
Figure 1.
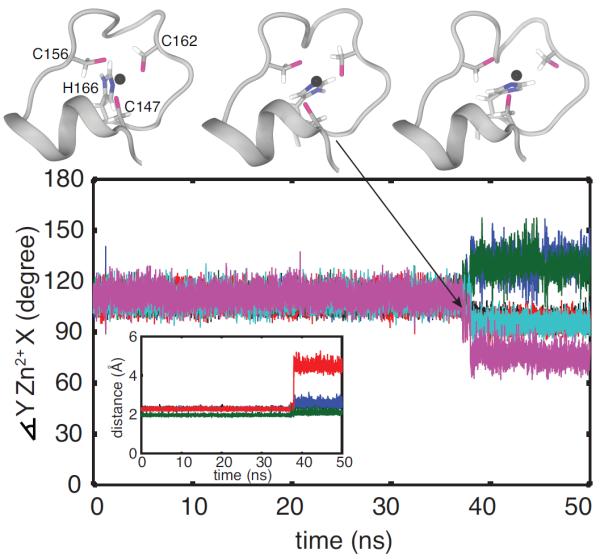
Unfolding of ZF2 during MD simulation of TTP. Conformations of the unfolding C-terminal zinc finger of TTP, sampled from an MD trajectory, are shown at top and correspond to t =37.130 ns (top, left), 37.145 ns (top, center) and 38.420 ns (top, right). Below, the geometry of the zinc coordination in the C-terminal zinc finger of TTP is monitored. In the main figure, the angles between the zinc ion and the zinc coordinating atoms are shown for the first 50 ns: ∡SC147-Zn2+-SC156 in black, ∡SC147-Zn2+-SC162 in blue, in red, ∡SC156-Zn2+-SC162 in green, in cyan and in magenta. In the inset, the distances between the zinc ion and the zinc coordinating atoms are shown: SC147-Zn2+ in black, SC156-Zn2+ in blue, SC162-Zn2+ in red and in green.