY 94 SUBFAMILY B POLYPEPTIDE1 [CYP94B1], CYP94B3, and CYP94C1). Moreover, with 8 of the 12 genes encoding JAZ proteins and the MYC2-encoding gene, the main regulators of JA-induced gene expression were present in this cluster. Altogether, this suggests an extensive crosstalk between ethylene and JA signaling in regulating the response of growing leaves to mild drought. Interestingly, JA response mutants such as coi and jin1, but not jar1, displayed an enhanced tolerance to long-term mild drought stress (Harb et al., 2010).
The fact that large gene clusters respond differently between accessions, but retain coexpression, suggests coregulation of the mild drought response of the genes in these clusters and hints at accession-specific differences in how the clusters are regulated. Assuming that genes in these clusters are important for the response to mild drought, the regulators of these clusters are potential key players in the adaptation of different accessions to mild drought.
Predictive Genes for Mild Drought in Growing Leaves
Based on the natural variation in our population, we also endeavored to identify a set of genes that could be used as predictors for the applied stress. Using a machine learning approach, a set of 283 genes was found to be valuable to predict with high accuracy whether a sample was subjected to mild drought or not. Transcriptional profiling of these genes may therefore indicate whether a sample was subjected to mild drought without a priori knowledge of the treatment.
The set of stress predictive genes was enriched for genes involved in drought responses; hence, many of these genes may also be important players in the mild drought response of young developing leaves. Within the 283-gene set were 11 “core” genes that showed very similar transcriptional responses in almost all accessions for their response to mild drought, suggesting that they play a pivotal role in the mild drought response. Three of these 11 genes encode proteins that mediate ABA responses: ABI FIVE BINDING PROTEIN1 (AFP1), HOMEOBOX7 (HB7), and PROTEIN PHOSPHATASE 2CA (AHG3/PP2CA) (Garcia et al., 2008; Rodrigues et al., 2013; Olsson et al., 2004), of which HB7 is known to be involved in the response of leaf growth to drought (Ré et al., 2014; Valdés et al., 2012; Olsson et al., 2004). Two other core genes encode EXPANSIN A 15 (EXPA15) and BETA-D-XYLOSIDASE4 (XYL4), both cell wall modifiers involved in growth responses. Cell wall modifications are important to control cell expansion in response to drought (Wu and Cosgrove, 2000; Moore et al., 2008; Minic et al., 2004), and genes encoding cell wall modifiers have previously been shown to be differentially expressed in response to mild drought in growing leaves (Harb et al., 2010; Clauw et al., 2015). Two other proteins encoded by core genes that regulate osmolyte concentrations were found to be up- and downregulated, respectively: Δ-1-PYRROLINE-5-CARBOXYLATE SYNTHASE1 (P5CS1) is the rate-limiting enzyme in proline biosynthesis, a well-known osmoprotectant (Strizhov et al., 1997; Szabados and Savouré, 2010), and SMALL AND BASIC INTRINSIC PROTEIN2 (SIP2) is suggested to be involved in phloem unloading of raffinose in sink leaves (Peters et al., 2010). Raffinose and other members of the raffinose family oligosaccharides are involved in stress tolerance and act as antioxidants. Furthermore, raffinose family oligosaccharides are part of the carbon storage system (ElSayed et al., 2014), possibly explaining why SIP2 is downregulated. The other four core response genes are involved in the synthesis of wax esters (FOLDED PETAL1) (Takeda et al., 2013), primary metabolism (PHOSPHOENOLPYRUVATE CARBOXYLASE KINASE1), stress-induced chloroplast disruption (CHLOROPLAST VESICULATION) (Wang and Blumwald, 2014), and Zn/Fe transport (IRON REGULATED TRANSPORTER3). Altogether, these 11 core genes are potential markers of the mild drought response in different genetic backgrounds.
However, an additional attempt to find predictive genes for the phenotypic response to mild drought was not successful. This is indicative of the complex relationship between gene expression and phenotype. Moreover, the large genetic variation in the set of accessions adds complexity to the task of identifying a clear gene expression-phenotype relation. Recently, a similar approach in maize RIL populations identified genes whose expression in young, dividing leaf tissue correlated to phenotypes of mature leaves such as length, width, and leaf area (Baute et al., 2015, 2016). It is likely that the lack of gene expression-phenotype correlation in natural accessions is due to higher genetic variation present in this population compared with a RIL population. In addition, maize (Zea mays) has been subjected to extensive selection through breeding, lowering the genetic variation.
Diversity of Potential Regulators of Gene Expression
To identify the genetic polymorphisms that are associated with the expression differences between genes, we conducted GWAS on gene expression (eGWAS). Among the associated loci for treatment-independent expression, the majority was located in the proximity of the regulated gene. These SNPs are mainly thought to be cis-regulatory in nature. The test for gene-environment interaction retrieved fewer associations, but interestingly all detected associations were located in trans, indicating that the natural variation in expression changes induced by the environment are mainly regulated in trans, while cis-regulatory variation affects expression changes independently of the environment. This observation is strengthened by the variance component analysis that indicated a clear role for trans-located SNPs for explaining the expression variation. Previous studies on RIL populations identified mainly cis variation to be associated with gene expression in general (West et al., 2007; Keurentjes et al., 2007), whereas the environment-dependent expression showed enrichment in trans regulators (Cubillos et al., 2014; Lowry et al., 2013; Snoek et al., 2013). This general trend corresponds to our results; however, the absence of cis regulators for environment-dependent expression is striking, especially because cis loci typically have stronger effects on gene expression compared with trans (Fusi et al., 2012). The explanation probably needs to be sought in the difference between the variation in RILs and natural accessions, where the latter will contain more rare alleles. Further functional characterization of the cis and trans loci is needed to elucidate why adaptations to environmental perturbations are affecting trans rather than cis elements and a dedicated comparative experiment could give insight into the difference between RIL populations and natural accessions.
The potential regulatory loci that were detected in the eGWAS were functionally diverse. As expected, the greatest density of cis loci was detected in a region up to 2 kb upstream of the transcribed region. The associated SNPs that were located in the gene body might affect gene regulation by interfering with intron-mediated transcriptional regulation (Rose, 2008; Schauer et al., 2009; Krizek, 2015), alterations in protein and hence turnover, or alternative splicing. Alternative splicing occurs in ∼60% of the intron-containing genes and is likely to play a role in plant growth, development, and responses to environmental changes (Staiger and Brown, 2013). Natural variation in alternative splicing has been shown for the proline synthesis gene P5CS1, resulting in variable proline levels (Kesari et al., 2012). SNPs were also detected further than 2 kb away from the gene and can be located in more distant enhancer regions or be simply in linkage with the causative SNP in the promoter element. Alternatively, it cannot be excluded that proximate trans loci play a role in expression regulation. Functionally related genes can colocalize and the regulators may thus be present in the proximity of such a gene cluster (Schweizer and Stein, 2011).
The associated trans loci that were associated with environment-specific changes in gene expression were located in transposable elements and protein-encoding genes, of which a minority (6%) encoded transcription factors. However, a large number of loci were positioned in noncoding regions and can thus play a role in shaping specific promoter elements, enhancers, or silencers of trans-regulatory genes, but they might as well be in linkage with the causal SNP. The low number of associated transcription factors as trans regulators was also observed in a genome-wide study in yeast (Yvert et al., 2003), where trans regulators were not enriched for transcription factors. Transposable elements are known to regulate gene expression as cis regulators, by containing promoter or other regulatory elements, or as trans regulators, by encoding, for example, small interfering RNA (Rebollo et al., 2012). They have also been suggested to play a role in the gene expression divergence between species (Hollister et al., 2011) and are therefore likely candidates to shape gene expression variation in the different accessions.
However, the lack of enrichment of specific regulatory processes among the identified potential regulators may suggest that natural variation in transcriptional regulation affects many different factors that are involved in a complex regulatory network. Moreover, it is likely that rare alleles are involved in adapting gene expression regulation to environmental conditions (M. Nordborg, personal communication). Our data suggest that cis loci are of little importance to the adaptation of transcriptional responses to the environment, whereas genetic variation in different types of trans regulators (direct or indirect) seems to be responsible for the observed variation in differential expression upon mild drought.
METHODS
Plant Growth Conditions and Experimental Setup
A collection of 98 accessions was grown in a growth chamber under controlled environmental conditions (21°C, 55% relative humidity, 16 h day/8 h night, and 110 to 120 μmol m−2 s−1 light intensity using a mixture of 2 mercury-vapor and 2 sodium vapor lamps). All accessions were bulked simultaneously. The last day of full proliferation varied between 8 and 10 DAS depending on the accession and was determined microscopically as the last day when no expanding cells (jigsaw-shaped cells) were observed at the leaf tip. Plants were subjected to a controlled drought treatment that started at 4 DAS (Clauw et al., 2015). Plant handling, irrigation, and imaging were performed using the plant phenotyping platform WIWAMxy (Skirycz et al., 2011b) (www.wiwam.com). The control-treated pots contained 2.2 g water per gram dry soil. The drought-treated pots initially contained 1.2 g water per gram dry soil, and their relative water content was further decreased to 0.7 g water per gram dry soil after 10 DAS. Experiments were conducted in batches of 16 accessions. Two reference accessions (Col-0 and Oy-0) were added to each batch to correct for batch effects. Experiments to obtain the growth-related phenotyping data were conducted in duplicate. Images of the rosette of each plant were taken daily until 22 DAS and analyzed for the PRA using an in-house developed phenotyping interface (http://www.psb.ugent.be/phenotyping/pippa). Size measurements of the third leaf were done at the transition from cell proliferation to cell expansion (10 to 11 DAS) and at maturity (23 DAS). For practical reasons, the mature third leaf was harvested 1 d later than the last PRA measurement at 22 DAS. To this end, the leaves were cut from the rosette, cleared in ethanol, and transferred to lactic acid before mounting on microscope slides. Measurements based on microscope images were done using ImageJ (http://imagej.nih.gov/ij/), and analysis of cell drawings made from the abaxial epidermis allowed for quantification of the pavement cell area, pavement cell number, and stomatal index (Andriankaja et al., 2012). All correlations with pavement cell number were conducted with exclusion of the outlier accession Ler-1. Phenotypic measurements were not corrected for water added by the automated watering system. The added water volume is likely to be affected by many different factors and was considered not to be a good proxy for plant water usage. In concert, the relation between percentage of reduction of rosette area upon drought versus the control rosette area corrected for water usage was found to be not significant (r = −0.08, Pearson correlation coefficient; P value= 0.40, t test), neither was the relation between the rosette area in control conditions and the water usage (r = 0.06, P value = 0.55).
Phenotyping Data Analysis
Because accessions were grown in separate batches, the phenotypic data were normalized to correct for batch effects. To this end, a mixed model was used with genotype, treatment, and their interaction as fixed factors. The batch effect was added to the model as a random factor:
 |
The least square means over the two replicates for each genotype and treatment were then used in the further analysis. These estimates did not show any clustering dependent on the batch (Supplemental Figure 10). The normalization was performed with the lmer function in the R package “lme4” (v. 1.1-6), and the least square means were obtained with the lsmeans function in the R package “lsmeans” (v. 2.10). All analyses were performed using R version 3.0.1.
Correlations are Pearson correlations calculated in R (version 3.0.1) using the standard cor.test function. Significance of correlations was assessed with the t test implemented in the cor.test function.
Transcriptome Analysis
Sampling
To ensure sufficient material for transcriptome analysis, 60 seedlings were grown per accession per treatment. Material of each accession was harvested in one experiment. Plants were harvested at the last day of full proliferation, as determined through microscopy analysis for each accession. The time of harvest therefore ranged from 8 to 10 DAS depending on the accession. Plants were flash-frozen in liquid nitrogen upon harvest. To prevent RNA degradation, RNAlater-ICE (Ambion) cooled at −70°C was added to the samples and was allowed to penetrate the tissue at −20°C for 5 d. The third leaf was collected by microdissection under a microscope. Samples were microdissected in a Petri dish on dry ice to keep the samples below room temperature. Dissected leaves were ground with a Retsch machine (Retsch) and 3-mm metal balls.
RNA Extraction
RNA was extracted with Trizol (Invitrogen) according to the manufacturer’s instructions. RNA samples were subjected to DNA digestion with an RNase-free DNase I Kit (Qiagen), and impurities were removed with the RNeasy Mini Kit (Qiagen).
RNA-Sequencing
The library was prepared using the TruSeq RNA Sample Preparation Kit v2 (Illumina). Briefly, poly(A)-containing mRNA molecules were reverse transcribed, double-stranded cDNA was generated, and adapters were ligated. After quality control using a 2100 Bioanalyzer (Agilent), clusters were generated through amplification using the TruSeq PE Cluster Kit v3-cBot-HS kit (Illumina) followed by sequencing on an Illumina HiSeq 2000 with the TruSeq SBS Kit v3-HS (Illumina). Sequencing was performed in paired-end mode with a read length of 50 nucleotides. The quality of the raw data was verified with FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/, version 0.9.1).
Due to the small sampling material (proliferating leaves) and resulting low amounts of RNA, transcriptomics data were obtained for only 89 of the 98 accessions (Supplemental Data Set 1).
Next, quality filtering was performed using the FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/, version 0.0.13): reads where globally filtered to only retain reads for which the base quality exceeds Q10 for at least 75% of the bases. Adapters were trimmed using cutadapt (adapter sequence GATCGGAAGAGCACACGTCTGAACTCCAGTCAC with at least a 10-nucleotide overlap; Martin, 2011). Next, 3′ trimming was performed to remove bases with a quality below Q20, ensuring a minimum length of 35 nucleotides remaining. Repairing was performed using a custom perl script. Reads were subsequently mapped to the Arabidopsis reference genome (TAIR10) using GSNAP (Wu and Nacu, 2010), version 2013-02-05, allowing maximally two mismatches. The concordantly paired reads that uniquely mapped to the genome were used for quantification on the gene level with htseq-count from the HTSeq.py python package (Anders et al., 2015).
Expression Data Normalization
RNA-seq data were normalized for library size using DESeq2 v.2.10 package for R 3.0.1 with default settings. Genes expressed in less than five samples were removed, leaving 23,460 genes. After transforming the normalized counts with the inverse hyperbolic sine (arcsinh), the genes with the 5% weakest coefficients of variation over the samples were removed, leaving 22,287 genes for further analysis. Differential expression (DE) was calculated as the mild drought minus the control normalized and arcsinh transformed expression values of a gene.
 |
Cluster Affinity Search Technique
The CAST clustering, as described by Ben-Dor et al. (1999), was performed on the normalized differential expression values of all 89 accessions for 22,287 genes, using the CAST implementation in MultiExperiment Viewer (MeV) version 4.8 (Saeed et al., 2003). Pearson correlation was selected as the distance metric, and the affinity threshold was set at 0.7, resulting in 5447 clusters. Clusters that contained more than 1000 genes were further subdivided by performing the CAST clustering at a 0.8 affinity threshold. One of the resulting clusters still contained more than 1000 genes and was further subdivided in smaller clusters at an affinity threshold of 0.9. Altogether, the clustering and subclustering resulted in 6102 clusters. Eight clusters were selected for follow-up based on size (containing at least 10 genes), GO enrichment of selected GO categories (selection based on the following keywords: drought, abiotic stress, osmotic, water deprivation, growth, leaf, cell cycle, cell expansion, and cell wall), and presence of at least one of the stress predictor genes (see below).
Stress Predictors
A two-class classification was performed on the normalized gene expression data under control and mild drought stress conditions. Herein, the samples under stress were set as the positive class, whereas the control samples were set as the negative class. To reduce the gene space of the data set and decrease the complexity of the classification model, we applied a filter feature selection technique (Saeys et al., 2007). One-way ANOVA tests were performed on every gene’s expression profile across accessions and treatments, using treatment as a factor (R version 3.1.0; Supplemental Data Set 8). FDR-adjusted P values were calculated using the Benjamini-Hochberg procedure (Benjamini and Hochberg, 1995), and only genes exhibiting a significant treatment effect at FDR = 0.05, dubbed “stress predictors,” were considered informative for the classification problem at hand.
Classification was performed by training a SVM using the filtered genes as classification features. SVMs are large margin classifiers and rely on kernel functions to solve nonlinear separability problems in a higher feature space. By transforming the data to a higher feature space, the classification problem can be solved using a linear model (Cortes and Vapnik, 1995). In this study, a linear kernel was used for classification. The software package scikit-learn v14 was used as a modeling framework (Pedregosa et al., 2011). The SVM cost parameter C was optimized in the interval [2−5, 215] with a step size of 22 (Chang and Lin, 2011). To ensure generalization properties and obtain an unbiased performance estimation, a stratified nested 10-fold cross-validation was performed (Kohavi, 1995; Varma and Simon, 2006). Inner cross-validation was used for model optimization, while outer cross-validation was performed solely for performance estimation. Given the number of positive and negative samples in each outer test fold, a confusion matrix was generated to assess the performance of the classification model. Parameter optimization and performance estimation were done using the F-score, which is the harmonic mean between sensitivity and precision:
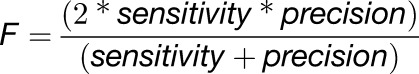 |
A final performance estimate was calculated as the average score over the different outer cross-validation folds. Next to the F-score, the area under the receiver operating characteristic curve (AUC) was also calculated (Fawcett, 2006). The AUC can be interpreted as the probability of ranking a randomly chosen positive sample higher than a randomly chosen negative sample. An AUC value of 1 implies a perfect separation of positive and negative samples, while a value of 0.5 implies random classification.
GO Enrichment
All GO enrichment analyses were conducted using the online tool PLAZA v. 3.0 (Proost et al., 2015).
GWAS
The GWAS was performed on the normalized mean values of the different phenotypes in control and mild drought conditions. The mapping panel consisted of 91 accessions for which genotypic data could be obtained. The genotypic data were based on whole-genome sequencing data of the different accessions (The 1001 Genomes Consortium, 2016) and covered ±4,000,000 SNPs. For accessions that were not sequenced, genotype information was imputed based on the 250-k SNP chip data (Horton et al., 2012). Imputations of genotypic information are known to have a negligible effect on the outcome of GWAS studies (Cao et al., 2011). Markers with a minor allele frequency of below 5% were excluded from the analysis. The GWAS was performed using a multitrait mixed model as described by Korte et al. (2012).
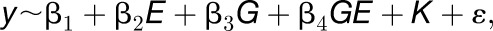 |
where y is a vector of phenotype values for n accessions, E is the treatment factor (control or mild drought stress), G the homozygous genotype of the respective marker, and GE the genotype by treatment interaction. K denotes the kinship matrix that is included as a random factor to correct for population structure. The kinship matrix is calculated using the genotype information as an identical-by-state matrix, where relatedness is based on shared alleles, as discussed by Kang et al. (2008). β denotes the respective regression coefficient of each term. To retrieve associations with the differential response against mild drought stress, the full model was tested against the model without GE (β4 = 0). A multiple testing correction was applied by dividing the 0.05 significance threshold by the number of markers with a minor allele frequency of at least 5%. For the third leaf area at proliferation, this resulted in a threshold of 7.53 –log10. Peak selection for the other phenotypes was done visually, resulting in the following –log10 P value cutoffs: 4.28 (rosette area), 5.46 (pavement cell area), 4.71 (pavement cell number), 4.42 (stomatal index), and 5.26 (leaf 3 area at maturity). All analyses were conducted using a custom R-script (R version 3.1).
Pseudo-heritabilities were calculated according to:
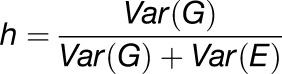 |
Var(G) and Var(E) are the genetic variation and environmental variation, respectively, as the variances estimated from the multitrait mixed model.
The phenotypic variance (Ve) explained by a single SNP marker in the multitrait mixed model was calculated according to:
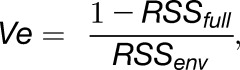 |
where RSSfull is the residual sum of squares of the full model, including the single SNP marker, and RSSenv is the residual sum of squares of the model without the single SNP marker.
eGWAS
The eGWAS was conducted with the same multitrait mixed model as used for the GWAS analysis. As phenotypic values, the gene expression data (log2 transcripts per million) was used. The trait-specific test (differential gene expression) was performed by testing the full model against the model without GE (β4 = 0). For the common test (treatment independent expression), the model without GE (β4 = 0) was tested against the model without G nor GE (β3 = 0 and β4 = 0). A fixed P value cutoff for all genes was set at 10−7. The model yielded results for 5557 of the 22,287 genes. For the remaining genes, the iterative model fitting was unable to find appropriate estimates of the model parameters. To create a high-confidence data set, a selection was conducted against inflated models by calculating the genomic inflation factor (λ) (Devlin and Roeder, 1999) and performing the D’Agostino test for normality (D’Agostino, 1986), as non-normal distributed data potentially lead to model inflation. The genomic inflation factor was calculated as the ratio between the median of the P values of the association of the markers with the trait (gene expression) and their expected median (0.5). Models with 𝜆 = 1 ± 0.1 were considered to be noninflated. The D’Agostino test on the gene expression values was performed as implemented in the R-package “moments” (version 0.14). Genes with a P value > 0.05 for the D’Agostino test in both treatments were considered to have normally distributed expression values and were subsequently selected as noninflated if they exhibited a genomic inflation factor 𝜆 in the range [0.9, 1.1]. This selection resulted in a high-confidence data set of 2433 genes. All analyses were conducted in R (version 3.1).
Variance Component Analysis
The relative contribution of environment, cis, trans, cis × environment, and trans × environment to gene expression variation was investigated using LIMIX (Lippert et al., 2014), which efficiently estimates variance components using linear mixed models. For each gene, the model was fitted as discussed by Sasaki et al. (2015), where SNPs located within 5 kb of the transcription start were considered to be cis.
Accession Numbers
RNA-seq data are available in the ArrayExpress database (www.ebi.ac.uk/arrayexpress) under accession number E-MTAB-5009.Raw phenotyping data are available through Zenodo (www.zenodo.org) under doi/10.5281/zenodo.147920.
Supplemental Data
Supplemental Figure 1. Correlations between Different Phenotypic Measurements in 98 Accessions.
Supplemental Figure 2. Rosette Images of Mild Drought-Tolerant and -Sensitive Accessions.
Supplemental Figure 3. Manhattan Plots for Each of the Single-Trait GWAS.
Supplemental Figure 4. Manhattan Plots Showing Significance of the Association of Each SNP with the Studied Phenotype.
Supplemental Figure 5. Manhattan Plots Showing Chosen SNPs (Red) for Gene Selection.
Supplemental Figure 6. Venn Diagram Showing the Overlap between “Stress Predictor” Genes and Differentially Expressed Genes in Mature Leaf Tissue upon Mild Drought Treatments (Baerenfaller et al., 2012; Harb et al., 2010; Des Marais et al., 2012) and Severe Drought (Huang et al., 2008; Matsui et al., 2008; Harb et al., 2010).
Supplemental Figure 7. Differential Expression Profile of Coexpression Clusters A-H.
Supplemental Figure 8. Histogram of Locations of SNPs Associated with Treatment-Independent Expression.
Supplemental Figure 9. Variance Component Analysis of Gene Expression Data.
Supplemental Figure 10. Phenotypic Measurements with Batches Indicated.
Supplemental Table 1. Heritability Estimates for Single-Trait GWAS.
Supplemental Table 2. Variance Explained by Single SNPs.
Supplemental Table 3. Genes Underlying the Peaks (1-4) of SNPs Associated with the third Leaf Area at Maturity.
Supplemental Data Set 1. Geographic Distribution of the 98 Accessions.
Supplemental Data Set 2. List of Genes within 10 kb of SNPs Associated with Rosette Area, Pavement Cell Area, Pavement Cell Number, and Stomatal Index.
Supplemental Data Set 3. Coexpression Clusters.
Supplemental Data Set 4. Genes Present in Coexpression Cluster A.
Supplemental Data Set 5. Genes Present in Coexpression Cluster C.
Supplemental Data Set 6. List of the Identified Stress Predictors.
Supplemental Data Set 7. Overview of trans Regulators of Differential Expression.
Supplemental Data Set 8. ANOVA Results Used for Selecting the “Stress Predicting” Genes.
Supplementary Material
Acknowledgments
We thank Magnus Nordborg for insightful comments and adjustments to the manuscript. We also thank Annick Bleys for the overall improvement of the quality of the text and Eriko Sasaki for assistance with the variance component analysis. The research leading to these results was funded by Ghent University (“Bijzonder Onderzoeksfonds Methusalem project” no. BOF08/01M00408), by the Interuniversity Attraction Poles Programme (IUAP P7/29 “MARS”) initiated by the Belgian Science Policy Office, and by the European Research Council under the European Community’s Seventh Framework Program (FP7/2007-2013) under ERC Grant [339341-AMAIZE]11. P.C. is indebted to the Agency for Innovation by Science and Technology for a predoctoral fellowship and S.D. to the Research Foundation-Flanders for a postdoctoral fellowship.
AUTHOR CONTRIBUTIONS
P.C., F.C, N.G., and D.I. designed the experiments and wrote the manuscript. P.C, T.V.D., L.D.M., M.V., and K.M. conducted the experiments. S.D. was involved in the image analyses. P.C. and D.H. analyzed the phenotyping data. P.C., F.C., and D.H. conducted the transcriptome data analyses. A.K. performed the different GWAS analyses. B.S. and S.M. designed and performed the classification modeling.
Glossary
- GWAS
genome-wide association studies
- DAS
days after stratification
- PRA
projected rosette area
- GA
gibberellic acid
- GO
Gene Ontology
- JA
jasmonic acid
- ABA
abscisic acid
- FDR
false discovery rate
- SVM
support vector machine
- AUC
area under the curve
- SNP
single nucleotide polymorphism
- RIL
recombinant inbred line
- CAST
cluster affinity search technique
Footnotes
Articles can be viewed without a subscription.
References
- Aguirrezabal L., Bouchier-Combaud S., Radziejwoski A., Dauzat M., Cookson S.J., Granier C. (2006). Plasticity to soil water deficit in Arabidopsis thaliana: dissection of leaf development into underlying growth dynamic and cellular variables reveals invisible phenotypes. Plant Cell Environ. 29: 2216–2227. [DOI] [PubMed] [Google Scholar]
- Allocco D.J., Kohane I.S., Butte A.J. (2004). Quantifying the relationship between co-expression, co-regulation and gene function. BMC Bioinformatics 5: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S., Pyl P.T., Huber W. (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andriankaja M., Dhondt S., De Bodt S., Vanhaeren H., Coppens F., De Milde L., Mühlenbock P., Skirycz A., Gonzalez N., Beemster G.T.S., Inzé D. (2012). Exit from proliferation during leaf development in Arabidopsis thaliana: a not-so-gradual process. Dev. Cell 22: 64–78. [DOI] [PubMed] [Google Scholar]
- Bac-Molenaar J.A., Vreugdenhil D., Granier C., Keurentjes J.J.B. (2015). Genome-wide association mapping of growth dynamics detects time-specific and general quantitative trait loci. J. Exp. Bot. 66: 5567–5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bac-Molenaar J.A., Granier C., Keurentjes J.J.B., Vreugdenhil D. (2016). Genome-wide association mapping of time-dependent growth responses to moderate drought stress in Arabidopsis. Plant Cell Environ. 39: 88–102. [DOI] [PubMed] [Google Scholar]
- Baerenfaller K., et al. (2012). Systems-based analysis of Arabidopsis leaf growth reveals adaptation to water deficit. Mol. Syst. Biol. 8: 606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baute J., Herman D., Coppens F., De Block J., Slabbinck B., Dell’Acqua M., Pè M.E., Maere S., Nelissen H., Inzé D. (2015). Correlation analysis of the transcriptome of growing leaves with mature leaf parameters in a maize RIL population. Genome Biol. 16: 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baute J., Herman D., Coppens F., De Block J., Slabbinck B., Dell’Acqua M., Pè M.E., Maere S., Nelissen H., Inzé D. (2016). Combined large-scale phenotyping and transcriptomics in maize reveals a robust growth regulatory network. Plant Physiol. 170: 1848–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beemster G.T.S., Fiorani F., Inzé D. (2003). Cell cycle: the key to plant growth control? Trends Plant Sci. 8: 154–158. [DOI] [PubMed] [Google Scholar]
- Ben-Dor A., Shamir R., Yakhini Z. (1999). Clustering gene expression patterns. J. Comput. Biol. 6: 281–297. [DOI] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 57: 289–300. [Google Scholar]
- Bouchabke O., Chang F., Simon M., Voisin R., Pelletier G., Durand-Tardif M. (2008). Natural variation in Arabidopsis thaliana as a tool for highlighting differential drought responses. PLoS One 3: e1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J., et al. (2011). Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat. Genet. 43: 956–963. [DOI] [PubMed] [Google Scholar]
- Chang C.-C., Lin C.-J. (2011). LIBSVM: A library for support vector machines. ACM Trans. Intell. Syst. Technol. 2: 27. [Google Scholar]
- Chardon F., Barthélémy J., Daniel-Vedele F., Masclaux-Daubresse C. (2010). Natural variation of nitrate uptake and nitrogen use efficiency in Arabidopsis thaliana cultivated with limiting and ample nitrogen supply. J. Exp. Bot. 61: 2293–2302. [DOI] [PubMed] [Google Scholar]
- Claeys H., Inzé D. (2013). The agony of choice: how plants balance growth and survival under water-limiting conditions. Plant Physiol. 162: 1768–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clauw P., Coppens F., De Beuf K., Dhondt S., Van Daele T., Maleux K., Storme V., Clement L., Gonzalez N., Inzé D. (2015). Leaf responses to mild drought stress in natural variants of Arabidopsis. Plant Physiol. 167: 800–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes C., Vapnik V. (1995). Support-vector networks. Mach. Learn. 20: 273–297. [Google Scholar]
- Cubillos F.A., Stegle O., Grondin C., Canut M., Tisné S., Gy I., Loudet O. (2014). Extensive cis-regulatory variation robust to environmental perturbation in Arabidopsis. Plant Cell 26: 4298–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Agostino R.B. (1986). Tests for the normal distribution. In Goodness-of-Fit Techniques, D’Agostino R.B., Stephens M.A., eds (New York: Marcel Dekker; ), pp. 367–420. [Google Scholar]
- Delker C., Pöschl Y., Raschke A., Ullrich K., Ettingshausen S., Hauptmann V., Grosse I., Quint M. (2010). Natural variation of transcriptional auxin response networks in Arabidopsis thaliana. Plant Cell 22: 2184–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Des Marais D.L., McKay J.K., Richards J.H., Sen S., Wayne T., Juenger T.E. (2012). Physiological genomics of response to soil drying in diverse Arabidopsis accessions. Plant Cell 24: 893–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin B., Roeder K. (1999). Genomic control for association studies. Biometrics 55: 997–1004. [DOI] [PubMed] [Google Scholar]
- Dhondt S., Wuyts N., Inzé D. (2013). Cell to whole-plant phenotyping: the best is yet to come. Trends Plant Sci. 18: 428–439. [DOI] [PubMed] [Google Scholar]
- Dombrecht B., Xue G.P., Sprague S.J., Kirkegaard J.A., Ross J.J., Reid J.B., Fitt G.P., Sewelam N., Schenk P.M., Manners J.M., Kazan K. (2007). MYC2 differentially modulates diverse jasmonate-dependent functions in Arabidopsis. Plant Cell 19: 2225–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois M., Van den Broeck L., Claeys H., Van Vlierberghe K., Matsui M., Inzé D. (2015). The ETHYLENE RESPONSE FACTORS ERF6 and ERF11 antagonistically regulate mannitol-induced growth inhibition in Arabidopsis. Plant Physiol. 169: 166–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois M., Skirycz A., Claeys H., Maleux K., Dhondt S., De Bodt S., Vanden Bossche R., De Milde L., Yoshizumi T., Matsui M., Inzé D. (2013). Ethylene Response Factor6 acts as a central regulator of leaf growth under water-limiting conditions in Arabidopsis. Plant Physiol. 162: 319–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbein A.D., Pan Y.T., Pastuszak I., Carroll D. (2003). New insights on trehalose: a multifunctional molecule. Glycobiology 13: 17R–27R. [DOI] [PubMed] [Google Scholar]
- ElSayed A.I., Rafudeen M.S., Golldack D. (2014). Physiological aspects of raffinose family oligosaccharides in plants: protection against abiotic stress. Plant Biol (Stuttg) 16: 1–8. [DOI] [PubMed] [Google Scholar]
- Fawcett T. (2006). An introduction to ROC analysis. Pattern Recognit. Lett. 27: 861–874. [Google Scholar]
- Flood P.J., Kruijer W., Schnabel S.K., van der Schoor R., Jalink H., Snel J.F.H., Harbinson J., Aarts M.G.M. (2016). Phenomics for photosynthesis, growth and reflectance in Arabidopsis thaliana reveals circadian and long-term fluctuations in heritability. Plant Methods 12: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier-Level A., Korte A., Cooper M.D., Nordborg M., Schmitt J., Wilczek A.M. (2011). A map of local adaptation in Arabidopsis thaliana. Science 334: 86–89. [DOI] [PubMed] [Google Scholar]
- Fu J., et al. (2009). System-wide molecular evidence for phenotypic buffering in Arabidopsis. Nat. Genet. 41: 166–167. [DOI] [PubMed] [Google Scholar]
- Fusi N., Stegle O., Lawrence N.D. (2012). Joint modelling of confounding factors and prominent genetic regulators provides increased accuracy in genetical genomics studies. PLOS Comput. Biol. 8: e1002330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia M.E., Lynch T., Peeters J., Snowden C., Finkelstein R. (2008). A small plant-specific protein family of ABI five binding proteins (AFPs) regulates stress response in germinating Arabidopsis seeds and seedlings. Plant Mol. Biol. 67: 643–658. [DOI] [PubMed] [Google Scholar]
- Gonzalez N., Vanhaeren H., Inzé D. (2012). Leaf size control: complex coordination of cell division and expansion. Trends Plant Sci. 17: 332–340. [DOI] [PubMed] [Google Scholar]
- Granier C., et al. (2006). PHENOPSIS, an automated platform for reproducible phenotyping of plant responses to soil water deficit in Arabidopsis thaliana permitted the identification of an accession with low sensitivity to soil water deficit. New Phytol. 169: 623–635. [DOI] [PubMed] [Google Scholar]
- Green R.A., Wollman R., Kaplan K.B. (2005). APC and EB1 function together in mitosis to regulate spindle dynamics and chromosome alignment. Mol. Biol. Cell 16: 4609–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harb A., Krishnan A., Ambavaram M.M.R., Pereira A. (2010). Molecular and physiological analysis of drought stress in Arabidopsis reveals early responses leading to acclimation in plant growth. Plant Physiol. 154: 1254–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckerman D., et al. (2016). Linear mixed model for heritability estimation that explicitly addresses environmental variation. Proc. Natl. Acad. Sci. USA 113: 7377–7382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry I.M., Dilkes B.P., Young K., Watson B., Wu H., Comai L. (2005). Aneuploidy and genetic variation in the Arabidopsis thaliana triploid response. Genetics 170: 1979–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M.H. (2002). Biogeography of Arabidopsis thaliana (L.) Heynh. (Brassicaceae). J. Biogeogr. 29: 125–134. [Google Scholar]
- Hollister J.D., Smith L.M., Guo Y.-L., Ott F., Weigel D., Gaut B.S. (2011). Transposable elements and small RNAs contribute to gene expression divergence between Arabidopsis thaliana and Arabidopsis lyrata. Proc. Natl. Acad. Sci. USA 108: 2322–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton M.W., et al. (2012). Genome-wide patterns of genetic variation in worldwide Arabidopsis thaliana accessions from the RegMap panel. Nat. Genet. 44: 212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotta T., Kong Z., Ho C.-M.K., Zeng C.J.T., Horio T., Fong S., Vuong T., Lee Y.-R.J., Liu B. (2012). Characterization of the Arabidopsis augmin complex uncovers its critical function in the assembly of the acentrosomal spindle and phragmoplast microtubule arrays. Plant Cell 24: 1494–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D., Wu W., Abrams S.R., Cutler A.J. (2008). The relationship of drought-related gene expression in Arabidopsis thaliana to hormonal and environmental factors. J. Exp. Bot. 59: 2991–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huard-Chauveau C., Perchepied L., Debieu M., Rivas S., Kroj T., Kars I., Bergelson J., Roux F., Roby D. (2013). An atypical kinase under balancing selection confers broad-spectrum disease resistance in Arabidopsis. PLoS Genet. 9: e1003766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hundertmark M., Hincha D.K. (2008). LEA (late embryogenesis abundant) proteins and their encoding genes in Arabidopsis thaliana. BMC Genomics 9: 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H.-G., Kim J., Kim B., Jeong H., Choi S.H., Kim E.K., Lee H.-Y., Lim P.O. (2011). Overexpression of FTL1/DDF1, an AP2 transcription factor, enhances tolerance to cold, drought, and heat stresses in Arabidopsis thaliana. Plant Sci. 180: 634–641. [DOI] [PubMed] [Google Scholar]
- Kang H.M., Zaitlen N.A., Wade C.M., Kirby A., Heckerman D., Daly M.J., Eskin E. (2008). Efficient control of population structure in model organism association mapping. Genetics 178: 1709–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katori T., Ikeda A., Iuchi S., Kobayashi M., Shinozaki K., Maehashi K., Sakata Y., Tanaka S., Taji T. (2010). Dissecting the genetic control of natural variation in salt tolerance of Arabidopsis thaliana accessions. J. Exp. Bot. 61: 1125–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney A.M., McKay J.K., Richards J.H., Juenger T.E. (2014). Direct and indirect selection on flowering time, water-use efficiency (WUE, δ (13)C), and WUE plasticity to drought in Arabidopsis thaliana. Ecol. Evol. 4: 4505–4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesari R., Lasky J.R., Villamor J.G., Des Marais D.L., Chen Y.-J., Liu T.-W., Lin W., Juenger T.E., Verslues P.E. (2012). Intron-mediated alternative splicing of Arabidopsis P5CS1 and its association with natural variation in proline and climate adaptation. Proc. Natl. Acad. Sci. USA 109: 9197–9202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keurentjes J.J.B., Fu J., Terpstra I.R., Garcia J.M., van den Ackerveken G., Snoek L.B., Peeters A.J.M., Vreugdenhil D., Koornneef M., Jansen R.C. (2007). Regulatory network construction in Arabidopsis by using genome-wide gene expression quantitative trait loci. Proc. Natl. Acad. Sci. USA 104: 1708–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohavi R. (1995). A study of cross-validation and bootstrap for accuracy estimation and model selection. In Proceedings IJCAI-95 (Montreal, Canada: Morgan Kaufmann), pp. 1137–1143. [Google Scholar]
- Korte A., Farlow A. (2013). The advantages and limitations of trait analysis with GWAS: a review. Plant Methods 9: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte A., Vilhjálmsson B.J., Segura V., Platt A., Long Q., Nordborg M. (2012). A mixed-model approach for genome-wide association studies of correlated traits in structured populations. Nat. Genet. 44: 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krizek B.A. (2015). Intronic sequences are required for AINTEGUMENTA-LIKE6 expression in Arabidopsis flowers. BMC Res. Notes 8: 556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y., Choi D., Kende H. (2001). Expansins: ever-expanding numbers and functions. Curr. Opin. Plant Biol. 4: 527–532. [DOI] [PubMed] [Google Scholar]
- Li Y., Huang Y., Bergelson J., Nordborg M., Borevitz J.O. (2010). Association mapping of local climate-sensitive quantitative trait loci in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 107: 21199–21204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.F., Liang H.M., Yang S.Y., Boch A., Clemens S., Chen C.C., Wu J.F., Huang J.L., Yeh K.C. (2009). Arabidopsis IRT3 is a zinc-regulated and plasma membrane localized zinc/iron transporter. New Phytol. 182: 392–404. [DOI] [PubMed] [Google Scholar]
- Lippert C., Casale F.P., Rakitsch B., Stegle O. (2014). LIMIX: genetic analysis of multiple traits. BioRxiv, http://dx.doi.org/10.1101/003905. [Google Scholar]
- Lowry D.B., Logan T.L., Santuari L., Hardtke C.S., Richards J.H., DeRose-Wilson L.J., McKay J.K., Sen S., Juenger T.E. (2013). Expression quantitative trait locus mapping across water availability environments reveals contrasting associations with genomic features in Arabidopsis. Plant Cell 25: 3266–3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn J.E., Delorge I., Figueroa C.M., Van Dijck P., Stitt M. (2014). Trehalose metabolism in plants. Plant J. 79: 544–567. [DOI] [PubMed] [Google Scholar]
- Lunn J.E., Feil R., Hendriks J.H.M., Gibon Y., Morcuende R., Osuna D., Scheible W.-R., Carillo P., Hajirezaei M.R., Stitt M. (2006). Sugar-induced increases in trehalose 6-phosphate are correlated with redox activation of ADPglucose pyrophosphorylase and higher rates of starch synthesis in Arabidopsis thaliana. Biochem. J. 397: 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z., Hu X., Cai W., Huang W., Zhou X., Luo Q., Yang H., Wang J., Huang J. (2014). Arabidopsis miR171-targeted scarecrow-like proteins bind to GT cis-elements and mediate gibberellin-regulated chlorophyll biosynthesis under light conditions. PLoS Genet. 10: e1004519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magome H., Yamaguchi S., Hanada A., Kamiya Y., Oda K. (2004). dwarf and delayed-flowering 1, a novel Arabidopsis mutant deficient in gibberellin biosynthesis because of overexpression of a putative AP2 transcription factor. Plant J. 37: 720–729. [DOI] [PubMed] [Google Scholar]
- Magome H., Yamaguchi S., Hanada A., Kamiya Y., Oda K. (2008). The DDF1 transcriptional activator upregulates expression of a gibberellin-deactivating gene, GA2ox7, under high-salinity stress in Arabidopsis. Plant J. 56: 613–626. [DOI] [PubMed] [Google Scholar]
- Martin M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17: 10–12. [Google Scholar]
- Matsui A., et al. (2008). Arabidopsis transcriptome analysis under drought, cold, high-salinity and ABA treatment conditions using a tiling array. Plant Cell Physiol. 49: 1135–1149. [DOI] [PubMed] [Google Scholar]
- Meijón M., Satbhai S.B., Tsuchimatsu T., Busch W. (2014). Genome-wide association study using cellular traits identifies a new regulator of root development in Arabidopsis. Nat. Genet. 46: 77–81. [DOI] [PubMed] [Google Scholar]
- Meimoun P., Gousset-Dupont A., Lebouteiller B., Ambard-Bretteville F., Besin E., Lelarge C., Mauve C., Hodges M., Vidal J. (2009). The impact of PEPC phosphorylation on growth and development of Arabidopsis thaliana: molecular and physiological characterization of PEPC kinase mutants. FEBS Lett. 583: 1649–1652. [DOI] [PubMed] [Google Scholar]
- Minic Z., Rihouey C., Do C.T., Lerouge P., Jouanin L. (2004). Purification and characterization of enzymes exhibiting β-D-xylosidase activities in stem tissues of Arabidopsis. Plant Physiol. 135: 867–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore J.P., Vicré-Gibouin M., Farrant J.M., Driouich A. (2008). Adaptations of higher plant cell walls to water loss: drought vs desiccation. Physiol. Plant. 134: 237–245. [DOI] [PubMed] [Google Scholar]
- Müller J., Aeschbacher R.A., Wingler A., Boller T., Wiemken A. (2001). Trehalose and trehalase in Arabidopsis. Plant Physiol. 125: 1086–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson A.S., Engström P., Söderman E. (2004). The homeobox genes ATHB12 and ATHB7 encode potential regulators of growth in response to water deficit in Arabidopsis. Plant Mol. Biol. 55: 663–677. [DOI] [PubMed] [Google Scholar]
- Paganelli L., Caillaud M.-C., Quentin M., Damiani I., Govetto B., Lecomte P., Karpov P.A., Abad P., Chabouté M.-E., Favery B. (2015). Three BUB1 and BUBR1/MAD3-related spindle assembly checkpoint proteins are required for accurate mitosis in Arabidopsis. New Phytol. 205: 202–215. [DOI] [PubMed] [Google Scholar]
- Pauwels L., Goossens A. (2011). The JAZ proteins: a crucial interface in the jasmonate signaling cascade. Plant Cell 23: 3089–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F., et al. (2011). Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 12: 2825–2830. [Google Scholar]
- Pereyra-Irujo G.A., Velázquez L., Lechner L., Aguirrezábal L.A.N. (2008). Genetic variability for leaf growth rate and duration under water deficit in sunflower: analysis of responses at cell, organ, and plant level. J. Exp. Bot. 59: 2221–2232. [DOI] [PubMed] [Google Scholar]
- Peters S., Egert A., Stieger B., Keller F. (2010). Functional identification of Arabidopsis ATSIP2 (At3g57520) as an alkaline α-galactosidase with a substrate specificity for raffinose and an apparent sink-specific expression pattern. Plant Cell Physiol. 51: 1815–1819. [DOI] [PubMed] [Google Scholar]
- Proost S., Van Bel M., Vaneechoutte D., Van de Peer Y., Inzé D., Mueller-Roeber B., Vandepoele K. (2015). PLAZA 3.0: an access point for plant comparative genomics. Nucleic Acids Res. 43: D974–D981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ré D.A., Capella M., Bonaventure G., Chan R.L. (2014). Arabidopsis AtHB7 and AtHB12 evolved divergently to fine tune processes associated with growth and responses to water stress. BMC Plant Biol. 14: 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebollo R., Romanish M.T., Mager D.L. (2012). Transposable elements: an abundant and natural source of regulatory sequences for host genes. Annu. Rev. Genet. 46: 21–42. [DOI] [PubMed] [Google Scholar]
- Rodrigues A., et al. (2013). ABI1 and PP2CA phosphatases are negative regulators of Snf1-related protein kinase1 signaling in Arabidopsis. Plant Cell 25: 3871–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas U., Cibrian-Jaramillo A., Ristova D., Banta J.A., Gifford M.L., Fan A.H., Zhou R.W., Kim G.J., Krouk G., Birnbaum K.D., Purugganan M.D., Coruzzi G.M. (2013). Integration of responses within and across Arabidopsis natural accessions uncovers loci controlling root systems architecture. Proc. Natl. Acad. Sci. USA 110: 15133–15138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose A.B. (2008). Intron-mediated regulation of gene expression. Curr. Top. Microbiol. Immunol. 326: 277–290. [DOI] [PubMed] [Google Scholar]
- Saeed A.I., et al. (2003). TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34: 374–378. [DOI] [PubMed] [Google Scholar]
- Saeys Y., Inza I., Larrañaga P. (2007). A review of feature selection techniques in bioinformatics. Bioinformatics 23: 2507–2517. [DOI] [PubMed] [Google Scholar]
- Sasaki E., Zhang P., Atwell S., Meng D., Nordborg M. (2015). “Missing” G x E variation controls flowering time in Arabidopsis thaliana. PLoS Genet. 11: e1005597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer S.E., Schlüter P.M., Baskar R., Gheyselinck J., Bolaños A., Curtis M.D., Grossniklaus U. (2009). Intronic regulatory elements determine the divergent expression patterns of AGAMOUS-LIKE6 subfamily members in Arabidopsis. Plant J. 59: 987–1000. [DOI] [PubMed] [Google Scholar]
- Schulze S., Schäfer B.N., Parizotto E.A., Voinnet O., Theres K. (2010). LOST MERISTEMS genes regulate cell differentiation of central zone descendants in Arabidopsis shoot meristems. Plant J. 64: 668–678. [DOI] [PubMed] [Google Scholar]
- Schweizer P., Stein N. (2011). Large-scale data integration reveals colocalization of gene functional groups with meta-QTL for multiple disease resistance in barley. Mol. Plant Microbe Interact. 24: 1492–1501. [DOI] [PubMed] [Google Scholar]
- Sewelam N., Kazan K., Thomas-Hall S.R., Kidd B.N., Manners J.M., Schenk P.M. (2013). Ethylene response factor 6 is a regulator of reactive oxygen species signaling in Arabidopsis. PLoS One 8: e70289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skirycz A., De Bodt S., Obata T., De Clercq I., Claeys H., De Rycke R., Andriankaja M., Van Aken O., Van Breusegem F., Fernie A.R., Inzé D. (2010). Developmental stage specificity and the role of mitochondrial metabolism in the response of Arabidopsis leaves to prolonged mild osmotic stress. Plant Physiol. 152: 226–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skirycz A., Claeys H., De Bodt S., Oikawa A., Shinoda S., Andriankaja M., Maleux K., Eloy N.B., Coppens F., Yoo S.-D., Saito K., Inzé D. (2011a). Pause-and-stop: the effects of osmotic stress on cell proliferation during early leaf development in Arabidopsis and a role for ethylene signaling in cell cycle arrest. Plant Cell 23: 1876–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skirycz A., et al. (2011b). Survival and growth of Arabidopsis plants given limited water are not equal. Nat. Biotechnol. 29: 212–214. [DOI] [PubMed] [Google Scholar]
- Snoek L.B., Terpstra I.R., Dekter R., Van den Ackerveken G., Peeters A.J.M. (2013). Genetical genomics reveals large scale genotype-by-environment interactions in Arabidopsis thaliana. Front. Genet. 3: 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staiger D., Brown J.W.S. (2013). Alternative splicing at the intersection of biological timing, development, and stress responses. Plant Cell 25: 3640–3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strizhov N., Abrahám E., Okrész L., Blickling S., Zilberstein A., Schell J., Koncz C., Szabados L. (1997). Differential expression of two P5CS genes controlling proline accumulation during salt-stress requires ABA and is regulated by ABA1, ABI1 and AXR2 in Arabidopsis. Plant J. 12: 557–569. [DOI] [PubMed] [Google Scholar]
- Szabados L., Savouré A. (2010). Proline: a multifunctional amino acid. Trends Plant Sci. 15: 89–97. [DOI] [PubMed] [Google Scholar]
- Takeda S., Iwasaki A., Matsumoto N., Uemura T., Tatematsu K., Okada K. (2013). Physical interaction of floral organs controls petal morphogenesis in Arabidopsis. Plant Physiol. 161: 1242–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1001 Genomes Consortium (2016). 1,135 genomes reveal the global pattern of polymorphism in Arabidopsis thaliana. Cell 166: 481–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisné S., Reymond M., Vile D., Fabre J., Dauzat M., Koornneef M., Granier C. (2008). Combined genetic and modeling approaches reveal that epidermal cell area and number in leaves are controlled by leaf and plant developmental processes in Arabidopsis. Plant Physiol. 148: 1117–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisné S., et al. (2013). Phenoscope: an automated large-scale phenotyping platform offering high spatial homogeneity. Plant J. 74: 534–544. [DOI] [PubMed] [Google Scholar]
- Tisné S., Barbier F., Granier C. (2011). The ERECTA gene controls spatial and temporal patterns of epidermal cell number and size in successive developing leaves of Arabidopsis thaliana. Ann. Bot. (Lond.) 108: 159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukaya H. (2002). Interpretation of mutants in leaf morphology: genetic evidence for a compensatory system in leaf morphogenesis that provides a new link between cell and organismal theories. Int. Rev. Cytol. 217: 1–39. [DOI] [PubMed] [Google Scholar]
- Valdés A.E., Overnäs E., Johansson H., Rada-Iglesias A., Engström P. (2012). The homeodomain-leucine zipper (HD-Zip) class I transcription factors ATHB7 and ATHB12 modulate abscisic acid signalling by regulating protein phosphatase 2C and abscisic acid receptor gene activities. Plant Mol. Biol. 80: 405–418. [DOI] [PubMed] [Google Scholar]
- Vanhaeren H., Inzé D., Gonzalez N. (2016). Plant growth beyond limits. Trends Plant Sci. 21: 102–109. [DOI] [PubMed] [Google Scholar]
- Vanhaeren H., Gonzalez N., Coppens F., De Milde L., Van Daele T., Vermeersch M., Eloy N.B., Storme V., Inzé D. (2014). Combining growth-promoting genes leads to positive epistasis in Arabidopsis thaliana. eLife 3: e02252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Houtte H., Vandesteene L., López-Galvis L., Lemmens L., Kissel E., Carpentier S., Feil R., Avonce N., Beeckman T., Lunn J.E., Van Dijck P. (2013). Overexpression of the trehalase gene AtTRE1 leads to increased drought stress tolerance in Arabidopsis and is involved in abscisic acid-induced stomatal closure. Plant Physiol. 161: 1158–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen H., Kliebenstein D.J., West M.A.L., Kim K., van Poecke R., Katagiri F., Michelmore R.W., Doerge R.W., St Clair D.A. (2007). Natural variation among Arabidopsis thaliana accessions for transcriptome response to exogenous salicylic acid. Plant Cell 19: 2099–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma S., Simon R. (2006). Bias in error estimation when using cross-validation for model selection. BMC Bioinformatics 7: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vile D., Pervent M., Belluau M., Vasseur F., Bresson J., Muller B., Granier C., Simonneau T. (2012). Arabidopsis growth under prolonged high temperature and water deficit: independent or interactive effects? Plant Cell Environ. 35: 702–718. [DOI] [PubMed] [Google Scholar]
- Wang S., Blumwald E. (2014). Stress-induced chloroplast degradation in Arabidopsis is regulated via a process independent of autophagy and senescence-associated vacuoles. Plant Cell 26: 4875–4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West M.A.L., Kim K., Kliebenstein D.J., van Leeuwen H., Michelmore R.W., Doerge R.W., St Clair D.A. (2007). Global eQTL mapping reveals the complex genetic architecture of transcript-level variation in Arabidopsis. Genetics 175: 1441–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright I.J., et al. (2004). The worldwide leaf economics spectrum. Nature 428: 821–827. [DOI] [PubMed] [Google Scholar]
- Wu Y., Cosgrove D.J. (2000). Adaptation of roots to low water potentials by changes in cell wall extensibility and cell wall proteins. J. Exp. Bot. 51: 1543–1553. [DOI] [PubMed] [Google Scholar]
- Wu T.D., Nacu S. (2010). Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26: 873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuyts N., Dhondt S., Inzé D. (2015). Measurement of plant growth in view of an integrative analysis of regulatory networks. Curr. Opin. Plant Biol. 25: 90–97. [DOI] [PubMed] [Google Scholar]
- Yvert G., Brem R.B., Whittle J., Akey J.M., Foss E., Smith E.N., Mackelprang R., Kruglyak L. (2003). Trans-acting regulatory variation in Saccharomyces cerevisiae and the role of transcription factors. Nat. Genet. 35: 57–64. [DOI] [PubMed] [Google Scholar]
- Zhou X.F., Jin Y.H., Yoo C.Y., Lin X.-L., Kim W.-Y., Yun D.-J., Bressan R.A., Hasegawa P.M., Jin J.B. (2013). CYCLIN H;1 regulates drought stress responses and blue light-induced stomatal opening by inhibiting reactive oxygen species accumulation in Arabidopsis. Plant Physiol. 162: 1030–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.