


Abstract
Tyrosyl DNA phosphodiesterase 2 (TDP2) is a multifunctional protein implicated in DNA repair, signal transduction and transcriptional regulation. In its DNA repair role, TDP2 safeguards genome integrity by hydrolyzing 5′-tyrosyl DNA adducts formed by abortive topoisomerase II (Top2) cleavage complexes to allow error-free repair of DNA double-strand breaks, thereby conferring cellular resistance against Top2 poisons. TDP2 consists of a C-terminal catalytic domain responsible for its phosphodiesterase activity, and a functionally uncharacterized N-terminal region. Here, we demonstrate that this N-terminal region contains a ubiquitin (Ub)-associated (UBA) domain capable of binding multiple forms of Ub with distinct modes of interactions and preference for either K48- or K63-linked polyUbs over monoUb. The structure of TDP2 UBA bound to monoUb shows a canonical mode of UBA-Ub interaction. However, the absence of the highly conserved MGF motif and the presence of a fourth α-helix make TDP2 UBA distinct from other known UBAs. Mutations in the TDP2 UBA-Ub binding interface do not affect nuclear import of TDP2, but severely compromise its ability to repair Top2-mediated DNA damage, thus establishing the importance of the TDP2 UBA–Ub interaction in DNA repair. The differential binding to multiple Ub forms could be important for responding to DNA damage signals under different contexts or to support the multi-functionality of TDP2.
INTRODUCTION
Tyrosyl DNA phosphodiesterase-2 (TDP2) is a multifunctional protein involved in a broad range of biological processes including DNA repair, gene transcription and signal transduction (1,2). The 5′-tyrosyl DNA phosphodiesterase activity of TDP2 enables excision of trapped Top2-DNA covalent complexes that block replication and transcription (1–3). Besides its well established role in the repair of Top2-mediated DNA damage, TDP2 (also known as ETS1-associated protein 2 (EAPII)) was reported to interact with an apoptosis-promoting transcription factor ETS1 and regulate its activity (4). TDP2 had also been named TTRAP (TRAF and TNF receptor-associated protein) for its function in apoptosis and inflammatory response, as it inhibits NFκB activation and enhances activation of MAPK/JNK/p38 (1,5).
Consistent with its varied roles, loss of TDP2 function has been linked to a number of disease manifestations including defective neuronal development, Parkinson's disease and cancer (1,6,7), and TDP2 up-regulation is implicated in resistance against topoisomerase inhibitors used as anti-cancer drugs (8). Moreover, the unique enzymatic activity of TDP2 is exploited by hepatitis B virus (HBV) and picornaviruses to remove covalently bound terminal proteins from the replicated viral genome during the viruses’ life cycle (9,10). Hence, mechanistic insights into TDP2 activity and its regulation are relevant for the development of a therapeutic strategy that targets TDP2 in a broad spectrum of human diseases.
Earlier structural studies showed that TDP2 consists of two domains (Figure 1), a small N-terminal domain and the C-terminal catalytic domain, the latter of which is responsible for the phosphodiesterase activity (11,12). While the structure and activity of the C-terminal catalytic domain have been extensively studied, the role of the N-terminal domain remains unknown, although it has been proposed to interact with ubiquitin (Ub) or Ub-like proteins based on its primary sequence (13) and structural homology to known Ub-associated (UBA) domains. The ubiquitin receptor family containing the three-helix bundle UBA domain has many structurally characterized members that are involved in various biological processes, including proteasomal protein degradation and DNA-damage signaling (14–16). Interestingly, the crystal structure of the full-length TDP2 from C. elegans (PDB ID: 4GEW) (Figure 1) showed an N-terminal domain consisting of four short α-helices, rather than the canonical tri-helix UBA structure (11). In addition, the TDP2 N-terminal domain lacks the ‘MGF’ sequence motif highly conserved among the three-helix UBA domains that makes critical hydrophobic interactions with Ub (14,15,17). It remained to be investigated whether the extra helix functions as an integral part of the core helical bundle in solution, and whether this domain indeed binds Ub. Nonetheless, the presence of a putative UBA domain raises possibilities for the versatile regulation of TDP2 activity mediated by interactions with ubiquitinated proteins.
Figure 1.

Structure of the full-length CeTDP2. Schematic and ribbon representations of the previously published structure of C. elegans TDP2 protein, PDB ID 4GEW (11).
Ubiquitination is an important post-translational modification that controls a myriad of biological processes. Either through monoUb or polyUb conjugation to substrate proteins, various downstream responses can be instigated (18–20). Many different types of Ub-binding domains (UBDs), including UBA, CUE, UIM, NZF, GAT and PAZ, mediate localization or modulation of activities of downstream effectors in response to ubiquitination signals (21). The diverse UBDs exhibit differential affinities toward distinct ubiquitination states, such as mono-Ub and poly-Ub with different linkage types. A linkage-selective polyUb-binding mode allows the proteins carrying the UBDs to function in distinct signaling pathways to bring about varied responses like endocytosis, DNA repair, apoptosis and proteasomal protein degradation/turnover (19,21). Alternatively, in the absence of clear preference for a particular polyUb linkage-type, a response to ubiquitin signals could be based on a temporal/spatial regulation of the Ub-UBD interactions (22).
In the present study, we examined the interactions of the N-terminal domain of TDP2 with various Ub species (monoUb, K48-linked diUb and K63-linked diUb) and show that it adopts an unusual 4-helix bundle UBA domain. Despite this variation in the UBA domain fold, the structure of the TDP2 UBA-monoUb complex based on NMR-derived restraints shows a mode of monoUb interaction similar to that observed for other UBAs (14,15,17). We find that TDP2 UBA binds ubiquitin specifically and with greater affinity for either K48- or K63-linked polyubiquitin over monoUb. Moreover, NMR analyses show that, in spite of similar affinities for K48- and K63-linked diUb, the mode of binding is different for these two linkage types. We hypothesize that the ability to bind to multiple forms of ubiquitin chains allows TDP2 to respond to distinct ubiquitination signals during DNA repair. In addition, the promiscuous binding might also be important for regulation of the differential roles of the multifunctional TDP2/TTRAP/EAPII protein.
MATERIALS AND METHODS
Cloning, expression and purification of TDP2 UBA domain
A codon-optimized gene for C. elegans TDP2 (corresponding to amino acid residues 41 to 100) was cloned into pE-SUMO vector (LifeSensors Inc.). His6-SUMO-CeTDP241-100 protein (hereafter called His6-SUMO-UBA) was expressed from Escherichia coli (E. coli) BL21(DE3) in Luria Broth (LB) by induction with 1.0 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) and post-induction growth for 16 h at 25°C. The E. coli cells were harvested by centrifugation and cell pellet lysed by sonication in lysis buffer [50 mM Tris-Cl (pH 7.4), 0.5 M sodium chloride, 10 mM imidazole and 0.5 mM phenylmethylsulfonyl fluoride (PMSF) as protease inhibitor (MP Biomedicals)]. After lysis, the supernatant was subjected to Ni2+-affinity chromatography and eluted with increasing concentrations of imidazole. Eluted His6-SUMO-UBA was subjected to cleavage with Ulp1 protease overnight at 4°C and the cleaved sample was injected onto a Superdex 200 (10/300) size-exclusion column (GE Healthcare). A second round of Ni2+-affinity chromatography was next used to further separate UBA from His6-SUMO that could not be separated by size-exclusion chromatography due to overlapping elution profiles. Finally, UBA was dialyzed against buffer A [25 mM sodium phosphate buffer (pH 6.5), 50 mM sodium chloride] while His6-SUMO was eluted with 250 mM imidazole, concentrated and dialyzed into buffer A as well (Supplementary Figure S1A).
15N labeled and 15N,13C labeled UBA were expressed in E. coli BL21(DE3) cells in M9 minimal medium supplemented with 15N labeled ammonium chloride (Cambridge Isotope laboratories Inc.) alone and with 15N labeled ammonium chloride and 13C labeled glucose (Cambridge Isotope laboratories Inc.), respectively. Protein production was induced with 1.0 mM IPTG, followed by post-induction growth for 24 h at 25°C. Proteins were purified as described above.
Site-directed spin labeling was done at two positions on TDP2 UBA for paramagnetic relaxation enhancement (PRE) NMR experiments. Met43 and Ser84 were individually mutated to cysteine using site-directed mutagenesis. All mutant expression constructs were confirmed by DNA sequencing. Proteins were expressed and purified as described above. A thiol-specific spin label (1-Oxyl-2,2,5,5-tetramethylpyrroline-3- methyl)methanethiosulfonate (MTSL) (Santa Cruz Biotechnology Inc.) was added to 8 times molar excess of the cysteine-tagged protein, incubated overnight at 4°C and completion of reaction monitored by a DTNB assay (23). Excess spin label was removed by dialysis into buffer A.
Cloning, expression and purification of mono and diUbiquitins
Human ubiquitin was expressed using a pET23a-based expression vector in E. coli Rosetta2(DE3) pLysS cells (EMD Millipore) in 2xYT broth by induction with 0.5 mM IPTG and post-induction growth for 4 h at 37°C. Cells were harvested by centrifugation. Purification of the protein was as described elsewhere (24) with purified protein dialyzed into buffer A (Supplementary Figure S1A). For 15N labeled ubiquitin, the protein was expressed similarly, but in M9 minimal medium supplemented with 15N labeled ammonium chloride (Cambridge Isotope laboratories Inc.) and post-induction growth at 37°C for 14 h.
For PRE experiments, site-directed spin labeling was done at Gly75 of ubiquitin, which was mutated to cysteine. Spin labeling with MTSL and sample preparation was done as described for TDP2 UBA.
Conjugation of monoUb to generate K48- or K63-linked diUbs was done as described elsewhere (24) with purified conjugated proteins dialyzed into buffer A. Plasmids for all conjugating enzyme expression, pET3d-E2-25K-C170S, pGEX-Ubc13, MBP-His6-TEV-MMS2 and pET21d-Ube1 were obtained from the Addgene plasmid repository. Expression and purification of conjugating enzymes was done as described elsewhere (24).
NMR spectroscopy and chemical shift assignments
All NMR data were collected at 25°C on 1 mM protein samples in 25 mM sodium phosphate buffer (pH 6.5), 50 mM sodium chloride, 5% 2H2O / 95% 1H2O. 1H,13C,15N-labeled TDP2 UBA protein was used for HSQC experiments and chemical shifts referenced to 4,4- dimethyl-4-silapentane-1-sulfonic acid (DSS). Backbone assignments were done with 1H,13C,15N-labeled UBA sample using standard experiments: HNCA, CBCA(CO)NH, HN(CO)CA, HNCACB and HNCO, all collected on a Bruker 900 MHz spectrometer equipped with a 5 mm TCI cryoprobe. Side-chain assignments were done with the same sample using standard experiments: 1H -13C HSQC, HCCH-COSY, H(CC)(CO)NH TOCSY and (H)CC(CO)NH TOCSY for aliphatic groups and HBCBCGCDCEHE and HBCBCGCDHD for aromatic sidechains on a Bruker 900 MHz spectrometer equipped with a 5 mm TCI cryoprobe. All spectra were processed with NMRPipe software (25). The PINE server (26) was used for automated backbone and sidechain assignments, followed by manual confirmation with the CcpNMR analyses software (27).
A PRE experiment was used to generate intermolecular distances involving the backbone amides of 15N-labeled TDP2 UBA and MTSL-labeled monoUb(G75C). Peak broadening in the presence of the MTSL-labeled monoUb(G75C) was recorded by a 2D 1H,15N HSQC experiment. After measurements of the paramagnetically labeled sample were completed, MTSL was reduced to the diamagnetic state by addition of 5-fold molar excess of ascorbic acid (6 mM). Similarly, the reciprocal experiment with15N-labeled monoUb and MTSL-labeled TDP2 UBA (M43C or S84C) was done. The procedure and equations for distance calculations were as described previously (23,28,29), with an evolution time of 11 ms.
NMR titration experiments using 2D 1H,15N HSQCs for obtaining dissociation constants between TDP2 UBA and Ub were done on a Bruker 700 MHz spectrometer equipped with a 1.7 mm TCI cryoprobe and SampleJet sample changer for automated sample loading and data acquisition through the IconNMRTM software. Every titration set consisted of 10 tubes in a 96-tube rack, each with sample volumes of 40 μl of 0.4 mM 15N labeled TDP2 UBA and increasing concentrations of Ub (either monoUb, K48-diUb or K63-diUb), as follows (mM): 0, 0.1, 0.2, 0.4, 0.8, 1.2, 1.6, 2.0, 2.4 and 3.2. For the reciprocal experiment, 0.4 mM 15N-labeled Ub (monoUb, proximal 15N-labeled K48 diUb, distal 15N-labeled K48 diUb, proximal 15N-labeled K63 diUb, distal 15N-labeled K63 diUb or SUMO) was titrated with increasing concentration of TDP2 UBA (in mM): 0, 0.1, 0.2, 0.4, 0.8, 1.2, 1.6, 2.0, 2.4 and 3.2. All titrations were conducted at 25°C. Spectra were processed with NMRPipe software (25) and analyzed with the CcpNMR analyses software (27). The peak shift data for identified residues were analyzed with Prism Graphpad 6.0 software and titration curves fit to the following equation for Kd estimation (23):
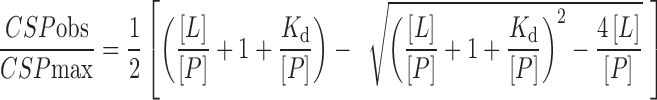 |
where, [L] and [P] are concentrations of ligand (unlabeled species) and protein (15N labeled), respectively. CSPobs and CSPmax are chemical shift perturbation values of selected 1H15N labeled residues at corresponding [L]/[P] molar ratio and at saturation, respectively.
For chemical shift perturbation mapping (Figures 2C and D, 6A–C and 7A–C), the combined CSP is defined as 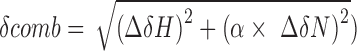 , (30), where
, (30), where 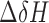 is 1H chemical shift change,
is 1H chemical shift change, 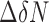 is 15N chemical shift change, and a scaling factor, α, was set at 0.15.
is 15N chemical shift change, and a scaling factor, α, was set at 0.15.
Figure 2.

TDP2 UBA binds specifically to ubiquitin. (A) Binding isotherms of TDP2 UBA titrated with increasing concentrations of monoUb, diUb (K48 or K63-linked) and SUMO, monitored by fluorescence anisotropy of Trp72 of TDP2 UBA. (B) HSQC spectrum of 15N labeled CeTDP2 UBA (black), superimposed over that of 15N CeTDP2 UBA in the presence of 6-fold molar excess of monoUb (red). Blue arrows trace a few of the significantly shifted peaks from their free to the bound position. (C) Per residue chemical shift perturbation map of monoUb on the left and residues with significant change (>2 σ0 corr) highlighted on the structure of monoUb on the right (PDB ID: 1D3Z). The solid red and dashed green lines cross the map at 1 and 2 σ0 corr (corrected standard deviation (44,67)), respectively. Peaks that vanished due to significant broadening during the titration are marked by black triangles on the graph. Red triangles on the graph represent the three proline residues that do not show NH peaks in the 2D 1H,15N HSQC experiment and were therefore excluded from the CSP analysis. Secondary structure elements of Ub are drawn on top of the graph. (D) Per residue chemical shift perturbation map of TDP2 UBA on the left and residues with significant change (>2 σ0 corr) highlighted on the structure of TDP2 UBA on the right. Red and green lines as well as black triangles are drawn as in panel C. Secondary structure elements of TDP2 UBA are drawn on top of the graph.
Figure 6.

Interaction of K48-linked diUb with UBA. (A) Per residue chemical shift perturbation map of 15N labeled distal Ub residues when K48-linked diUb was titrated with increasing concentrations of CeTDP2 UBA. The red line denotes cut-off for significance; set at 1 standard deviation (1 σ) from all the weighted averaged chemical shift values. Peaks that vanished during the titration are represented by black triangles on the graph and prolines, which are excluded from this analysis, indicated with red triangles. (B) Per residue chemical shift perturbation map of 15N labeled proximal Ub residues when K48-linked diUb was titrated with increasing concentrations of CeTDP2 UBA. Significance denoted by a red line defined as in A. Prolines and amino acids with signals that disappeared are represented by red and black triangles, respectively. (C) Per residue chemical shift perturbation map of 15N labeled CeTDP2 UBA residues when it was titrated with increasing concentrations of K48-linked diUb. Significance as in A. (D) A hypothetical model of one molecule of TDP2 UBA binding to one molecule of K48-linked diUb. Significantly shifted residues for TDP2 UBA and Ub are colored in red with some of these residues that determine the interaction surface on each Ub moiety labeled. Peaks that disappeared upon titration are colored in blue on both UBA and Ub moieties.
Figure 7.

Interaction of K63-linked diUb with UBA. (A) Per residue chemical shift perturbation map of 15N labeled distal Ub residues when K63-linked diUb was titrated with increasing concentrations of CeTDP2 UBA. The red line denotes the cut-off for significance; set at 1 standard deviation (1 σ) from all the weighted averaged chemical shift values. Peaks that vanished during the titration are represented by black triangles on the graph. (B) Per residue chemical shift perturbation map of 15N labeled proximal Ub residues when K63-linked diUb was titrated with increasing concentrations of CeTDP2 UBA. Significance as in A. (C) Per residue chemical shift perturbation map of 15N labeled CeTDP2 UBA residues when it was titrated with increasing concentrations of K63-linked diUb. Significance as in A. (D) A hypothetical model of one molecule of TDP2 UBA binding to one molecule of K63-linked diUb. Significantly shifted residues for TDP2 UBA and Ub are colored in red with some of these residues that determine the UBA interaction surface on each Ub moiety labeled. Peaks that disappeared upon titration are colored in blue on both UBA and Ub moieties.
NMR data deposition
The backbone and side-chain NMR chemical shift assignment data are available in the Biological Magnetic Resonance Bank (BMRB), entry 26771.
Docking of TDP2 UBA and monoUb using HADDOCK and modeling of the diubiuqitin complexes
HADDOCK 2.1 (High Ambiguity Driven protein-protein DOCKing) (31) in combination with CNS (32) was used to generate CeTDP2 UBA:ubiquitin complexes. The atomic coordinates for TDP2 UBA and ubiquitin were obtained from PDB entries 4GEW and 1D3Z, respectively. The model was calculated by using ambiguous interaction restraints (AIRs) and 85 unambiguous PRE-derived distance restraints. In addition, the restraints from the hydrogen bonds published for ubiquitin (PDB 1D3Z) were used to maintain its fold. TDP2 or ubiquitin amino acids with amide chemical shift perturbation (CSP) values greater than two standard deviations above the average upon forming the complex and with solvent accessibility >40% were defined as ‘active’. The neighboring amino acids of ‘active’ residues with solvent accessibility >40% were defined as ‘passive’ residues. AIRs restrict ‘active’ residues to be within 3.0 Å of any atoms of the binding partner's ‘active’ or ‘passive’ residues, as described in (31). Flexible residues were also defined, which include ‘active’ and ‘passive’ residues as well as amino acids that are up to two sequence positions away from them in either the N- or C-terminal direction.
PRE-derived distance restraints, which define the distance between the unpaired electron center of spin label MTSL and an amide proton of the binding partner, were obtained by comparing HSQC spectra acquired on the complex in its diamagnetic (achieved by addition of 6 mM ascorbic acid, which was 5X molar excess of the MTSL labeled species) and paramagnetic states. Three sets of PRE experiments were performed on samples of 15N-labeled ubiquitin mixed with MTSL-labeled TDP2 UBA with either M43C or S84C spin labeled and of 15N-labeled TDP2 UBA mixed with MTSL-labeled ubiquitin G75C; an evolution time of 11 ms was used in all cases. PRE-derived distance restraints were categorized into two distance ranges, 1.8–23 Å or > 23 Å, as determined by their Ipara/Idia intensity ratio, as described in (28,29). The cutoff ratio was determined to be 85% for 15N-labeled ubiquitin:MTSL-labeled TDP2 UBA M43C or MTSL-labeled TDP2 UBA S84C (Supplementary Figure S5). For 15N-labeled TDP2 UBA:MTSL-labeled ubiquitin G75C, the cutoff was determined to be 60% due to the flexibility of the C-terminal tail (Supplementary Figure S5).
One thousand structures were generated in the first step of rigid body docking, followed by energy minimization. The top scoring 200 structures were used for semi-flexible refinement in torsion angle space, followed by water refinement, scoring and clustering of all 200 structures. During semi-flexible simulated annealing, atoms at the interface were allowed to move, but constrained by AIRs and unambiguous PRE-derived distance restraints. After water refinement, TDP2 UBA:Ub complexes were clustered using a 5.5 Å cut-off criterion and requiring a minimum of four structures per cluster, resulting in an ensemble of 188 structures with an average backbone RMSD value of 1.97 ± 0.41 Å for this primary cluster compared to its lowest energy structure. A second cluster, consisting of 8 structures, shows a similar overall binding mode to the primary cluster with an average backbone RMSD of 2.3 Å, but with less favorable binding energy statistics (Table 1). The 10 lowest energy structures in the primary cluster was energy minimized by Schrödinger (www.schrodinger.com) after replacing the MTSL-labeled Cys with the wild-type amino acid type (Met43 for TDP2 UBA or Gly75 for ubiquitin).
Table 1. Structure statistics for the two clusters obtained from HADDOCK modeling of TDP2 UBA-monoUb complex.
| Cluster number from HADDOCK | 1 | 2 |
|---|---|---|
| Structures in the cluster | 188* | 8$ |
| RMSD from lowest energy structure | 1.9 ± 0.4 Å | 3.0 ± 0.4 Å |
| Van der Waals energy | −41.2 ± 8.9 | −28.9 ± 7.6 |
| Electrostatic energy | −171.3 ± 66.8 | −132.0±48.8 |
| Desolvation energy | 2.1 ± 6.5 | 1.2 ± 2.5 |
| Restraint violation energy | 4.5 ± 1.4 | 4.5 ± 1.8 |
| Buried surface area | 1344.6 ± 146.9 Å2 | 1068.7 ± 195.7 Å2 |
Starting structure for TDP2 UBA: 4GEW; Starting structure for Ubiquitin:1D3Z.
Ambiguous restraints (TDP2 UBA): 16; Ambiguous restraints for Ubiquitin:13.
Unambiguous restraints (TDP2 UBA): 60; Unambiguous restraints for Ubiquitin:25.
*Cluster with highest number of structures (188) was chosen as the final model for Tdp2 UBA-Ub complex.
$The cluster with 8 structures was similar in overall topology to the cluster with 188 structures, with lowest energy structures from the two clusters differing by 2.3 Å over all backbone atoms.
The hypothetical models for K48- and K63-diUb complexes with TDP2 UBA shown in Figures 6 and 7 were made manually in PyMOL software (33) and energy minimization performed with Schrödinger.
Steady state tryptophan fluorescence anisotropy
Trp72 in TDP2 UBA was exploited for tryptophan fluorescence experiments on a Cary Eclipse Fluorescence spectrophotometer (Varian Inc.). An excitation wavelength of 290 nm was used and emission recorded at 340 nm. The G-factor was calculated once for every set of titrations and incorporated as follows to calculate anisotropy (r):
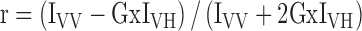 |
For each titration set, the TDP2 UBA concentration was fixed at 10 μM and ligands (monoUb, SUMO, K48-linked diUb or K63-linked diUb) were titrated as follows (μM): 0, 0.01, 0.1, 1.0, 10, 50, 100, 500, 1000 and 2000 (each in a reaction volume of 100 μl).
Human TDP2, wild type and mutants, transfection into DT40 cells
Human TDP2 (HsTDP2)-expressing DT40 cells were generated as described (34). To generate an expression construct for HsTDP2 with the amino acid residues 1–100 deleted (del 1–100), polymerase chain reaction (PCR) was run using the pair of primers 5′-GAGGCGATCGCCATGAGCCCATCTGAAGATACTCAGCAAGAA-3′ and 5′- GCGACGCGTCAATATTATATCTAAGTTGCACAGAAGACCCCA -3′; and the expression plasmid of HsTDP2, as template. HsTDP2 with the point mutation (Phe62 to Arg (F62R)) was amplified from the expression plasmid of HsTDP2 using 4 primers 5′-AGGAAGCATATGGAGTTGGGGAGTTGCCTGGA-3′, 5′-TGCAACGGATCCAATCAGGGCAAAACCCACAC-3′, 5′-GCTCTGAACTCCTACCGCGAGCCTCCGGTGGA-3′ and 5′-TCCACCGGAGGCTCGCGGTAGGAGTTCAGAGC-3′. The PCR products were confirmed by sequencing and then cloned into the pCMV-DDK vector. To generate DT40 cells expressing HsTDP2 del 1–100 or F62R, expression plasmids were linearized and transfected into TDP2−/−/− cells as described (34). The Neomycin resistance colonies were selected and Western blotting was performed to confirm protein expression (Supplementary Figure S6A and B). DT40 cells were lysed with radioimmunoprecipitation assay (RIPA) buffer and the HsTDP2 WT, del 1–100 or F62R protein expression levels were determined using the primary mouse monoclonal anti-flag antibody (F3165, Sigma). The nuclear and cytoplasmic fractions of DT40 cells were isolated using NE-PER nuclear and cytoplasmic extraction reagent (Thermo scientific). The final Western blot was read by Odyssey infrared imaging system (LI-COR).
DT40 FACS analyses
DT40 cells were continuously treated with 1 μM etoposide for 24 h. Harvested cells were fixed with 70% ethanol before re-suspension in phosphate-buffered saline containing 50 μg/ml propidium iodide and 25 μg/ml RNase. Samples were then subjected to analysis on an LSRFortessa cell analyzer from BD Biosciences (Franklin Lakes, NJ, USA).
Etoposide sensitivity assay
Assays were performed as described (35). Briefly, cells were seeded in a 384-well plate, treated with etoposide at the indicated concentrations for 72 h. Viability was measured with the ATPlite 1-step kit (PerkinElmer, Waltham, MA, USA).
HsTDP2 localization in Drosophila S2 cell
The Gateway cloning system (Invitrogen) was used to construct the Drosophila S2 cell expression plasmids. HsTDP2 coding sequences (full length construct: amino acid 2–362, catalytic domain construct: amino acid 103–362) were amplified with a forward primer containing an attB1 site and a reverse primer containing an attB2 site. The PCR fragments were recombined with a pDONR221 vector using BP clonase to produce entry plasmids pENT-HsTDP2 and pENT-HsTDP2cat. pENT-HsTDP2_F62R with Phe62 in the UBA domain replaced by Arg was produced by QuikChange site-directed mutagenesis (Agilent Technologies). A putative nuclear localization signal (NLS) sequence was identified at amino acid 20 to 26 (PEVKKRR) based on Kosugi et al. (36). To disrupt the NLS activity, Lys23, Arg25 and Arg26 was replaced with Asn, Asp and Pro, respectively, in pENT-HsTDP2 by site-directed mutagenesis (36). The resulting entry plasmid is named pENT-HsTDP2_KRR. The entry plasmids were recombined using LR clonase with pAGW, a Gateway S2 cell transformation vector for expression of N-terminal EGFP-tagged protein under the control of the actin 5C promotor, to produce S2 cell expression plasmids pAGW-HsTDP2, pAGW-HsTDP2cat, pAGW-HsTDP2_F62R and pAGW-HsTDP2_KRR.
Drosophila S2 cells were cultured at 22–24°C in M3 medium (Sigma-Aldrich) supplemented with 10% insect media supplement (IMS) (Sigma-Aldrich), 2% heat-inactivated fetal bovine serum (FBS) (GIBCO) and 1% PenStrep (GIBCO). Transfection of S2 cells with each expression plasmid was performed as previously described using dimethyl dioctadecyl ammonium bromide (DDAB) (Sigma-Aldrich) (37). Transfected cells were cultured in serum-free M3 medium containing IMS and PenStrep for three days at 22–24°C in a 12-well plate. On day four, a Concanavalin A (Sigma-Aldrich)-coated coverglass was placed in each well after cells were gently suspended. The cells were allowed to adhere to the coverglass for 2 h. Cells on the coverglasses were fixed as described by Rogers et al. (38). Briefly, the coverglasses were immersed in a fixation solution (90% methanol, 3% paraformaldehyde (EM Sciences), 5 mM sodium carbonate, pH9.0) pre-chilled to −80°C for 10 min. The samples were rehydrated in PBST (0.05% Triton X-100, 1x PBS) for 5 min at room temperature, and blocked with 5% normal goat serum (Jackson ImmunoResearch) in PBST for 30 min at 4°C. The fixed cells were incubated with anti-αTubulin antibody (DM1A at 1:500, Sigma-Aldrich) or anti-GFP antibody (ab290 at 1:1000, Abcam) overnight at 4°C. The samples were then washed twice with PBST for 5 min, incubated with Alexa555-anti-mouse IgG and Alexa488-anti-rabbit IgG antibodies (Invitrogen) at 1:500 dilution for 1.5 h at room temperature, washed twice with PBST for 5 min, incubated with 4′,6-diamidino-2-phenylindole (DAPI) at 2.5 μg/ml in PBST for 10min, and rinsed with PBST. The coverglasses were mounted upside-down on slide glasses with DABCO mounting media (23.3 mg/ml 1,4-diazabicydo[2,2,2]octane (DABCO), 90% glycerol and 1xPBS). The images were acquired on a Zeiss LSM710 laser-scanning confocal microscope system with 100×/1.46 oil α Plan-Apochromat objective lens. Scanned images were analyzed by ZEN (Carl Zeiss) and ImageJ (NIH: http://imagej.nih.gov/ij/index.html).
RESULTS
TDP2 N-terminal domain is a 4-helix bundle UBA that binds ubiquitin but not SUMO
The crystal structure of the full-length C. elegans TDP2 (CeTDP2) revealed a small N-terminal domain consisting of four α-helices, with structural similarity to the three-helix UBA domains (11). To test whether this putative UBA domain of TDP2 interacts with Ub or Ub-like proteins, we performed NMR titration and tryptophan fluorescence anisotropy analyses (Figure 2A, B, Supplementary Figure S1B and S3). We used CeTDP2 (41-100) for most of our biochemical and structural analyses, as it gave the best 2D 15N HSQC spectral profile when compared to zebrafish and human TDP2 N-terminal domain constructs under the conditions used in our NMR experiments (Supplementary Figure S2). Residue assignment of the 2D 15N HSQC peaks of CeTDP2 UBA was achieved to 95% completeness (Supplementary Figure S2C) by using the experiments listed in Materials and Methods; only the three residues at the N-terminus (Ser41, Ser42 and Met43) were missing signals.
In the 2D 1H,15N HSQC spectrum of 15N-labeled CeTDP2 (41–100), a number of peaks shifted significantly upon addition of unlabeled monoUb (Figure 2B). Reciprocally, addition of unlabeled CeTDP2 (41–100) caused significant changes in the spectrum of 15N-labeled Ub (Supplementary Figure S3A). Similar changes were observed upon addition of the corresponding region (residues 1–110) of unlabeled human TDP2 (HsTDP2) to 15N-labeled Ub (Supplementary Figure S3B), suggesting the conserved function of TDP2 N-terminal domain across species. In contrast, we observed no changes in the HSQC spectrum of 15N-labeled CeTDP2 (41–100) upon addition of unlabeled SUMO, even at a SUMO concentration as high as 8 times molar excess of CeTDP2 (41-100) concentration (Supplementary Figure S1C and data not shown). Similarly, fluorescence anisotropy of the single tryptophan residue (Trp72) in CeTDP2 (41–100) increased upon titration with Ub and plateaued at 50–100 μM, while it showed an unsaturable gradual increase at higher SUMO concentrations, likely as a result of non-specific interactions (Figure 2A). Taken together, these data show that the N-terminal domain of TDP2 selectively binds Ub and is indeed a UBA domain.
While the first three helices of the TDP2 UBA form the canonical structure of a typical UBA domain, the presence of a 4th helix as observed in the full length CeTDP2 structure is a unique feature of the TDP2 UBA domain. Multiple sequence alignment (MSA) and PSIPRED secondary structure prediction (39) of TDP2 UBAs (Figure 3A) from a number of organisms support the presence of a 4th helix across species. The secondary structure prediction based on backbone chemical shifts by TALOS+ (40) also suggests that the fourth helix of TDP2 UBA is indeed present in solution (Figure 3B). A similar feature at the C-terminus of the three-helix bundle is missing from other structurally characterized UBAs to date. The unique 4th helix of TDP2 UBA appears to play important roles in Ub binding, especially to polyUb (see below).
Figure 3.

TDP2 UBA has an extra fourth helix in solution. (A) Multiple sequence alignment (MSA) of TDP2 UBAs from 11 organisms (Danio rerio, Homo sapiens, Mus musculus, Callithrix jacchus, Gallus gallus, Sus scrofa, Xenopus laevis, Rattus norvegicus, Bos taurus, Macaca mulatta and Caenorhabditis elegans). Alignment was carried out with Clustal Omega (12,68) and this alignment was used in PSIPRED (39) to predict secondary structure, which is shown at the bottom of the alignment. H, helix; C, coil. A red box highlights the CeTDP2 UBA sequence, for which the residue numbering is shown at the top. (B) TALOS+ (40) secondary structure prediction based on backbone chemical shifts of CeTDP2 UBA. Helices are shown as spiral ribbons and labeled.
Helices 1 and 3 of the TDP2 UBA contact hydrophobic patches of Ub
Hydrophobic Ub surfaces, including the patches centered on Ile44 or Phe4, are typically involved in Ub-UBA interactions (19). To identify the residues of Ub involved in TDP2 UBA binding, previously determined backbone chemical shifts of Ub (41) were used to assign the peaks in the HSQC spectrum of 15N Ub in buffer A (see Materials and Methods). A per residue chemical shift perturbation plot (Figure 2C) indicated that hydrophobic residues Leu8, Ile44, and Val70 that cluster on a surface of Ub lined with β-strands 3, 4, and 5 were significantly perturbed upon addition of TDP2 UBA. A second surface comprising exposed residues Phe4 and Thr14 on β-strands 1 and 2, respectively, were also significantly perturbed. Thus, the surface patches of Ub typically involved in the canonical UBA-Ub interactions also mediate the binding of Ub to CeTDP2 UBA.
A chemical shift perturbation plot (Figure 2D) summarizes changes in the 2D 15N HSQC spectrum for 15N-labeled CeTDP2 UBA upon addition of six times molar excess of Ub, and identified the residues on the CeTDP2 UBA surface involved in Ub binding. The most significantly perturbed residues (Ile55, Thr56, Ala57, Lys75, Lys76, Leu78, Asp79, Val80, Phe81 and Tyr82) map to helices 1 and 3 (Figure 2D, right panel). Smaller perturbation was observed for residues in helices 2 and 4. Notably however, helix 4, which is an additional feature in CeTDP2 UBA, had three non surface-exposed residues (Ala90, Ala93 and Ala94) that shifted slightly above significance level (above 0.15 ppm as defined in the Figure 2D legend), likely indicating a proximity-induced conformational change in response to Ub-binding by the core UBA tri-helix. This observation supports the notion that the fourth α-helix is an integral part of the TDP2 UBA. Overall, the chemical shift perturbation analyses indicate that the binding interface of CeTDP2 UBA to monoUb involves residues on helices 1 and 3 of the UBA and the residues on the Phe4 patch (comprising of Gln2, Phe4 and Thr14) and Ile44 patch (including residues Leu8, Ile44 and Val70) of Ub.
TDP2 UBA binds monoUb in a canonical manner, but with non-conserved key residues
To further characterize the interaction between TDP2 UBA and Ub, we undertook docking of the structured N-terminal region of CeTDP2 (PDB ID: 4GEW) with Ub (PDB ID: 1D3Z) using HADDOCK 2.1 (42) and 29 AIRs (Supplementary Table S1) derived from the chemical shift perturbation plots (Figure 2C and D), as described in Materials and Methods. These initial HADDOCK runs yielded nine sets of CeTDP2 UBA-monoUb conformations (Supplementary Table S2), with one of them being similar to the canonical binding mode (Figure 4A for complex of Ede1 UBA and Ub) and the others non-canonical. To resolve this ambiguity, we supplemented the AIRs with 85 unambiguous long-range distance restraints obtained by PRE experiments with site-directed spin labeling either at CeTDP2 UBA M43C or Ub G75C (Figure 4D and Supplementary Figure S5). These PRE-derived distance restraints successfully drove the solution to an exclusively single overall conformation (Figure 4B), defined by two sets of clusters (backbone r.m.s.d. of 2.3 Å between the clusters) in the final 200 best structures (Table 1). The output statistics for different sets of HADDOCK inputs are compared in Supplementary Table S2. We validated the final model by using 15 PRE restraints from 15N-labeled ubiquitin:MTSL-labeled TDP2 UBA S84C that were not included in the HADDOCK modeling runs (Supplementary Table S3). The model agreed well with all validation PRE data. These results were achieved without alteration to the secondary structures of TDP2 UBA or Ub, as expected by chemical shift indices calculated from backbone chemical shifts of these proteins within the complex (data not shown).
Figure 4.

The TDP2 UBA-Ub complex adopts a canonical conformation. (A) The solution structure of the Ede1 UBA-Ub complex (PDB 2G3Q), published previously (15), is shown as a representative UBA-Ub complex. The UBA is colored in a rainbow spectrum from the N- to C-terminus and Ub in teal. Gly47 from the Ub loop that inserts between helix 1 and 3 of the UBA is highlighted in magenta. (B) An ensemble of 10 best scored TDP2 UBA-Ub complex models by HADDOCK (31), shown in a similar orientation and color scheme as in A. (C) HADDOCK model showing side chains in the interaction surface between TDP2 UBA (grey and red) and Ub (teal and purple). (D) HADDOCK model of the TDP2 UBA-Ub complex, as in C but rotated about the horizontal axis, showing the MTSL labeling sites on Ub (Gly75) or UBA (Met43 and Ser84) that were used for modeling and validation. Gly75 on Ub and the proximal residues to it on the UBA whose HN signals were attenuated in the PRE experiment are colored in orange. Likewise, Met43 on the UBA and the proximal residues to it on Ub are colored in magenta, and Ser84 and its proximal residues in green.
The docking model shows that the TDP2 UBA-monoUb complex is in a canonical conformation consistent with all other known structures of UBA-Ub complexes. The r.m.s.d. between backbone atoms of the Ede1 UBA-Ub complex and TDP2 UBA-Ub complex is 1.6 Å. However, despite the similar overall arrangement, the key residues involved in the interactions are different. As seen in the case of Ede1 UBA, the canonical conformation typically has the loop containing Gly47 of Ub making close contacts (3–6 Å) with the highly conserved hydrophobic side-chains of the MGF residues in the loop between helix 1 and helix 2 of UBA. In agreement to this, the TDP2 UBA loop between helix 1 and helix 2 is placed at a similar distance from Gly47 of Ub. However, TDP2 UBA lacks the MGF motif and the interaction is instead mediated by an ITA sequence (Ile55, Thr56, Ala57) between helix 1 and 2. The ITA patch, along with Tyr82 located at the center of the TDP2 UBA-Ub interface on helix 3, forms an important interaction surface that makes hydrophobic contacts with the Ile44 hydrophobic patch of Ub (Figure 4C).
TDP2 UBA preferentially binds diUbs over monoUb
To assess the ability of TDP2 to distinguish monoUb from its chained forms in vitro, we determined the TDP2 UBA binding affinities to monoUb and diUb (K48 and K63 linked) by fluorescence anisotropy (FA) and NMR titration experiments. It is important to note that our analyses were conducted with untagged proteins; hence the binding in solution was free of non-physiological tag effects that have sometimes led to experimental artifacts (43). The TDP2 UBA bound monoUb with a dissociation constant of 40 ± 16 μM (mean ± S.D) (by FA) or ∼120 μM ± 10 μM (mean ± S.E.M) (by NMR) (Figures 2A, 5D and F). In the reciprocal NMR experiment, in which 15N labeled Ub was titrated with TDP2 UBA, the estimated Kd was 100 ± 2 μM (Figure 5E and F). In comparison, TDP2 UBA bound K48-linked and K63-linked diUbs with Kd values of 8.4 ± 4.0 μM and 9.1 ± 4.2 μM, respectively (by FA) (Figure 2A) or 6.7 ± 2.1 μM and 10.2 ± 3.5 μM, respectively (by NMR) (graphical representation in Figure 5F). Due to the slow to intermediate exchange nature of binding of TDP2 UBA to diUbs, we used only a handful of surface exposed peaks at the interaction interface that were still traceable during the course of titration to estimate a Kd value (Figure 5, Supplementary Figure S4A and B).
Figure 5.

TDP2 UBA binds Ubs with different modes of binding. (A) Chemical shift titration profiles of TDP2 UBA Thr56 for binding with monoUb and diUbs (K48 or K63-linked) at UBA:Ub ratios of 1:0, 4:1, 2:1, 1:1, 1:2, 1:3, 1:4 and 1:6 show shifting of the peaks in the same direction for all, but in fast exchange for monoUb (red) and slow-intermediate exchange for diUbs (blue and green). (B) Chemical shift titration profiles of TDP2 UBA Gly83 for binding with monoUb and diUbs (K48 or K63-linked) show shifts in different directions. (C) Chemical shift titration profiles of CeTDP2 UBA Trp72 for binding with monoUb and diUbs (K48- or K63-linked) show no shift for monoUb binding, but shifts in different directions for the two diUbs. (D) The combined CSPs of significantly shifted peaks from 15N labeled CeTDP2 UBA at indicated molar ratios with monoUb. The range of Kd values calculated for the chosen residues is shown on the graph. (E) CSPs of significantly shifted peaks from 15N labeled monoUb titrated with increasing concentration of CeTDP2 UBA. The range of Kd values calculated for the chosen residues is shown on the graph. (F) Binding affinity averages from chemical shift titrations of different combinations of CeTDP2 UBA and Ubs are shown, with error bars for standard error of mean. Text labels inside each bar indicate the protein that was 15N labeled in that experiment. Ligand for the corresponding binding experiment is shown as a schematic cartoon label under the X-axis. The bottom half of the panel shows what each schematic cartoon represents. The kinked and straight black bars between two Ub moieties stands for the K48- and K63-linkage, respectively. The cartoons do not depict the actual binding poses (they are not meant to show that UBA only interacts with the distal Ub moieties of diUbs).
Our Kd values of UBA-Ub interactions are comparable to those observed for other UBAs, with a reported Kd range of ∼10–500 μM for monoUb and ∼0.03–9.0 μM for diUbs (21). We observed a 5- to 10-fold higher affinity for diUbs compared to monoUb, although no significant difference in affinity for K48- versus K63-linkage was observed. Thus, we conclude that TDP2 UBA indeed prefers diUbs to monoUb in vitro and can discriminate both of these forms of diUb from monoUb.
TDP2 UBA binds monoUb and diUb differently
Analyses of the NMR chemical shift perturbation of TDP2 UBA upon interactions with monoUb and diUbs showed distinct modes of peak shifts in their 2D 1H,15N HSQC spectra (Figure 5A, B, C and Supplementary Figure S9). Titration with monoUb caused a gradual shift in peak positions (red) until saturation was achieved. This behavior characterizes the fast exchange regime and is typically observed for weaker protein–ligand interactions (44). In agreement with this, TDP2 UBA interacts with monoUb with an affinity of ∼100 μM. In contrast, titration with either K48- or K63-linked diUb showed a pattern of intermediate exchange, whereby peaks shifted and broadened initially, and then disappeared due to extreme broadening, to ultimately reappear at a final position corresponding to the bound state (blue and green peaks in Figure 5A). The intermediate exchange regime typically indicates tighter binding compared to systems in fast exchange, which is consistent with our Kd values for diUbs from NMR (calculated from some of the peaks that were still observable throughout the titration) and fluorescence anisotropy experiments. Of note, the 1H,15N HSQC spectrum of TDP2 UBA showed extensive peak broadening upon binding to either K48- or K63-linked diUb (Supplementary Figure S9), with some of the peaks not reappearing even at 6 times molar excess of diUb over TDP2 UBA. This could indicate multiple states for these amide groups in the TDP2 UBA-diUb complexes, or possibly additional Ub molecules binding at higher stoichiometry.
The CSP analyses also suggest that UBA amino acids are in different chemical environments when interacting with monoUb or the diUbs. Some of the residue peaks, including Gly83 and Trp72, shift in altogether different directions in the 1H,15N HSQC 2D spectrum (Figure 5B and C). Interestingly, while Gly83 is perturbed upon interaction with all forms of Ub, the Trp72 peak shifts only upon binding to diUbs. Trp72 is an absolutely conserved (Figure 3) residue in the loop between helices 2 and 3 and its perturbation indicates the involvement of the ‘back’ side of UBA in binding one Ub moiety of diUb, while the ‘front’ side that comprises the canonical binding surface of helix 1 and 3 interacts with the 2nd Ub. Also outside the canonical Ub-binding interface of UBA, the binding of TDP2 UBA to diUbs (Figures 6C and 7C) shows extensive perturbation for the 4th helix residues (88–97), either through significant peak shifting or peak broadening and disappearance in the 2D 1H,15N HSQC spectra. These observations suggest that, compared to its monoUb-binding, diUb interactions by TDP2 UBA involves broader interfaces with involvement of a greater number of UBA domain amino acids.
TDP2 UBA has differential modes of binding for K48- versus K63-linked di-Ubiquitin
Although TDP2 UBA binds the two types of diUbs with similar apparent affinities, titration with K48 versus K63-linked diUb showed distinct chemical shift perturbation patterns (blue versus green peaks in Figure 5B and C). Thus, we sought to better characterize the contacts that individual Ub moieties of the two diUbs make with UBA to understand the mechanism of differential binding. To do this, we prepared diUb samples in which each Ub moiety (either proximal or distal) was selectively 15N labeled. ‘Proximal’ Ub has its Lys48 or Lys63 side-chain conjugated with the carboxyl terminus of the ‘distal’ Ub. NMR CSP analyses for titration of the variously labeled diUb with unlabeled TDP2 UBA displayed fast to intermediate exchange kinetics with extensive peak broadening and reappearance of some peaks, while some signals did not reappear even at high UBA concentrations. In addition, a subset of resonances demonstrated slow exchange in which they disappeared from the free state position and reappeared at a distinct bound state position. Nonetheless, some of the peaks that were significantly shifted and could still be located were used to assess the contribution of these amino acids to binding for the individual Ub moieties.
The spectra and corresponding CSP maps highlight how distal and proximal Ub moieties contribute to the interaction with the UBA (Figures 6 and 7 and Supplementary Figure S4C–G). As mentioned above, the bound state assignments could only be obtained for those peaks that either did not vanish completely during the titration or were proximal to their free state, allowing them to be reasonably traced to their bound state. In the case of the K48-linkage (Figure 6A and B), the CSP patterns are similar between proximal and distal Ub, suggesting that overall the interaction surface on both Ubs involve similar surface elements, especially β2, the β3–β4 loop and β5. Nonetheless, detailed inspection reveals differences, including a greater involvement of amino acids from β1 and the following loop for the proximal Ub and significant shifting for Ile44 from the proximal, but not distal Ub (Figure 6A and B). Moreover, the distal and not proximal His68 signal is lost when the UBA is at half-molar ratio, whereas the opposite trend is observed for Ile44 and Val70 (Supplementary Figure S4D and E). Thus, the two Ub components are distinct in their interactions with the UBA. Interestingly, proximal Ub from K48 diUb (Supplementary Figure S4D) more closely mimicked the behavior of monoUb (Supplementary Figure S4C), suggesting that it binds to the ‘front’ UBA surface, comprised of amino acids from α1 and α3.
In the case of the K63-linkage (Figure 7A, B and Supplementary Figure S4F and G), the spectra and corresponding CSP maps both show clear differences for the distal and proximal Ub moieties, with distal Ub having more significantly perturbed residues. At half-molar ratio UBA addition, Ile44 and Val70 are both lost from the free state position for the distal Ub; these amino acids are observable in the proximal Ub (Supplementary Figure S4F and G). Similarly, Ile13 and Thr14 from proximal Ub are not as significantly perturbed as in distal Ub (Figure 7A and B). By contrast, signals from Phe4 and His68, which form a hydrophobic binding surface separate from that centered on Ile44, are lost in proximal, but not distal Ub (Supplementary Figure S4F and G). Similarly, Gln2 shows large perturbations specifically in the proximal Ub of K63-linked diUb (Figure 7B). Thus, in contrast to K48 diUb, the distal Ub (and not proximal Ub) from K63 diUb more closely mimicked the behavior of monoUb (Supplementary Figure S4G). These observations suggest that the canonical Leu8-Ile44-Val70 hydrophobic patch from distal Ub binds to the ‘front’ UBA surface, while a non-canonical Phe4-based patch from proximal Ub bind to the ‘back’ UBA surface.
Taken together, the TDP2 UBA recognizes the two types of diUbs differently, with distinct interaction interface and preference for binding to proximal or distal Ub.
The UBA domain is integral to the DNA repair function of TDP2
Our NMR analyses showed that TDP2 has a UBA domain that preferentially recognizes poly-Ub chains. In light of this specific interaction and conservation of the UBA domain at the N-termini of known TDP2 orthologs, we asked whether the UBA domain is required for TDP2 function. As the best characterized role of TDP2 is removing stalled Top2 at the 5′ terminus of DNA breaks (3), we tested whether the UBA domain is required for this function using viability assays to measure cellular sensitivity to the Top2 poison etoposide. TDP2−/−/− DT40 cells (35,45) were transfected with an expression vector carrying human TDP2 gene constructs, either the full-length wild-type (WT), N-terminally truncated (del1-100), F62R point mutant or the F62R/V35R/R56D triple mutant and were subjected to a survival assay against etoposide as described previously (34,45) (Figure 8A). Phe62 of HsTDP2 was chosen for mutation because it corresponds to the surface exposed Tyr82 of CeTDP2, which was the most significantly perturbed residue in our NMR CSP analyses upon interaction with Ub (Figures 2D, 4C and 9B). Two other residues, Val35 and Arg56, were also chosen for the triple mutation as they correspond to the CeTDP2 Ile55 and Lys76 residues, respectively, which also showed significant CSP, and are at the ubiquitin interface (Figure 4C).
Figure 8.

TDP2 UBA-Ub interaction partially determines response to Top2 poison, but is not involved in localization of TDP2 to the nucleus. (A) Cell survival curves in the presence of increasing concentrations of etoposide are compared for DT40 clones lacking TDP2 (TDP2−/−/−) and those complemented by WT HsTDP2 or HsTDP2 with complete N-terminal domain (residues 1–100) deletion transfections. Four different clones of the N-terminal deletion are shown for reproducibility purpose. (B) Same as A, but instead of complete N-terminal deletion, only F62R mutation in the UBA-Ub interaction surface was tested for survival compared to WT and TDP2-deleted clones. Results from two clones of the F62R transfection are shown. (C) Wild-type HsTDP2 and HsTDP2 mutants (HsTDP2cat, HsTDP2 F62R and HsTDP2 KKR) were N-terminally tagged with EGFP and individually expressed in Drosophila S2 cells to investigate localization of the molecules in cells. Wild-type HsTDP2 is imported and accumulates in the nucleus (EGFP-HsTDP2), whereas HsTDP2cat is not (EGFP-HsTDP2cat). The F62R mutation in the UBA domain does not affect nuclear localization of HsTdp2 (EGFP-HsTDP2 F62R). A triple-mutation (K23N, R25D and R26P: KRR) in the potential nuclear localization signal (NLS) sequence within the N-terminal region of HsTDP2 inhibits nuclear import and accumulation (EGFP-HsTDP2 KKR). Anti-EGFP and anti-Tubulin antibodies were used to stain EGFP-fusion proteins and microtubules, respectively. DNA was stained with DAPI. Each panel shows the maximum intensity projections of multiple z sections scanned through the thickness of each cell. Bar: 10 μm.
Figure 9.

TDP2 does not have the MGF motif in loop1, conserved in other UBAs. (A) MSA of UBA domains from 17 different UBA-containing proteins. All these UBAs have been structurally characterized and their corresponding PDB IDs are listed on the right. MGF motif in loop 1 and important residues for Ub interaction in helix 3 are highlighted. CeTDP2, lacking the MGF motif, is boxed in red. The only other protein with significant dissimilarity in the MGF motif, mouse RSGI RUH, is boxed in black. Secondary structure schematic on top is representative of the four-helix bundle present in CeTDP2 UBA structure. (B) The 4th helix of TDP2 UBA may provide an extended surface for Ub binding. Hydrophobic residues forming and adjoining the ITA patch in TDP2 UBA (left) and MGF patch as well as hydrophobic residues on helix 3 in Ede1 UBA (right) are highlighted in red and labeled. α-helices are numbered for both structures.
As reported earlier (35,45), TDP2−/−/− cells are hypersensitive to etoposide and the WT HsTDP2 transfection rescued this hypersensitivity (Figure 8A and B). However, HsTDP2 with deletion of the N-terminal 100 residues (including UBA) only partially rescued the etoposide hypersensitivity, suggesting that the catalytic domain alone is not sufficient for the full activity of TDP2 (Figure 8A). Importantly, both the F62R single mutant and F62R/V35R/R56D triple mutant also gave partial rescue, with the survival rate slightly higher than the level observed for the del1-100 construct (Figure 8B and Supplementary Figure S6C). The phenotype of the F62R-expressing cells was also compared to that of WT HsTDP2-expressing and non-complemented TDP2−/−/− DT40 cells by propidium iodide (PI) based flow cytometry analyses of cell viability upon etoposide treatment (Supplementary Figure S7). The populations of high intensity PI labeled cells after 24 h etoposide treatment were significantly higher for TDP2−/−/− cells compared to the WT HsTDP2-expressing cells. The cells expressing F62R mutant showed intermediate distributions between the two, recapitulating the results observed with the clonogenic survival assays. Thus, binding of the UBA domain to Ub indeed plays an important role in the repair of Top2-mediated DNA damages by TDP2.
The repair of Top2-mediated damages on chromosomal DNA would require that TDP2 function in the nucleus. We therefore tested whether the TDP2 UBA-Ub interaction plays a role in the nuclear localization of TDP2, using an in vitro system (Figure 8C). HsTDP2 fused to EGFP was ectopically expressed in Drosophila S2 cells, which have no known endogenous TDP2 homolog. The WT HsTDP2 localized to the nucleus, whereas the N-terminally deleted construct containing only the catalytic domain (EGFP-HsTDP2cat) was not actively transported into the nucleus. We identified a ‘class 2′ nuclear localization signal (46) in the N-terminal flexible region of HsTDP2; disruption of this motif by mutations (K23N/R25D/R26P) abrogated nuclear localization similar to the N-terminal deletion. Importantly, TDP2 F62R showed a similar localization pattern as WT (Figure 8C), indicating that the UBA-Ub interaction is not required for nuclear localization of TDP2. Consistent results were obtained when we biochemically examined subcellular localization of HsTDP2 WT, del 1–100 and F62R constructs overexpressed in the DT40 cells (Supplementary Figure S6A and B). Taken together, our results suggest that the defect in Ub-binding compromises the DNA repair function of TDP2 by affecting its behavior after nuclear entry.
DISCUSSION
Structures of TDP2 UBA in complex with monoUb and diUbs
TDP2 UBA binds monoUb in the canonical conformation commonly observed for other tri-helix UBA domains, and it also binds to both K48- and K63-linked diUb with higher affinities compared to that for monoUb. We observed clear differences in the spectra and CSP patterns for the complexes formed with monoUb or diUb, as well as differences between the two Ub moieties for both K48- and K63-linked diUbs. These observations indicate that instead of two UBA domains from separate molecules binding equivalently to each Ub moiety within a diUb chain, as observed for Ubiquilin 1 UBA (47), the TDP2 UBA likely fits between two Ub moieties for both types of diUb chains. This sandwiching of a ubiquitin receptor domain between two Ub moieties of a K48-linked chain is distinct from the mechanism used by the proteasome Ub receptors hRpn10/S5a and Rpn1/PSMD2, which instead provide multiple Ub-binding sites that each contact one Ub moiety (48,49).
Given the different conformational preferences of the K48- versus K63-linked Ub chains (50), key binding residues of TDP2 UBA likely adopt different conformations depending on the type of Ub molecule it binds to, which is supported by the significantly different NMR chemical shifts of the bound states for different forms of Ub (Figure 5B and C). Based on the identity of strongly perturbed residues in the CSP analyses of distal versus proximal Ub of K48- and K63-linked species, we have generated hypothetical models for TDP2 UBA binding with either of the diUbs (Figures 6D and 7D and Supplementary Figure S8A and C), which depict a 1:1 stoichiometry of binding (UBA : diUb).
Several UBA domains have been reported to have distinct preference toward K48-linked diUb over K63-linked diUb. Structures of these UBAs in complex with K48-linked diUb, including those for hHR23A UBA2 (30) (Supplementary Figure S8B) and Mud1 UBA (17), show an arrangement in which the UBA is tightly sandwiched between two Ub moieties. The distal Ub moiety binds to the ‘front’ side of UBA (α-helices 1 and 3) whereas the proximal Ub, whose K48 sidechain is linked to the C-terminus of the distal Ub, contacts the ‘back’ side of UBA (α-helix 2). Our data for TDP2 UBA binding to K48-linked diUb suggests a novel binding mode, in which the distal Ub packs against the ‘back’ UBA side that is formed by both helix 2 and helix 4 while the proximal Ub packs against the ‘front’ UBA side. In this configuration, Leu8 and His68 from distal Ub interact with amino acids from the ‘back’ side of the UBA while proximal Ub I44 and V70 form optimal contacts to the ‘front’ UBA side (Figure 6D and Supplementary Figure S8C).
We also modeled K63-linked diUb bound to TDP2 UBA with the distal Ub moiety serving as the primary binding site for interaction with the UBA, where the canonical binding patch formed by Leu8-Ile44-Val70 of the distal Ub is bound to the ‘front’ side of UBA (Figure 7D). Although the proximal Ub of K63-linked diUb showed more modest CSP than the distal Ub, we observed large spectral changes for Gln2, Phe4, Glu64, Thr66 and His68 in proximal Ub. Accordingly, the proximal Ub was modeled to position its Phe4-patch facing the UBA (Figure 7D and Supplementary Figure S8A). Notably, the large Phe4 CSP for K63 proximal Ub is the only instance when we observe a significant shift in this residue. This Phe4-patch surface is also used by K63 diUb to bind the myosin VI MyUb domain (51); the surface formed by Phe4, Phe45, Ala46, Lys48 and Thr66 in proximal ubiquitin contacts the MyUb helix 1. The observation is also consistent with the important role of the Ub residue Phe4 in endocytosis (52), which was shown to be mediated by binding to K63-linked polyUbs (53).
TDP2 UBA is distinct from other known UBAs
Typically, the UBA domains are characterized by the presence of a MGF motif in the loop between helix 1 and helix 2 (Figure 9A), with Gly at the second position being particularly well conserved. The MGF motif forms a hydrophobic patch and interacts with the Gly47-containing loop of Ub. Notably, the C. elegans TDP2 UBA lacks an MGF motif and instead has ‘ITA’ at the corresponding position in the loop between helix 1 and helix 2 (Figure 9A). The first Ile55 of this stretch is conserved among TDP2 orthologs with either a hydrophobic Ile or Val residue (Figure 3A), while the second and third positions are occupied by smaller amino acids. Regardless of the lack of MGF motif, this ‘ITA’ residue cluster is involved in Ub binding (CSP, Figure 2D). In typical UBAs, Phe or an alternative hydrophobic residue at the third position of the MGF motif is positioned in the middle of the ‘front’ Ub-binding face and a spatially adjoining hydrophobic surface is formed by an Ile, Leu or Val side chain (e.g. Leu1377 and Leu1378 of Ede1 UBA) from the end of helix 3 (Figure 9A and B right panel). In TDP2 UBA, a hydrophobic patch at the end of helix 3 is created by a strictly conserved aromatic residue (Tyr82 in C. elegans TDP2 and Phe62 of HsTDP2), which plays a critical role in Ub interaction (Figures 2D, 3A and 9B left panel). The replacement of MGF with ITA in TDP2 UBA in Loop1 between helix 1 and helix 2, even with the uniquely conserved aromatic residue at the end of helix 3, leads to an overall reduction in hydrophobicity on the ‘front’ face of the UBA. We hypothesize that the 4th helix in close proximity to the canonical core 3-helix bundle serves to extend the binding surface of TDP2 UBA to include hydrophobic residues highlighted on the 4th Helix, 2nd Helix and Loop1 in Figure 9B, thus compensating for the otherwise reduced hydrophobic surface due to the missing MGF motif.
Sequence alignment identifies another UBA missing the MGF motif, the mouse RSGI RUH (PDB ID: 1WJ7), a domain of Hook1 microtubule-tethering protein that interacts with Rab GTPase family proteins and links endocytic membrane trafficking to the microtubule cytoskeleton (54,55), even though the Gly residue is still conserved in RSGI RUH. Interestingly, this is also the only other structurally characterized UBA, besides TDP2 UBA, which has the extra fourth helix in proximity of the UBA tri-helix bundle. Mouse RSGI RUH has an extra helix at the N-terminus, although the function of this extra helix is unknown. As hypothesized for TDP2 UBA, the extra helix could mediate an extended binding surface. However, the orientation of the extra helix is significantly different than that observed for TDP2 UBA.
TDP2 UBA also differs from other UBAs in the absolute conservation of the Trp residue in the loop between helix 2 and 3, at position 72 in C. elegans TDP2. Trp72 is positioned on the opposite side from the ‘front’ Ub-binding face that includes Tyr82. Interestingly, although no CSP for the conserved Trp72 was observed upon binding monoUb (Figure 2D), its involvement becomes apparent in interaction of TDP2 UBA with diUb, especially with the K63 linkage (Figure 7C). The Trp72 peak shifts significantly upon K63 diUb binding (Figure 5C), which indicates direct contacts made by the ‘back’ side of UBA with Ub or a conformational change, such as a breathing motion for this loop to better accommodate binding to this form of Ub. Thus, TDP2 digresses from known UBAs in several unique features that are likely essential for TDP2 specific activity in vivo.
The UBA domain is important for regulating TDP2 function
The C-terminal catalytic domain of TDP2 is necessary and sufficient for the 5′-tyrosyl DNA phosphodiesterase activity in vitro (11,12). However, we have shown in this study that the UBA domain is also required for optimal function in cells. In TDP2−/−/− DT40 cells, we found that the surface exposed residues within the UBA domain of HsTDP2 important in the interactions with Ub are needed for fully rescuing the cellular hypersensitivity to the Top2 poison etoposide. Thus, the UBA-Ub interaction is important for full TDP2 activity in the removal of stalled Top2 cleavage complexes from broken DNA ends. How does the UBA domain help TDP2 in its DNA repair function?
One likely scenario is that the TDP2 UBA-Ub interactions assist in localizing TDP2 to the site of activity. We have shown that HsTDP2 has a NLS N-terminal to the UBA domain, which is responsible for the active transport of TDP2 into the nucleus. Thus, we hypothesize that nuclear pool of TDP2 is further recruited to Top2-mediated DNA damages through interaction with ubiquitinated proteins on the chromosome. There are multiple possibilities for the ubiquitinated target molecule, which are not necessarily mutually exclusive: (i) Poly-ubiquitination of histones, by a K63-linked Ub chain in particular, in the vicinity of DNA double strand break (DSB) is known to recruit DNA repair factors such as 53BP1, BRCA1 and RAD18 (56,57). Thus, TDP2 may be one such protein responding to DSB signals; (ii) It has been shown that the TDP2-mediated DNA repair is particularly important during transcription (6). Actively transcribed chromatin regions are marked by histone mono-ubiquitination (58,59). Thus, the TDP2 UBA-Ub interaction might facilitate localization of TDP2 near the site of transcription; (iii) The repair of an abortive Top2 cleavage complexes requires unfolding and degradation of the Top2 molecules covalently attached to DNA termini before TDP2 can act on the phosphotyrosyl bond (60). Indeed, Top2 and especially the Top2β isozyme, is poly-ubiquitinated when its DNA cleavage-rejoining cycle is poisoned and is subjected to proteasome-dependent degradation (61–65). As the poly-ubiquitination signal for protein degradation is typically K48-linked, TDP2 could be recruited to the site of Top2-mediated damage via direct interaction of the UBA domain with the polyUb chain.
We have shown that TDP2 UBA promiscuously binds multiple forms of Ub. Although we observed a preference for diUbs in vitro, this preference could be countered by the abundance of monoUb over Ub chains in vivo (66). It is also possible that TDP2 UBA binds with appreciable affinity to a type of poly-Ub chain not tested in this study. Thus, an important question is how specificity is achieved in binding to the biologically significant target(s). The differential distribution of Ub species across subcellular sites, cytosolic and nuclear (66), could provide a spatial level of regulation of binding. Additionally, TDP2 may associate with other protein factors based on Ub-independent interactions, which can be further modulated by ubiquitination of the target protein. Even though TDP2 UBA binds to the two poly-Ub forms tested (K48- and K63-linked) with similar affinities in vitro, their distinct binding modes may confer selectivity in the presence of additional interactions with a target protein. In any case, it is plausible that binding of different types of Ubs to the TDP2 UBA domain serves as regulatory switch for recruiting TDP2 and for modulating its intrinsic activities, one of which could be brought about by a distinct conformational cues being relayed to the TDP2 active site.
In summary, we have shown in this study that TDP2 has a unique UBA domain capable of binding several forms of Ub in distinct fashions. Given the multiple biological functions TDP2 has been implicated for, the broad binding specificity of TDP2 UBA may be important for interactions with various targets to elicit different responses. Insights obtained from our analyses of TDP2 UBA-Ub interactions will help further study in vivo roles of TDP2.
AVAILABILITY
The backbone and side-chain NMR chemical shift assignment data are available in the Biological Magnetic Resonance Bank (BMRB), entry 26771.
Supplementary Material
Acknowledgments
The authors thank the Minnesota NMR center for training and assistance during data collection with the high field NMR spectrometers, and Dr Joseph Muretta for training and help with fluorescence experiments on the Cary Eclipse Fluorescence spectrophotometer (Varian Inc.) in the Biophysical Technology Center at UMN. The authors also thank Dr Shar-Yin (Naomi) Huang (Laboratory of Molecular Pharmacology and Developmental Therapeutics Branch, National Cancer Institute, NIH) for insightful suggestions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [GM095558 and GM118047 to H.A., Intramural Program of the National Cancer Institute, Center for Cancer Research Z01 BC 006150 to Y.P. and ZIA BC011627 to K.J.W.]. Funding for open access charge: National Institutes of Health [GM095558 and GM118047 to H.A.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Li C., Sun S.-Y., Khuri F.R., Li R. Pleiotropic functions of EAPII/TTRAP/TDP2: Cancer development, chemoresistance and beyond. Cell Cycle. 2011;10:3274–3283. doi: 10.4161/cc.10.19.17763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pommier Y., Huang S.-Y.N., Gao R., Das B.B., Murai J., Marchand C. Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2) DNA Repair (Amst) 2014;19:114–129. doi: 10.1016/j.dnarep.2014.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cortes Ledesma F., El Khamisy S.F., Zuma M.C., Osborn K., Caldecott K.W. A human 5′-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature. 2009;461:674–678. doi: 10.1038/nature08444. [DOI] [PubMed] [Google Scholar]
- 4.Pei H., Yordy J.S., Leng Q., Zhao Q., Watson D.K., Li R. EAPII interacts with ETS1 and modulates its transcriptional function. Oncogene. 2003;22:2699–2709. doi: 10.1038/sj.onc.1206374. [DOI] [PubMed] [Google Scholar]
- 5.Pype S., Declercq W., Ibrahimi A., Michiels C., Van Rietschoten J.G., Dewulf N., de Boer M., Vandenabeele P., Huylebroeck D., Remacle J.E. TTRAP, a novel protein that associates with CD40, tumor necrosis factor (TNF) receptor-75 and TNF receptor-associated factors (TRAFs), and that inhibits nuclear factor-kappa B activation. J. Biol. Chem. 2000;275:18586–18593. doi: 10.1074/jbc.M000531200. [DOI] [PubMed] [Google Scholar]
- 6.Gómez-Herreros F., Schuurs-Hoeijmakers J.H.M., McCormack M., Greally M.T., Rulten S., Romero-Granados R., Counihan T.J., Chaila E., Conroy J., Ennis S., et al. TDP2 protects transcription from abortive topoisomerase activity and is required for normal neural function. Nat. Genet. 2014;46:516–521. doi: 10.1038/ng.2929. [DOI] [PubMed] [Google Scholar]
- 7.Zucchelli S., Vilotti S., Calligaris R., Lavina Z.S., Biagioli M., Foti R., De Maso L., Pinto M., Gorza M., Speretta E., et al. Aggresome-forming TTRAP mediates pro-apoptotic properties of Parkinson's disease-associated DJ-1 missense mutations. Cell Death Differ. 2009;16:428–438. doi: 10.1038/cdd.2008.169. [DOI] [PubMed] [Google Scholar]
- 8.Do P.M., Varanasi L., Fan S., Li C., Kubacka I., Newman V., Chauhan K., Daniels S.R., Boccetta M., Garrett M.R., et al. Mutant p53 cooperates with ETS2 to promote etoposide resistance. Genes Dev. 2012;26:830–845. doi: 10.1101/gad.181685.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Virgen-Slane R., Rozovics J.M., Fitzgerald K.D., Ngo T., Chou W., van der Heden van Noort G.J., Filippov D.V., Gershon P.D., Semler B.L. An RNA virus hijacks an incognito function of a DNA repair enzyme. Proc. Natl. Acad. Sci. U.S.A. 2012;109:14634–14639. doi: 10.1073/pnas.1208096109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Königer C., Wingert I., Marsmann M., Rösler C., Beck J., Nassal M. Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc. Natl. Acad. Sci. U.S.A. 2014;111:E4244–E4253. doi: 10.1073/pnas.1409986111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi K., Kurahashi K., Gao R., Tsutakawa S.E., Tainer J.A., Pommier Y., Aihara H. Structural basis for recognition of 5′-phosphotyrosine adducts by Tdp2. Nat. Struct. Mol. Biol. 2012;19:1372–1377. doi: 10.1038/nsmb.2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schellenberg M.J., Appel C.D., Adhikari S., Robertson P.D., Ramsden D.A., Williams R.S. Mechanism of repair of 5′-topoisomerase II-DNA adducts by mammalian tyrosyl-DNA phosphodiesterase 2. Nat. Struct. Mol. Biol. 2012;19:1363–1371. doi: 10.1038/nsmb.2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kurz T., Özlü N., Rudolf F., O'Rourke S.M., Luke B., Hofmann K., Hyman A.A., Bowerman B., Peter M. The conserved protein DCN-1/Dcn1p is required for cullin neddylation in C. elegans and S. cerevisiae. Nature. 2005;435:1257–1261. doi: 10.1038/nature03662. [DOI] [PubMed] [Google Scholar]
- 14.Ohno A., Jee J., Fujiwara K., Tenno T., Goda N., Tochio H., Kobayashi H., Hiroaki H. Structure of the UBA domain of Dsk2p in complex with ubiquitin: molecular determinants for ubiquitin recognition. Structure. 2005;13:521–532. doi: 10.1016/j.str.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 15.Swanson K.A., Hicke L., Radhakrishnan I. Structural basis for monoubiquitin recognition by the Ede1 UBA domain. J. Mol. Biol. 2006;358:713–724. doi: 10.1016/j.jmb.2006.02.059. [DOI] [PubMed] [Google Scholar]
- 16.Tse M.K., Hui S.K., Yang Y., Yin S.-T., Hu H.-Y., Zou B., Wong B.C.Y., Sze K.H. Structural analysis of the UBA domain of X-linked inhibitor of apoptosis protein reveals different surfaces for ubiquitin-binding and self-association. PLoS One. 2011;6:e28511. doi: 10.1371/journal.pone.0028511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trempe J.-F., Brown N.R., Lowe E.D., Gordon C., Campbell I.D., Noble M.E.M., Endicott J.A. Mechanism of Lys48-linked polyubiquitin chain recognition by the Mud1 UBA domain. EMBO J. 2005;24:3178–3189. doi: 10.1038/sj.emboj.7600797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ikeda F., Dikic I. Atypical ubiquitin chains: new molecular signals. EMBO Rep. 2008;9:536–542. doi: 10.1038/embor.2008.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dikic I., Wakatsuki S., Walters K.J. Ubiquitin-binding domains - from structures to functions. Nat. Rev. Mol. Cell Biol. 2009;10:659–671. doi: 10.1038/nrm2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dikic I., Dötsch V. Ubiquitin linkages make a difference. Nat. Struct. Mol. Biol. 2009;16:1209–1210. doi: 10.1038/nsmb1209-1209. [DOI] [PubMed] [Google Scholar]
- 21.Hicke L., Schubert H.L., Hill C.P. Ubiquitin-binding domains. Nat. Rev. Mol. Cell Biol. 2005;6:610–621. doi: 10.1038/nrm1701. [DOI] [PubMed] [Google Scholar]
- 22.Grabbe C., Husnjak K., Dikic I. The spatial and temporal organization of ubiquitin networks. Nat. Rev. Mol. Cell Biol. 2011;12:295–307. doi: 10.1038/nrm3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen X., Walters K.J. Identifying and studying ubiquitin receptors by NMR. Methods Mol. Biol. 2012;832:279–303. doi: 10.1007/978-1-61779-474-2_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pickart C.M., Raasi S. Controlled synthesis of polyubiquitin chains. Methods Enzymol. 2005;399:21–36. doi: 10.1016/S0076-6879(05)99002-2. [DOI] [PubMed] [Google Scholar]
- 25.Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 26.Bahrami A., Assadi A.H., Markley J.L., Eghbalnia H.R. Probabilistic interaction network of evidence algorithm and its application to complete labeling of peak lists from protein NMR spectroscopy. PLoS Comput. Biol. 2009;5:e1000307. doi: 10.1371/journal.pcbi.1000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vranken W.F., Boucher W., Stevens T.J., Fogh R.H., Pajon A., Llinas M., Ulrich E.L., Markley J.L., Ionides J., Laue E.D. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 28.Battiste J.L., Wagner G. Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry. 2000;39:5355–5365. doi: 10.1021/bi000060h. [DOI] [PubMed] [Google Scholar]
- 29.Liang B., Bushweller J.H., Tamm L.K. Site-directed parallel spin-labeling and paramagnetic relaxation enhancement in structure determination of membrane proteins by solution NMR spectroscopy. J. Am. Chem. Soc. 2006;128:4389–4397. doi: 10.1021/ja0574825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Varadan R., Assfalg M., Raasi S., Pickart C., Fushman D. Structural determinants for selective recognition of a Lys48-linked polyubiquitin chain by a UBA domain. Mol. Cell. 2005;18:687–698. doi: 10.1016/j.molcel.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 31.Dominguez C., Boelens R., Bonvin A.M.J.J. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 32.Brünger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S., et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 33.Schrödinger L.L.C. The PyMOL Molecular Graphics System. Version 1.5.0.4. [Google Scholar]
- 34.Zeng Z., Sharma A., Ju L., Murai J., Umans L., Vermeire L., Pommier Y., Takeda S., Huylebroeck D., Caldecott K.W., et al. TDP2 promotes repair of topoisomerase I-mediated DNA damage in the absence of TDP1. Nucleic Acids Res. 2012;40:8371–8380. doi: 10.1093/nar/gks622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang S.-Y.N., Murai J., Dalla Rosa I., Dexheimer T.S., Naumova A., Gmeiner W.H., Pommier Y. TDP1 repairs nuclear and mitochondrial DNA damage induced by chain-terminating anticancer and antiviral nucleoside analogs. Nucleic Acids Res. 2013;41:7793–7803. doi: 10.1093/nar/gkt483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kosugi S., Hasebe M., Matsumura N., Takashima H., Miyamoto-Sato E., Tomita M., Yanagawa H. Six classes of nuclear localization signals specific to different binding grooves of importin alpha. J. Biol. Chem. 2009;284:478–485. doi: 10.1074/jbc.M807017200. [DOI] [PubMed] [Google Scholar]
- 37.Han K. An efficient DDAB-mediated transfection of Drosophila S2 cells. Nucleic Acids Res. 1996;24:4362–4363. doi: 10.1093/nar/24.21.4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rogers S.L., Rogers G.C., Sharp D.J., Vale R.D. Drosophila EB1 is important for proper assembly, dynamics, and positioning of the mitotic spindle. J. Cell Biol. 2002;158:873–884. doi: 10.1083/jcb.200202032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McGuffin L.J., Bryson K., Jones D.T. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 40.Shen Y., Delaglio F., Cornilescu G., Bax A. TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cornilescu G., Marquardt J.L., Ottiger M., Bax A. Validation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase. J. Am. Chem. Soc. 1998;120:6836–6837. [Google Scholar]
- 42.de Vries S.J., van Dijk M., Bonvin A.M.J.J. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010;5:883–897. doi: 10.1038/nprot.2010.32. [DOI] [PubMed] [Google Scholar]
- 43.Sims J.J., Haririnia A., Dickinson B.C., Fushman D., Cohen R.E. Avid interactions underlie the Lys63-linked polyubiquitin binding specificities observed for UBA domains. Nat. Struct. Mol. Biol. 2009;16:883–889. doi: 10.1038/nsmb.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Williamson M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013;73:1–16. doi: 10.1016/j.pnmrs.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 45.Zeng Z., Cortes Ledesma F., El Khamisy S.F., Caldecott K.W. TDP2/TTRAP is the major 5′-tyrosyl DNA phosphodiesterase activity in vertebrate cells and is critical for cellular resistance to topoisomerase II-induced DNA damage. J. Biol. Chem. 2011;286:403–409. doi: 10.1074/jbc.M110.181016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kosugi S., Hasebe M., Matsumura N., Takashima H., Miyamoto-Sato E., Tomita M., Yanagawa H. Six Classes of Nuclear Localization Signals Specific to Different Binding Grooves of Importin. J. Biol. Chem. 2008;284:478–485. doi: 10.1074/jbc.M807017200. [DOI] [PubMed] [Google Scholar]
- 47.Zhang D., Raasi S., Fushman D. Affinity makes the difference: Nonselective interaction of the UBA domain of Ubiquilin-1 with monomeric ubiquitin and polyubiquitin chains. J. Mol. Biol. 2008;377:162–180. doi: 10.1016/j.jmb.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang N., Wang Q., Ehlinger A., Randles L., Lary J.W., Kang Y., Haririnia A., Storaska A.J., Cole J.L., Fushman D., et al. Structure of the s5a:k48-linked diubiquitin complex and its interactions with rpn13. Mol. Biol. Cell. 2009;35:280–290. doi: 10.1016/j.molcel.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shi Y., Chen X., Elsasser S., Stocks B.B., Tian G., Lee B.-H., Shi Y., Zhang N., de Poot S.A.H., Tuebing F., et al. Rpn1 provides adjacent receptor sites for substrate binding and deubiquitination by the proteasome. Science. 2016;351:aad9421. doi: 10.1126/science.aad9421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suryadinata R., Roesley S.N.A., Yang G., Sarčević B. Mechanisms of generating polyubiquitin chains of different topology. Cells. 2014;3:674–689. doi: 10.3390/cells3030674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He F., Wollscheid H.-P., Nowicka U., Biancospino M., Valentini E., Ehlinger A., Acconcia F., Magistrati E., Polo S., Walters K.J. Myosin VI contains a compact structural motif that binds to ubiquitin chains. Cell Rep. 2016;14:2683–2694. doi: 10.1016/j.celrep.2016.01.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sloper-Mould K.E., Jemc J.C., Pickart C.M., Hicke L. Distinct functional surface regions on ubiquitin. J. Biol. Chem. 2001;276:30483–30489. doi: 10.1074/jbc.M103248200. [DOI] [PubMed] [Google Scholar]
- 53.Mukhopadhyay D., Riezman H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science. 2007;315:201–205. doi: 10.1126/science.1127085. [DOI] [PubMed] [Google Scholar]
- 54.Krämer H., Phistry M. Genetic analysis of hook, a gene required for endocytic trafficking in drosophila. Genetics. 1999;151:675–684. doi: 10.1093/genetics/151.2.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luiro K., Yliannala K., Ahtiainen L., Maunu H., Jarvela I., Kyttala A., Jalanko A. Interconnections of CLN3, Hook1 and Rab proteins link Batten disease to defects in the endocytic pathway. Hum. Mol. Genet. 2004;13:3017–3027. doi: 10.1093/hmg/ddh321. [DOI] [PubMed] [Google Scholar]
- 56.Lukas J., Lukas C., Bartek J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nature. 2011;13:1161–1169. doi: 10.1038/ncb2344. [DOI] [PubMed] [Google Scholar]
- 57.Jackson S.P., Durocher D. Regulation of DNA damage responses by ubiquitin and SUMO. Mol. Biol. Cell. 2013;49:795–807. doi: 10.1016/j.molcel.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 58.Minsky N., Shema E., Field Y., Schuster M., Segal E., Oren M. Monoubiquitinated H2B is associated with the transcribed region of highly expressed genes in human cells. Nature. 2008;10:483–488. doi: 10.1038/ncb1712. [DOI] [PubMed] [Google Scholar]
- 59.Fuchs G., Hollander D., Voichek Y., Ast G., Oren M. Cotranscriptional histone H2B monoubiquitylation is tightly coupled with RNA polymerase II elongation rate. Genome Res. 2014;24:1572–1583. doi: 10.1101/gr.176487.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gao R., Schellenberg M.J., Huang S.-Y.N., Abdelmalak M., Marchand C., Nitiss K.C., Nitiss J.L., Williams R.S., Pommier Y. Proteolytic degradation of topoisomerase II (Top2) enables the processing of Top2·DNA and Top2·RNA covalent complexes by tyrosyl-DNA-phosphodiesterase 2 (TDP2) J. Biol. Chem. 2014;289:17960–17969. doi: 10.1074/jbc.M114.565374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang A., Lyu Y.L., Lin C.-P., Zhou N., Azarova A.M., Wood L.M., Liu L.F. A protease pathway for the repair of topoisomerase II-DNA covalent complexes. J. Biol. Chem. 2006;281:35997–36003. doi: 10.1074/jbc.M604149200. [DOI] [PubMed] [Google Scholar]
- 62.Nitiss J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer. 2009;9:338–350. doi: 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alchanati I., Teicher C., Cohen G., Shemesh V., Barr H.M., Nakache P., Ben-Avraham D., Idelevich A., Angel I., Livnah N., et al. The E3 ubiquitin-ligase Bmi1/Ring1A controls the proteasomal degradation of Top2alpha cleavage complex - a potentially new drug target. PLoS One. 2009;4:e8104. doi: 10.1371/journal.pone.0008104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Isik S., Sano K., Tsutsui K., Seki M., Enomoto T., Saitoh H., Tsutsui K. The SUMO pathway is required for selective degradation of DNA topoisomerase IIbeta induced by a catalytic inhibitor ICRF-193(1) FEBS Lett. 2003;546:374–378. doi: 10.1016/s0014-5793(03)00637-9. [DOI] [PubMed] [Google Scholar]
- 65.Mao Y., Desai S.D., Ting C.Y., Hwang J., Liu L.F. 26 S proteasome-mediated degradation of topoisomerase II cleavable complexes. J. Biol. Chem. 2001;276:40652–40658. doi: 10.1074/jbc.M104009200. [DOI] [PubMed] [Google Scholar]
- 66.Kaiser S.E., Riley B.E., Shaler T.A., Trevino R.S., Becker C.H., Schulman H., Kopito R.R. Protein standard absolute quantification (PSAQ) method for the measurement of cellular ubiquitin pools. Nat. Methods. 2011;8:691–696. doi: 10.1038/nmeth.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schumann F.H., Riepl H., Maurer T., Gronwald W., Neidig K.-P., Kalbitzer H.R. Combined chemical shift changes and amino acid specific chemical shift mapping of protein-protein interactions. J. Biomol. NMR. 2007;39:275–289. doi: 10.1007/s10858-007-9197-z. [DOI] [PubMed] [Google Scholar]
- 68.Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.