

Abstract
Lonidamine (LND) was initially introduced as an antispermatogenic agent. It was later found to have anticancer activity sensitizing tumors to chemo-, radio-, photodynamic-therapy and hyperthermia. Although the mechanism of action remained unclear, LND treatment has been known to target metabolic pathways in cancer cells. It has been reported to alter the bioenergetics of tumor cells by inhibiting glycolysis and mitochondrial respiration, while indirect evidence suggested that it also inhibited L-lactic acid efflux from cells mediated by members of the proton-linked monocarboxylate transporter (MCT) family and also pyruvate uptake into the mitochondria by the mitochondrial pyruvate carrier (MPC). Recent studies have demonstrated that LND potently inhibits MPC activity in isolated rat liver mitochondria (Ki 2.5 μM) and cooperatively inhibits L-lactate transport by MCT1, MCT2 and MCT4 expressed in Xenopus laevis oocytes with K0.5 and Hill Coefficient values of 36–40 μM and 1.65–1.85, respectively. In rat heart mitochondria LND inhibited the MPC with similar potency and uncoupled oxidation of pyruvate was inhibited more effectively (IC50 ~7 μM) than other substrates including glutamate (IC50 ~20 μM). LND inhibits the succinate-ubiquinone reductase activity of respiratory Complex II without fully blocking succinate dehydrogenase activity. LND also induces cellular reactive oxygen species through Complex II and has been reported to promote cell death by suppression of the pentose phosphate pathway, which resulted in inhibition of NADPH and glutathione generation. We conclude that MPC inhibition is the most sensitive anti-tumour target for LND, with additional inhibitory effects on MCT-mediated L-lactic acid efflux, Complex II and glutamine/glutamate oxidation.
Keywords: Lonidamine, tumor acidification, tumor bioenergetics, 31P and 1H magnetic resonance spectroscopy, monocarboxylate transporter, mitochondrial pyruvate carrier, electron transport chain, xenografts, melanoma, breast cancer, prostate cancer, ovarian cancer, melphalan, doxorubicin
Graphical Abstract
Introduction
Lonidamine (LND), first introduced in 1979 as an antispermatogenic agent (1), has limited antineoplastic activity as a single agent but has exceptional potential in modulating the activities of conventional chemotherapeutic agents such as N-mustard alkylating agents (2–7) and anthracyclines (8) as well as hyperthermia (9–11), radiation therapy (12, 13) and photodynamic therapy (14); it may also enhance response to targeted therapeutics such as vemurafenib. The most critical property of LND is its selective activity against a broad range of tumors with little to no effect on normal tissues provided that doses are below a threshold level of ~400 mg/m2 (oral or i.v. doses) (15, 16). At such doses LND causes selective intracellular cytosolic acidification of tumors while diminishing tumor ATP levels.
Current evidence indicates that LND inhibits lactate export by the proton-linked monocarboxylate transporter(s) (MCT) and pyruvate uptake into mitochondria via the mitochondrial pyruvate carrier (MPC), whereas inhibition of respiration involves both diminished mitochondrial uptake of pyruvate via the MPC as well as inhibition of the mitochondrial electron-transport chain at Complex II and perhaps also Complex I, in both instances at the ubiquinone reduction step. There is also evidence that the drug may indirectly inhibit hexokinase (17–20) as well as possibly other glycolytic and pentose shunt enzymes as a result of cytosolic acidification. LND produces a substantial increase in total tumor lactic acid levels with most of the lactate being trapped in the cytosol as indicated by a pronounced decrease in intracellular pH (pHi); there is also a slight decrease in extracellular pH (pHe) reflecting a small extent of leakage of lactate through the MCT (6–8). However, direct evidence for LND inhibition of the MPC, any of the four MCT isoforms known to transport lactic acid (21) as well as inhibition of mitochondrial electron-transport has until recently been lacking. In this review article, we present data addressing these issues.
Contemporary Background
In 1981, Floridi et al. (22) reported that LND inhibited respiration as well as aerobic and anaerobic glycolysis in Ehrlich ascites tumor cells but had no effect on normal rat sertoli cells. They attributed the selective inhibition of glycolysis in tumor cells to LND binding to and inhibiting mitochondrial bound hexokinase that the Pederson lab had shown (23) existed mainly in tumor cells. In 1982, Floridi et al. (24) demonstrated that LND affected respiration of Ehrlich ascites cells only in the uncoupled state but not in the coupled state. Scatchard analysis indicated two classes of binding sites in uncoupled mitochondria, a high affinity site with dissociation constant (Kd) of 3.2 μM and a weaker but more highly populated site with Kd of 45 μM. Binding of LND to normal liver mitochondria was qualitatively similar but with lower affinity than binding to tumor mitochondria. Further studies of LND inhibition of the mitochondrial electron transport chain of Ehrlich ascites cells were reported by Floridi and Lehninger in 1983 (25). They concluded that LND was bound to mitochondria in state 4 but did not inhibit respiration in this state except at concentrations above 200 μM. Half-maximal inhibition of respiration of FCCP (carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone)-stimulated tumor mitochondria occurred at 32 μM LND with almost complete inhibition at LND concentrations greater than 200 μM. They concluded that LND shifted mitochondrial NAD(P) into a more oxidized steady state. Subsequent addition of an uncoupler or ADP, which would normally induce respiration, increased LND binding and inhibition of reduction of NAD(P) by mitochondrial dehydrogenases. The precise mechanism of interaction with the mitochondrial electron transport chain was not clearly defined although the authors noted that LND appears to affect the state 4 to state 3 transitions. It is noteworthy that the authors recognized that LND inhibits the reduction of NAD(P) by various NAD-linked substrates (pyruvate + malate, α-ketoglutarate and glutamate) and that it inhibits succinate dehydrogenase at some point prior to the reduction to ubiquinone.
In 1995 Ben-Horin et al. (26) reported 31P and 13C NMR (nuclear magnetic resonance) spectroscopic studies of isolated perfused MCF7 breast cancer cells immobilized by encasement in calcium alginate beads. By incorporating Pi (inorganic phosphate) into the perfusate, the authors were able to detect two Pi resonances, which they used to simultaneously monitor both the pHi and pHe, respectively. LND produced a decrease in pHi with no effect on pHe. Furthermore, 13C NMR demonstrated that this decrease in pHi was accompanied by accumulation of lactic acid in the intracellular compartment, and depletion of extracellular lactate. The evidence clearly pointed to inhibition of lactate export (i.e., via MCTs) as a key mechanism responsible for LND-induced tumor acidification. Rather than inhibiting glycolysis, the accumulation of intracellular lactate indicated that LND was stimulating glycolysis, although kinetic inhibition of certain steps along the glycolytic pathway might still be occurring since it is well known that enzymes such as phosphofructokinase are subject to allosteric H+ inhibition. These investigators also noted that LND decreased NTP (nucleoside triphosphate) levels in the tumor, which also decreased phospholipid metabolites as a consequence of diminution of choline and ethanolamine kinase activities. The decrease in the bioenergetic status of the tumors was attributed to the effect of LND on mitochondrial metabolism that had been reported by Floridi et al. (25).
Mardor et al. (27) combined NMR studies of MCF7 cells with diffusion-weighted imaging of perfused cells, a method that eliminates signals from metabolites in the perfusate. This ability to distinguish between intracellular and extracellular compartments further supported the conclusions of Ben-Horin et al. (26).
These NMR studies of breast cancer cells were confirmed and extended by studies of 9L glioma cells and 9L glioma xenografts by Ben-Yoseph et al. (28). Using 31P NMR, these investigators demonstrated that LND produced intracellular acidification and de-energization both in culture and in vivo and showed that the in vivo effects were selective for the tumor with no effect on skeletal muscle or brain.
Selective Intracellular Acidification and De-energization of Various Human Cancer Xenografts
In 2000 (29) and 2001 (30), Zhou et al. demonstrated by 31P NMR studies of DB-1 melanoma xenografts that LND had a similar but more pronounced and sustained effect on tumor pHi and NTP levels as α-cyano-4′-hydroxycinnamate (CHC), an agent that Halestrap et al. (21, 31) had demonstrated to be a potent inhibitor of the MCT and the MPC. Zhou et al. (30) showed that the effects of CHC were specific for the tumor with no effect on the metabolism of the skeletal muscle, liver or brain.
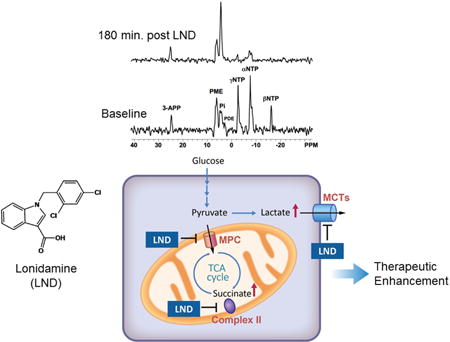
In 2013, Nath et al. (7) reported that administration of 100 mg/kg LND i.p as a solution in pH 8.3 tris-glycine buffer (the method used by Ben-Yoseph et al. (24)) monitored by 31P NMR spectroscopy produced a sustained intracellular acidification of DB-1 melanoma xenografts with pHi decreasing from 6.90 ± 0.05 to 6.33 ± 0.10 (p < 0.001) during 3 hr post-injection (Figure 1 and Table 1). DB-1 tumors have a relatively high rate of glycolysis as defined by a significant rate of lactic acid production. As such, it is possible that MCT transport of lactate becomes limiting. This in concert with a slow clearance of extracellular lactate might provide a rationale for the slightly acidic basal pHi observed in the DB-1 melanoma tumor model. The acidification was accompanied by a decrease in NTP levels and an increase in Pi levels that were sustained over the same period of time as the intracellular acidification of the tumor. The βNTP/Pi ratio, that reflects the bioenergetic state of the tumor, decreased by 66.8 ± 5.7% (p < 0.001) 3 hr post-LND injection (Figure 1 and Table 1). The pHe of the tumor decreased slightly from 7.00 ± 0.04 to 6.80 ± 0.07, p > 0.05 (Figure 1 and Table 1). CHC had produced similar effects, but the acidification and de-energization of the tumor were of substantially shorter duration. 1H MRS (magnetic resonance spectroscopy) of lactate demonstrated a 3-fold increase in lactic acid levels in the tumor after LND treatment providing further evidence that glycolytic metabolism was not inhibited although kinetic effects were not excluded (Figure 1 and Table 1). Nath et al. (7) demonstrated that acidification and de-energization were selective for the tumor with no change in pHi, pHe or NTP/Pi of skeletal muscle or brain and only a slight transient decrease in pHi of the liver 20 min post-LND injection and a decrease in NTP/Pi of the liver 40 min post-LND. Ben-Yoseph et al. (28) also demonstrated selective acidification and de-energization of gliomas in rats with no significant effects on the muscle or brain. Selectivity for tumors probably results from their higher glycolytic capacity although perfused DB-1 melanoma cells derive 54% of their energy from oxidative metabolism and 46% by glycolysis as shown by Shestov et al. (32).
Figure 1. The intracellular pH (pHi), extracellular pH (pHe), bioenergetics and lactate profiles of human melanoma xenograft after lonidamine (LND) administration.

A) In vivo localized (Image Selected In vivo Spectroscopy-ISIS) 31-Phosphorus magnetic resonance spectroscopy (31P MRS) spectra of a human melanoma xenograft grown subcutaneously in nude mice (lower) pre- and (upper) 180 min post administration of LND (100 mg/kg, i.p.). Resonance assignments are as follows, 3-APP (3-aminopropylphosphonate); PME (phosphomonoesters); Pi (inorganic phosphate); PDE (phosphodiesters); γNTP (γ nucleoside-triphosphate), αNTP (α nucleoside-triphosphate), and βNTP (β nucleoside-triphosphate). Decrease in βNTP levels and the corresponding increase in Pi following LND administration (Upper spectrum of panel A) indicating impaired energy metabolism. B) pHi, pHe profiles as a function of time. C) The changes of bioenergetics (βNTP/Pi) (ratio of peak area) relative to baseline D) Change in tumor lactate as a function of time, inset picture showing lactate spectra using 1H MRS with Hadamard Selective Multiquantum coherence transfer pulse sequence in human melanoma xenografts. Area under the curve was compared to baseline at each time point and was normalized to baseline levels as a function of time in response to LND (100 mg/kg; i.p.) administered at time zero. The values are presented as mean ± S.E.M. When not displayed, S.E.M. values were smaller than the symbol size. Part of this research was originally published in NMR Biomed (7) and modified in the current review manuscript.
Table 1.
Summary of ΔpHi, ΔpHe and ΔβNTP/Pi of human tumor xenografts measured by 31P magnetic resonance spectroscopy (31P MRS) following lonidamine (LND) treatment.
| Parameter | DB-1 melanoma Xenografts (n = 15) | BT-474 breast cancer xenografts (n = 3) | HCC1806 breast cancer xenografts (n = 3) | A2780 ovarian cancer xenografts (n = 3) | LNCaP prostate cancer xenografts (n = 3) |
|---|---|---|---|---|---|
| ΔpHi | 0.60 ± 0.1 P < 0.05 |
0.44 ± 0.14 P = 0.05 |
0.54 ± 0.23 P < 0.05 |
0.56 ± 0.10 P < 0.05 |
0.47 ± 0.11 P < 0.05 |
| ΔpHe | 0.20 ± 0.07 P < 0.05 |
0.22 ± 0.07 P = 0.05 |
0.28 ± 0.19 P = 0.05 |
0.34 ± 0.23 P = 0.05 |
0.30 ± 0.13 P > 0.05 |
| ΔβNTP/Pi | 66.8 ± 5.7% P < 0.05 |
70.0 ± 0.12% P < 0.05 |
77.0 ± 0.09% P < 0.05 |
70.0 ± 0.22% P < 0.05 |
77.5 ± 0.04% P < 0.05 |
In 2015 Nath et al (8) demonstrated by 31P MRS that 100 mg/kg LND has similar effects on two breast carcinoma lines--the triple negative HCC1806 subline that lacks estrogen, progesterone and Her2/Neu receptors and the BT-474 line that expresses each of these receptors- and also in human prostate (LNCaP) and ovarian carcinoma xenografts (A2780). Each of these tumors exhibited a rapid decrease in pHi, a small decrease in pHe, and a concomitant monotonic decrease in bioenergetics (βNTP/Pi) over a 2–3 h period (Table 1).
Potentiation of Chemotherapeutic Efficacy of Nitrogen Mustard and Doxorubicin
Nath et al. (7) also demonstrated that LND potentiated the activity of melphalan (7.5 mg/kg; i.v.) (Figure 2 and Table 2) in DB-1 melanoma xenografts and also enhanced the activity of doxorubicin in DB-1 melanoma (7.5 mg/kg and 10 mg/kg; i.v.) and in HCC1806 (12 mg/kg; i.v.) breast cancer xenografts (Table 2). Panel D in figure 1 demonstrated that it takes more than 40 min to maximize effects on pHi after administration of LND (100 mg/kg; i.p.). Chemotherapeutic agents such as nitrogen mustards and doxorubicin were administered by i.v. injection 40 min post injection of LND in order to maximize tumor acidification prior to drug delivery. In the case of N-mustards (6, 7, 33), LND-induced potentiation of chemotherapeutic activity most likely due to three known effects: 1) increased concentrations of the active intermediate cyclic aziridinium ion, 2) decreased concentrations of competing nucleophiles such as hydroxide and glutathione, whose production is diminished by decreased activity of gluthathione-S-transferase under acidic conditions, and 3) decreased DNA repair due to acid inhibition of O6-alkyltransferase (34–36). The enhancement of tumor response to doxorubicin probably resulted from cation-trapping of the weakly basic anthracycline in the tumor (8). The pHi was more acidic than the pHe (reversing the translammelar pH gradient generally observed in tumors (37–39), and a weak base was, therefore, accumulating by a cation-trapping mechanism – i.e., the neutral free base was entering the cell by diffusion, becoming protonated once it entered the cell and, thereby, being trapped in the cell because it became positively charged and could not diffuse out. However, the LND-induced decrease in tumor NTP levels may also contribute to the enhanced activity of doxorubicin by decreasing multi-drug resistance of the tumor to anthracyclines, an energy dependent process that pumps drugs out of the tumor cell (40).
Figure 2. Representative growth delay experiments performed on DB-1 human melanoma xenografts in nude mice.

Animals were treated on Day 0 as follows: Control (sham i.p. tris/glycine buffer + sham i.v. PBS), LND (100 mg/kg i.p.), melphalan (7.5 mg/kg i.v.), lonidamine (LND) + melphalan. Mice were treated on day 0 as follows: Control (sham intraperitoneal Tris/glycine buffer + sham intravenous phosphate-buffered saline (PBS)), lonidamine (LND), melphalan, LND + melphalan. This allows each tumor to serve as its own control. As such, day 0 is equal to 100% for each tumor. In LND + melphalan cohorts melphalan was injected 40 min. after the injection of LND in order to optimize intracellular acidification prior to injection of melphalan. The values shown are means ± standard error of the mean (SEM) of n = 4 animals in the control and LND groups, n = 3 animals in the melphalan and LND + melphalan groups. When not shown, error bars are smaller than the symbol size. The data yielded tumor growth delays in a representative experiment of 1.1 ± 0.1, 6.6 ± 0.0 and 19.9 ± 2.0 days for LND alone, melphalan alone and LND + melphalan, respectively. This research was originally published in NMR Biomed (7).
Table 2.
Summary of estimated growth delay (T-C), doubling time (Td) and surviving fraction as a percentage {100×exp[−(T−C)×B], with bootstrap 95% confidence interval (CI)}, via the log cell kill method, by experiment and treatment arm in DB-1 human melanoma and HCC 1806 breast cancer xenografts.
| Experiment | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||
| Treatment | DB-1 Melanoma xenografts treated with Melphalan (7.5 mg/kg; i.v.) |
DB-1 Melanoma xenografts treated with Doxorubicin (7.5 mg/kg; i.v.) |
DB-1 Melanoma xenografts treated with Doxorubicin (10 mg/kg; i.v.) |
HCC1806 Breast cancer xenografts treated with Doxorubicin (12 mg/kg; i.v.) |
||||||||
|
| ||||||||||||
| T-C (days) |
Td (days) |
Surviving fraction (%) |
T-C (days) |
Td (days) |
Surviving fraction (%) |
T-C (days) |
Td (days) |
Surviving fraction (%) |
T-C (days) |
Td (days) |
Surviving fraction (%) |
|
| Lonidamine (100 mg/kg; i.p.) | 1.99 | 6.50 | 81 (46, 158) | 4.89 | 4.79 | 49 (27, 82) | 2.34 | 5.01 | 72 (45, 121) | 2.31 | 3.16 | 60 (20, 204) |
| Melphalan | 7.62 | 5.91 | 41 (13, 108) | - | - | - | - | - | - | - | - | - |
| Doxorubicin | - | - | - | 4.58 | 4.19 | 47 (23, 83) | 5.43 | 5.04 | 47 (28, 81) | 6.70 | 3.16 | 23 (11, 52) |
| Lonidamine + Melphalan | 17.75 | 5.32 | 10 (4, 28) | - | - | - | - | - | - | - | - | - |
| Lonidamine + Doxorubicin | - | - | - | 18.2 | 4.26 | 5 (2, 14) | 28.57 | 5.20 | 2 (1, 10) | 13.39 | 3.16 | 5 (3, 10) |
Hyperglycemia Induced Selective Intracellular Acidification and De-energization and Sensitization to Melphalan
Nath et al (6) later undertook studies to determine whether the addition of exogenous glucose to mice implanted with DB-1 melanoma can further increase LND-induced tumor acidification by increasing lactate production and whether hyperglycemia can increase the tumor response to melphalan. Comparative data on the normal liver, skeletal muscle and brain were obtained to determine whether LND has a selective effect on tumors and to delineate possible mechanisms underlying this selectivity (6). These studies pointed to the potential utility of nitrogen mustards and LND in the systemic treatment of disseminated melanoma and other malignancies. Hyperglycemia resulted in improvement of the bioenergetic state of the tumor that diminished the cytotoxic effect of melphalan (6). Since the MCT is the main mechanism by which the DB-1 melanoma (7) and most other tumors (8) maintain intracellular pH homeostasis, inhibition of this co-transporter could lead to tumor acidification unless the excess pyruvate was depleted by mitochondrial oxidation (see below). Delivery of high levels of exogenous glucose (26 mM) to the tumor decreased the pHi to 6.17 and increased lactate levels ~6 fold. Without exogenous glucose, pHi was 6.33 and lactate had increased ~3 fold from baseline following LND administration (7). Tumor cells also maintain pH homeostasis as a result of the buffering capacity of CO2. As carbonic acid has a pKa of 6.4, this lack of acidification can be attributed to the high buffering capacity of CO2 produced from oxidative phosphorylation and/or introduced via the Cl−/HCO3− exchanger. The extent of tumor cell kill by melphalan in the presence of glucose diminished (cell kill = 62.8% compared to 89.4% in the absence of exogenous glucose). This study indicated that administration of exogenous glucose should be avoided in the clinic.
The currently available evidence strongly indicates that LND inhibits lactate export by the MCT but precisely which isozyme of MCT is being inhibited remains to be determined. The mechanism responsible for the selective ability of LND to decrease NTP levels in tumors with minimal to no effect on normal tissues remains to be elucidated. The proposal that LND inhibits mitochondrial electron transport requires validation and demonstration of the site of inhibition. These issues have been recently addressed and will be summarized in this review. The rationale of the present study was to determine whether the extent of tumor acidification could be increased by infusing high concentrations of glucose into the mouse to induce an increased rate of glycolysis, resulting in higher levels of lactate and increased acidification, and further resulting in greater efficacy of response. We believe that our hypothesis was false for several reasons: 1) pH balance is highly buffered, so even though we greatly increased the production of lactate, infusion of glucose failed to decrease pH for this reason, and, 2) For most tissues with adequate perfusion, normal physiological levels of glucose (~5 mM) are not limiting to transport of this substrate into cells, so that transport of glucose is at Vmax in these cells under basal conditions and is not influenced by higher levels of glucose. Thus, the increase in lactate production detected in these experiments probably originates from the regions of the tumors whose glucose levels are limited by low access to glucose under normoglycemic conditions. At this point, since these regions of tumor tissue are no longer biochemically limited in their energy production from glucose, they might be able to better resist the effects of chemotherapy, as compared to the more vulnerable state in which glucose was limited. For these reasons we failed to detect a greater decrease in pH levels in the tumor and observed decreased efficacy in response to chemotherapy under hyperglycemic conditions. Furthermore, the improved tumor energetics mitigated any increase in DNA alkylation resulting from acute hyperglycemia.
Mechanism of Action of Lonidamine (LND) at the Level of Mitochondrial Pyruvate Carrier
Inhibition of lactate export also might not suffice to cause intracellular acidification since lactate is in equilibrium with pyruvate, which could be exported to the mitochondria where it could be converted to acetyl-CoA by pyruvate dehydrogenase and then be completely oxidized via the TCA cycle. If this occurred, inhibition of mitochondrial oxidation of pyruvate would be essential to cause tumor acidification. Thus, under hyperglycemic conditions with addition of a mitochondrial inhibitor such as meta-iodobenzyl guanidine (MIBG), a Complex I inhibitor, tumor acidification has been noted by Kuin et al. (41), Burd et al. (42) and by Zhou et al. (29). However, MIBG cannot be used in the clinic at the therapeutic doses required to produce tumor acidification. Similar effects might be produced by metformin that is a Food and Drug Administration (FDA) approved Complex I inhibitor (43–49). However, Zhou et al. (30) also found that CHC produced selective acidification and de-energization of DB-1 melanomas, and Halestrap et al. (21, 31) had shown that this agent inhibits both MCT1 and MCT4 while also inhibiting the MPC, thus blocking oxidation of pyruvate. However, CHC inhibits lactate export and oxidation for relatively short periods of time and is not approved for clinical use, whereas LND has been used extensively in Europe and Canada and with special FDA permission in the US. This drug produces a much more robust and sustained effect on tumor pHi and NTP levels. Nath et al. (6, 7) proposed that LND also inhibited both the MPC and the MCT and Nancolas et al. (50) provided direct proof of this hypothesis. These authors demonstrated that LND induced inhibition of MPC activity in isolated liver mitochondria and also inhibited the activity of MCT1, MCT2 and MCT4 expressed in Xenopus laevis oocytes. It is particularly noteworthy that they showed the IC50 for MPC inhibition was about an order of magnitude lower than for inhibition of the MCTs. Thus, the MPC inhibition was likely to be playing a critical role in this process even though previous studies have attributed tumor acidification exclusively to inhibition of the MCTs (50) (Figure 3).
Figure 3. Lonidamine (LND) inhibits pyruvate transport into mitochondria.

In Panel A) pyruvate transport into liver mitochondria was assayed directly at 9°C using [1-14C]-pyruvate as described under the methods section of Nancolas et al (50). Data are presented as Means ± S.E.M. for 3 separate mitochondrial preparations. In each experiment, four replicates were performed at each LND concentration and the average value taken to calculate the extent of inhibition as percentage of control (no LND). Data were fitted to the standard inhibition equation (methods section of Nancolas et al (50)) to give a derived inhibitory constant (Ki) value of 2.5 ± 0.1 μM. The absolute rate of pyruvate (60) μM uptake in the absence of LND was 0.303 ± 0.032 nmol/mg protein in 45s. Panel B) shows data for the inhibition of uncoupled pyruvate oxidation by isolated rat heart mitochondria at 30°C measured using an oxygen electrode as described under the methods section of Nancolas et al (50). Mean data (± S.E.M.) are presented for 3 separate mitochondrial preparations. The absolute rate of pyruvate oxidation in the absence of LND was 87.9 ± 5.8 nmol O2 per mg protein per min. The data were fitted to an equation that assumes oxidation of pyruvate is set by the activity of pyruvate dehydrogenase (PDH) which in turn is controlled by the rate of pyruvate transport relative to that of PDH as described under methods section of Nancolas et al (50). The Ki value for LND of 2.5 μM derived from Panel A was employed and the Vmax of PDH and MPC activity (expressed as % control rate of oxygen consumption and ± S.E.) were then calculated by least squares regression analysis to be 127 ± 6.3 and 233 ± 9.1, respectively. This research was originally published in Biochem J (50).
Lonidamine (LND) Inhibits Uncoupled Mitochondrial Pyruvate Oxidation more Potently than Glutamate + Malate or Succinate Oxidation
Nancolas et al (50) demonstrated that pyruvate oxidation by uncoupled mitochondria was inhibited by LND more potently than glutamate + malate or succinate oxidation. The effect of LND on uncoupled pyruvate oxidation by rat heart mitochondria was significantly greater than on the oxidation of other respiratory substrates, which is entirely consistent with a dominant effect of the inhibitor on the MPC as opposed to the respiratory chain (Figure 4).
Figure 4. Lonidamine (LND) inhibits oxidation of glutamate, 2-oxoglutarate and succinate by uncoupled rat heart and liver mitochondria less potently than pyruvate transport.

Rates of oxidation by isolated rat liver A) or heart B) mitochondria at 30°C were measured using an oxygen electrode as described under the methods section of Nancolas et al. (50). Data are expressed as the percentage of rates in the absence of LND to allow better comparison between the different substrates and the lines drawn were fitted by FigSys using a Bezier Spline function. For heart mitochondria, data are presented for a single representative experiment with absolute rates of pyruvate + malate, glutamate + malate, 2-oxoglutarate + malate and succinate + rotenone oxidation in the absence of LND of 84, 83, 67 and 212 nmol O2 per mg protein per min, respectively. For liver mitochondria A), mean data (± S.E.M.) are presented for 3 separate mitochondrial preparations. The absolute rates of glutamate + malate and succinate (+ rotenone) oxidation in the absence of LND were 67.9 ± 3.0 and 109 ± 7.8 nmol O2 per mg protein per min, respectively. This research was originally published in Biochem J (50).
Liver mitochondria, unlike heart mitochondria, do not oxidize pyruvate rapidly, and the MPC does not exert much control over its oxidation (50). However, uncoupled respiration in the presence of either glutamate + malate or succinate (in the presence of rotenone) is rapid, and the data of figure 3 confirm that LND inhibits both with IC50 values of 25 and 150 μM, respectively.
Mechanism of Action of Lonidamine (LND) at the Level of Monocarboxylate Transporter
Full exploitation of LND’s role as a sensitizer in cancer therapy requires delineation of its detailed mechanism of action, which has been the subject of considerable controversy. The initial claim that LND inhibited glycolysis at the level of mitochondrial hexokinase II was based on decreases in extracellular lactic acid, which appear artifactual in light of strong evidence that this drug was inhibiting lactate export from tumor cells (19, 22). However, the evidence for inhibition of the MCT, while convincing, was indirect with definitive identification of which isoform(s) of MCT were involved and their affinities for the drug yet to be identified. Because of the similarity of LND to CHC in its ability to selectively lower the pHi and NTP levels of melanoma, Nath et al. (7) suggested that LND also inhibits the mitochondrial pyruvate carrier (MPC). There was strong evidence for MCT inhibition by CHC (30) but none for LND until Nancolas et al. (50) recently published their findings. Finally, as noted above, there was also additional evidence to those presented by Floridi and Lehninger (25) that LND also inhibits respiration, but the site and detailed mechanism of inhibition remained to be determined.
Inhibition of lactate export was based on indirect evidence of retention of lactate by tumor cells. However, the proposal that inhibition of the MCT was responsible for this decrease was purely hypothetical. Nancolas et al. recently provided the first direct proof that this is, in fact, the case and identified specific MCT isoforms involved. Transport through MCT1, MCT2, and MCT4 are all inhibited cooperatively with similar K0.5 values (36–40 μM) and Hill Coefficients (1.65–1.85), but only MCT1 and MCT4 are detected in DB-1 melanomas at approximately equal levels (50). However, MCT1 and MCT4 have Km values for lactate of about 4.5 mM and 22 mM, respectively (51). Therefore, the dominant effect on inhibition of lactate export by LND can probably be attributed to inhibition of MCT1 (Figure 5).
Figure 5. Lonidamine (LND) inhibits the proton-linked monocarboxylate carriers MCT1, MCT2 and MCT4.

[U-14C]-L-lactate uptake into Xenopus laevis oocytes expressing the MCT isoform indicated was determined as described in Methods. Panel A) shows the absolute rates of L-lactate uptake while Panels B–D) show the effects of increasing concentrations of LND on rates of L-lactate uptake into oocytes expressing MCT1, MCT2 or MCT4 as indicated. Rates are expressed as a percentage of the control (no LND) after subtraction of the uptake by water-injected oocytes. Each data point represents mean data ± S.E.M. for 10–45 individual oocytes and data were fitted to the equation for cooperative inhibition using FigSys as described in the methods section of Nancolas et al (50). The derived values for K0.5 and the Hill Coefficient (n) are indicated on each plot (± S.E. of the fit shown). This research was originally published in Biochem J (50).
Mechanism of Action of Lonidamine (LND) at the Level of Complex II of Electron Transport Chain (ETC)
Using the liquid chromatography-selected reaction monitoring/mass spectrometry (LC-SRM/MS) technique, Guo et al (52) recently demonstrated that in DB-1 cells treated with LND, the levels of succinate and α-ketoglutarate increased whereas levels of citrate, fumarate and malate decreased (Figure 6). The oxidation of succinate to fumarate is catalyzed by the succinate dehydrogenase activity of Complex II. Floridi and Lehninger (25) first recognized that LND inhibited mitochondrial respiration. They attributed this to inhibition of the electron-transfer chain, but could not unambiguously identify the specific locus of inhibition. The cellular accumulation of succinate and inhibition of succinate dependent mitochondrial respiration indicate that Complex II is one of the sites of this activity (Figure 7). Complex II or succinate dehydrogenase (SDH) consists of four basic components delineated as SDHA, which oxidizes succinate to fumarate while reducing FAD to FADH2, SDHB which contains three iron-sulfur clusters, and SDHC and SDHD, contain a heme moiety and reduce ubiquinone (Q) to ubiquinone dihydride (QH2), respectively (Figure 7). With the aid of electron acceptor dyes that specifically accept electrons from certain components of Complex II (Figure 7), Guo et al. have demonstrated that SDHA and SDHB are not significantly inhibited by LND. Hence, the inhibitory effect of LND on SDH must be on the SDHC – SDHD moiety involving transfer of electrons from the iron sulfur clusters to ubiquinone. A more extensive description of the effects of LND on Complex II is presented by Guo et al. (52)
Figure 6. Lonidamine (LND) alters TCA cycle intermediates.

LND treatment (150 μM) in DB-1 melanoma cells increases the intracellular levels of lactate, α-ketoglutarate and succinate with concurrent reduction in the levels of citrate, fumarate and malate. Part of this research was originally published in J Biol Chem (52) and modified in the current review manuscript.
Figure 7. Lonidamine (LND) inhibits the SQR activity of Complex II.

A schematic representation of Complex II subunits and enzyme activities. Complex II contains four subunits: succinate dehydrogenase A (SDHA; flavoprotein), succinate dehydrogenase B (SDHB; iron-sulfur subunit), succinate dehydrogenase C (SDHC; integral membrane protein) and succinate dehydrogenase D (SDHD; cytochrome b small subunit). Flavin adenine dinucleotide (FAD) cofactor bound to SDHA obtains electrons from succinate oxidation. The electrons are then passed to the Fe-S clusters of SDHB and finally to the ubiquinone reduction site within SDHC and SDHD where ubiquinone (Q) is reduced to ubiquinol (QH2). LND inhibits the succinate ubiquinone reductase (SQR) activity of Complex II, whereas it has minimal effect on the SDH of Complex II. SQR activity was measured by electron transfer from succinate to decylubiquinone and 2,6-dichlorophenolindophenol (DCPIP). SDH of Complex II was determined by the electron transfer from succinate to iron-sulfur cluster and finally to phenazine methosulfate (PMS) and 2-(4,5-dimethyl-2-thiazolyl)-3,5-diphenyl-2H-tetrazolium bromide (MTT). Part of this research was originally published in J Biol Chem (52).
Other Effects of Lonidamine (LND)
Lonidamine (LND) Induces Reactive Oxygen Species (ROS) Formation and Cell Death
It has been reported that Complex II is a source of ROS from either the reduced FAD or ubiquinone site when downstream components of the ETC are blocked (52–54). Guo et al. (52) have quantified the level of ROS by the dichlorodihydrofluorescein (DCF) fluorescence technique to examine whether LND induces intracellular ROS generation. The ROS generated upon LND treatment can be partially reduced by 3-nitropropionic acid (3-NPA) and further enhanced by thenoyltrifluoroacetone (TTFA), indicating that LND induces ROS at a site within Complex II downstream of SDHA. Guo et al. (52) have also shown that LND induces substantial cell death after treatment of DB-1 cells for 24 h or 48 h.
Lonidamine (LND) Reduces Cellular Levels of Glutathione and NADPH and the Pentose Phosphate Pathway (PPP)
Guo et al. (52) compared the cellular levels of GSH and antioxidants in LND and TTFA-treated DB-1 tumor cells. LND caused a 40% drop in GSH levels at a concentration of 150 μM and above. In contrast, TTFA (50 or 200 μM) caused a modest reduction of 6%, indicating that this effect is independent of Complex II inhibition. Consistent with the reduced GSH levels, the levels of NADPH as well as the NADPH/NADP+ ratio were both decreased after LND-treatment but not after TTFA-treatment. The pentose phosphate pathway (PPP) is an important source of NADPH (55) required for the GSH-reductase mediated reduction of glutathione disulfide (GSSG) to GSH (56).
Role of Lonidamine (LND) as Inhibitor of Hexokinase
LND has been reported to be an inhibitor of hexokinase (17–20), which catalyzes the first step of glycolysis. Therefore, it is possible that both the PPP and glycolysis are inhibited in DB-1 cells. In support of this hypothesis, the cellular concentration of 6-phosphogluconate (6-PG), an important PPP metabolite, was markedly decreased in LND treated cells (52). In addition, a time-course for the incorporation of [13C6]-glucose into PPP metabolites (determined by Liquid chromatography–mass spectrometry (LC-MS)) revealed that incorporation into the M+6 isotopologue, [13C6]-6-PG as well as the glycolytic metabolite [13C6]-fructose-1,6-bisphosphate (Fru-1,6-BP) was significantly delayed. These data, together with the reduced levels of 6-PG, suggest that flux into the PPP was greatly reduced by LND, possibly through inhibition of hexokinase. Thus, the reduced NADPH and glutathione (GSH) levels in LND treated cells resulted, in part, from inhibition of the PPP (52).
Ben-Horin et al. (26) co-administered 2-deoxyglucose (2DG), a well-documented inhibitor of hexokinase, and used the 2DG proton NMR resonance to evaluate possible competitive inhibition by LND for which no evidence was found. However, failure to find such indirect evidence for hexokinase inhibition does not definitively rule it out. The authors also concluded that the apparent inhibition of glycolysis by LND was really an artifact caused by using extracellular lactate as the indicator of glycolytic activity. They concluded that inhibition of the MCT rather than inhibition of hexokinase was the cause of the drop in extracellular lactate produced by LND.
Ben-Yoseph et al. (28) claimed that the lack of effects of LND on either soluble or particulate (i.e., mitochondrial-bound) hexokinase or on relative flux through the PPP of cultured 9L cells (monitored by GC-MS analysis of [1,6-13C2, 6,6-2H] glucose) confirmed that LND was not inhibiting mitochondrial hexokinase. This argument appears inconclusive since hexokinase produces glucose-6-phosphate that could enter either the pentose shunt or the glycolytic pathway and need not affect the relative flux through the two pathways, so direct evidence of the absence of hexokinase inhibition by LND appears to have been missing.
In summary, Floridi reported that LND inhibited hexokinase II (19, 20). Ben-Horin et al. (26) found no evidence of any effect of LND on the hexokinase-2DG complex, suggesting that LND may not interact with hexokinase II but not directly proving that this inhibition did not occur. Ben-Yoseph et al. (28) found that LND did not change relative flux through the PPP vs. glycolysis and suggested that this contradicted the claim of Floridi et al. (19, 20), but relative flux through the PPP and glycolysis would not be changed if hexokinase II was inhibited since hexokinase acts upstream of glucose-6-phosphate and would affect both of these pathways equivalently. The data of Guo et al. (52) suggest substantial decreases in flux through the PPP and glycolytic pathways, which may be consistent with inhibition of hexokinase II but does not prove it since inhibition of these pathways could result from other mechanisms such as acid inhibition of specific enzymes in the PPP and glycolytic pathways. Sadeghi et al. (18) have reported the inhibition of hexokinase with 600 μM LND in prostate cancer cells using glucose 6-phosphate dehydrogenase (G6PD)-coupled assay. Cervantes-Madrid et al. (17) have also suggested the inhibitory effects of LND on hexokinase based on genetic manipulation of hexokinase mRNA.
Role of 13C Metabolic Modeling to Study the Effects of Lonidamine (LND)
To understand the regulation of the cancer metabolic network and to mechanistically evaluate the mechanism of action of LND, quantitative models have been used to interrogate altered metabolic pathways. To quantitatively model the metabolic pathways related to energy and other intermediary metabolism, Shestov et al. (32, 58) have used 13C metabolic modeling techniques called Bonded Cumomer and Fragmented Cumomer Analysis for analysis of 13C NMR and LC-MS data, respectively. These techniques provide a detailed picture of metabolism at the level of in vivo enzyme activities, whole pathways and the integrated systems level. A three-compartment metabolic model which included extracellular medium, cell cytoplasm and the mitochondria was used to fit 13C steady state or time courses of labeled metabolites to determine the metabolic and inter-compartmental transport fluxes. The main intermediary metabolic pathways including glycolysis, TCA cycle, PPP, α-ketoglutarate-glutamate and oxaloacetate-aspartate exchange (to model malate-aspartate shuttle), pyruvate carboxylase activity, anaplerosis at the succinyl-CoA level, pyruvate recycling through malic enzyme, glutaminolysis, reductive carboxylation and lactate dehydrogenase activity were included in the melanoma bionetwork. To express the model mathematically, Shestov et al. have used: 1) mass balances for the total metabolite concentration in medium, cytosol, and mitochondria and 2) 13C isotopomer mass balance for labeled metabolites. Transport of the main nutrients utilized by cancer cells were included in the model: the perfused labeled glucose, lactate and glutamine were transported from the extracellular medium to the cancer cells and vice versa assuming reversible non-steady-state Michaelis-Menten transport kinetics through corresponding transporters. Isotopomer balance equations were derived for all bonded cumomers of orders 1, 2, and 3 of participating metabolites (32, 57, 58). Fine structure multiplets were completely described by each metabolite’s bonded cumomers (32, 57, 58) of order 1, 2, and 3 using connection matrices between bonded cumomers and 13C fine multiplets (32, 57, 58). The use of bonded and fragmented cumomer techniques leads to a reduced number of equations compared to a model including all possible isotopomers while retaining all the NMR- and MS-measurable isotopomer information. The errors for the obtained fluxes and other metabolic parameters were estimated using Monte Carlo simulations with experimental noise levels (59). By using these modeling techniques Guo et al. (52) were able to determine the effects of LND on the activities of various key metabolic pathways, e.g., glutaminolysis and reductive carboxylation in cancer cells for the first time to the best of our knowledge, which are discussed in the next section.
Lonidamine (LND) Increases the Contribution of Glutamine to the TCA Cycle through Oxidative Metabolism and Reduces Glutamine-Dependent Reductive Carboxylation
Many cancer cells depend on glutamine to sustain bioenergetics, building blocks and redox balance. To determine whether LND altered flux from glutamine into TCA cycle and production of citrate through reductive carboxylation, Guo et al. (52) used [U-13C5, U-15N2] glutamine tracing combined with a three-compartment metabolic flux modeling to demonstrate that the glutaminolysis pathway as mentioned in the above section was significantly higher in LND treated cells (increase from 0.9 mM/h to 2.1 mM/h with LND treated cells). The enhanced glutamine utilization in LND treated cells was underscored by their increased glutamine uptake during 12 h incubation. Guo et al. (52) also showed that DB-1 cells metabolize glutamine through reductive carboxylation as demonstrated by the enhanced formation of malate and fumarate M+3 isotopologues and the citrate M+5 isotopologue when U-13C labeled glutamine was used as a tracer. LND lowered flux through the reductive carboxylation pathway by nearly 50%, from 2.2 mM/h in control to 1.2 mM/h with LND treatment. In contrast, the reductive carboxylation flux was unchanged in TTFA-treated cells, indicating that this effect is independent of Complex II inhibition.
13C NMR Experiments to Study Effects of Lonidamine (LND)
We have performed a 13C NMR study of DB-1 melanoma cells cultured in flasks. The cells confluent on the T-150 flasks were treated with either vehicle or 150 μM LND for 6 hr in DMEM with glucose substituted by 8 mM [1,6-13C2] glucose. 1.5×107 cells from each flask were harvested using 0.05% trypsin-EDTA (Gibco, Thermo Fisher Scientific Inc., Philadelphia, PA). Water-soluble metabolites were extracted using a perchloric acid extraction procedure (60). The dried metabolites were dissolved in 300 μl of D2O, containing 0.05% (w/v) trimethylsilyl propionic acid (Sigma Aldrich, St. Louis, MO) as an internal reference and inserted into a 5 mm Shigemi NMR tube (Wilmad-Lab Glass, Inc, Vineland, NJ). 13C NMR experiments were performed (~15 hr) on a 400 MHz NMR spectrometer (Agilent Technologies Inc., Wilmington, DE) with proton decoupling and nuclear Overhauser enhancement. Figure 8 shows the 13C NMR spectra of vehicle- and LND-treated DB-1 cells. The intracellular C3-lactate increased 4-fold after LND treatment. This result is consistent with LND blocking lactate export by inhibiting the monocarboxylate transporters (50). The TCA cycle metabolite C4- and C3-glutamate did not change significantly after treatment with LND. However, the alanine level increased 2-fold after treatment. Alanine can enter mitochondria independently of the MPC and thus provide a source of pyruvate to maintain TCA cycle intermediates (61). An analysis is in progress to quantitatively evaluate detailed metabolic fluxes (32) in response to LND under these conditions.
Figure 8. 13C NMR of DB-1 melanoma cells with lonidamine (LND) treatment.

13C NMR spectra of DB-1 melanoma cells after 6 hr incubation in 8 mM [1,6-13C2] glucose containing medium without A) and with B) LND. Resonance assignments are as follows: C4-Glu (C4-glutamate), C3-Glu (C3-glutamate), C3-Lac (C3-lactate), C3-Ala (C3-alanine), TSP (trimethylsilyl propionic acid). Graphical abstract
Effects of Lonidamine (LND) on L-lactate Output by DB-1 Melanoma cells
Nancolas et al. detected (50) inhibition of the MPC at low concentrations of LND resulting in a decrease in pyruvate oxidation and an increase in L-lactate. Higher LND concentrations were required for inhibition of the MCTs, decreasing L-lactic acid cellular efflux with a corresponding rise in levels of intracellular [L-lactate]. They found that 1–10 μM LND increased L-lactate output, consistent with MPC inhibition, and this occurred without a detectable increase in intracellular lactate concentration. However, higher LND concentrations, 40 μM and 150 μM, caused intracellular [L-lactate] to increase by two-fold and eight-fold, respectively, with a corresponding decrease in L-lactate output, consistent with MCT inhibition as a result of intracellular acidification that inhibited glycolytic enzyme activity (52).
Lonidamine (LND) Toxicity
Price et al. (15, 16) reported that dogs administered single doses of 400 mg/m2 LND intravenously and 1200 mg/m2 orally twice daily exhibited signs of acute hepatic and pancreatic toxicity with one quarter of these animals showing increases in alanine amino transferase (ALT) activity. However, no toxicity was noted at half this oral dose. Also, a clinical trial of LND chronically administered orally to humans for treatment of benign prostate hyperplasia was terminated when six out of more than eight hundred patients exhibited elevations in liver ALT and AST enzymes. Thus, reports of liver toxicity after prolonged treatment with LND at elevated levels can probably be attributed to accumulation of the drug in the liver, but this might be avoided by single intravenous administration of doses of 400 mg/m2 or less. There have also been reports of myalgia (16% in a breast cancer study (62–64) and testicular pain (65–67) which again may be due to higher levels of LND reaching the plasma following oral administration. Myalgia can be avoided by bed rest; the absence of effects on skeletal muscle in mouse studies may be attributed to the animals being sedated throughout the experiments. We suspect that the transient effects on liver pHi and NTP/Pi resulted from transient accumulation of LND in the liver that was quickly washed out or detoxified (7). LND pharmacokinetic studies have been investigated in advanced breast cancer (68) and lung cancer patients (69) as part of phase II evaluation of the LND with wide variation in plasma concentration. Detailed Phase II clinical trials of LND have been performed in patients with breast cancer (70–73), lung cancer (74–76), ovarian cancer (77–80), and head neck cancer (81–83). Phase III clinical trials have also been performed on metastatic breast cancer (84–88) and lung cancer (89–93) patients. Overall, treatment with LND appears to be free of significant toxicity to normal tissues and such toxicity that is encountered can be ameliorated or avoided by intravenous administration of proper doses (6, 7). Tumor selectivity and low toxicity to normal tissues are critical characteristics that make LND an attractive agent for treatment of cancer by potentiating the activity of other agents (6, 7).
Conclusions and Perspectives
The most avid site for LND activity appears to be the MPC with an IC50 of 2.5 μM. The sites with the next highest affinity for LND are the MCTs with MCT1, MCT2 and MCT4 all exhibiting K0.5 values of 36–40 μM. Simultaneous inhibition of the MPC and MCT1 as well as MCT4 is essential for intracellular tumor acidification, but inhibition of the MPC may suffice to produce tumor de-energization. Further contributions to tumor de-energization appear to result from inhibition of the ETC (Electron Transport Chain) at Complex II and perhaps also Complex I, since both sites involve ubiquinone reduction, but these effects occur at higher LND concentrations. Other effects on hexokinase, other glycolytic and PPP enzymes are probably secondary to tumor acidification.
Selective effects of LND on tumors compared to other potential targets probably result from dependence of most tumors on glycolytic metabolism, but the exact mechanism of specificity is still not fully known.
The primary clinical utility of LND derives from its selective effect on tumors, producing intracellular acidosis and depletion of NTP pools. This potentiates the response of tumors to a variety of cancer drugs such as N-mustards and anthracyclines, and also enhances the effect of hyperthermia, radiation therapy and photodynamic therapy. A variety of mechanism may be responsible for these potentiation effects including cation trapping of weak bases, stabilization of active intermediates like aziridinium ions by acid, inhibition of glutathione-transferase and O6-alkyltransferase and perhaps other DNA repair enzymes by acid as well diminution of multidrug resistance pumps by decreases in tumor energy levels.
Key problems that remain to be addressed are production of LND under GMP conditions since Angelini Pharmaceuticals in Rome, Italy, the sole commercial source of this drug, stopped producing it in 2006. In addition, utilization of LND in the US requires IND approval by the FDA, which has previously been granted for a number of clinical trials (68, 94, 95). Finally, even though LND is a potent enhancer of the activity of a number of potent anti-cancer agents, potentially less toxic (and patentable) “targeted-tumor agents” are replacing traditional chemotherapy. Another problem remaining to be addressed is the limited solubility of LND at neutral pH. Oral delivery has led to variable results; more soluble derivatives that can be administered by intravenous administration are needed to accurately control the dosing schedules. More study may be required to determine why LND is so selective for tumor cells even though it inhibits isolated uncoupled liver and heart mitochondria.
Highlights.
Mitochondrial pyruvate carrier (MPC) has highest affinity for lonidamine (LND).
Followed by monocarboxylate transporters (MCTs) with MCT1, MCT2 and MCT4.
Inhibition of the MPC and MCTs is essential for lowering intracellular pH (pHi).
Inhibition of the MPC may suffice to produce tumor de-energization.
De-energization also involves inhibition of the ETC (Electron Transport Chain).
Acknowledgments
Support for this project was provided by NIH grants R01-CA129544 and R01-CA172820.
Abbreviations
- LND
lonidamine
- MIBG
meta-iodobenzyl guanidine
- FCCP
carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone
- GSH
glutathione
- ROS
reactive oxygen species
- SDH
succinate dehydrogenase
- SQR
succinate-ubiquinone reductase
- TCA
tricarboxylic acid
- TTFA
4,4,4-trifluoro-1-(2-thienyl)-1,3-butanedione
- QH2
ubiquinone dihydride
- NAD
nicotinamide adenine dinucleotide
- FAD
flavin adenine dinucleotide
- MRS
magnetic resonance spectroscopy
- MCT
monocarboxylate transporter
- MPC
mitochondrial pyruvate carrier
- ETC
electron transport chain
- PPP
pentose phosphate pathway
- pHi
intracellular pH
- pHe
extracellular pH
- i.p
intraperitoneal
- i.v
intravenous
- s.c
subcutaneous
- NTP
nucleoside triphosphate
- CHC
α-cyano-4-hydroxycinnamic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cioli V, Bellocci B, Putzolu S, Malorni W, Demartino C. Anti-spermogenic activity of lonidamine (AF-1890) in rabbit. Ultramicroscopy. 1980;5(3):418–418. [Google Scholar]
- 2.Atema A, Buurman KJ, Noteboom E, Smets LA. Potentiation of DNA-adduct formation and cytotoxicity of platinum-containing drugs by low pH. Int J Cancer. 1993;54(1):166–72. doi: 10.1002/ijc.2910540126. [DOI] [PubMed] [Google Scholar]
- 3.Jahde E, Glusenkamp KH, Klunder I, Hulser DF, Tietze LF, Rajewsky MF. Hydrogen ion-mediated enhancement of cytotoxicity of bis-chloroethylating drugs in rat mammary carcinoma cells in vitro. Cancer Res. 1989;49(11):2965–72. [PubMed] [Google Scholar]
- 4.Jahde E, Glusenkamp KH, Rajewsky MF. Nigericin enhances mafosfamide cytotoxicity at low extracellular pH. Cancer Chemother Pharmacol. 1991;27(6):440–4. doi: 10.1007/BF00685157. [DOI] [PubMed] [Google Scholar]
- 5.Jahde E, Roszinski S, Volk T, Glusenkamp KH, Wiedemann G, Rajewsky MF. Metabolic response of AH13r rat tumours to cyclophosphamide as monitored by pO2 and pH semi-microelectrodes. Eur J Cancer. 1992;29A(1):116–22. doi: 10.1016/0959-8049(93)90587-6. [DOI] [PubMed] [Google Scholar]
- 6.Nath K, Nelson DS, Heitjan DF, Zhou R, Leeper DB, Glickson JD. Effects of hyperglycemia on lonidamine-induced acidification and de-energization of human melanoma xenografts and sensitization to melphalan. NMR Biomed. 2015;28(3):395–403. doi: 10.1002/nbm.3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nath K, Nelson DS, Ho AM, Lee SC, Darpolor MM, Pickup S, et al. 31P and 1H MRS of DB-1 melanoma xenografts: lonidamine selectively decreases tumor intracellular pH and energy status and sensitizes tumors to melphalan. NMR Biomed. 2013;26(1):98–105. doi: 10.1002/nbm.2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nath K, Nelson DS, Heitjan DF, Leeper DB, Zhou R, Glickson JD. Lonidamine induces intracellular tumor acidification and ATP depletion in breast, prostate and ovarian cancer xenografts and potentiates response to doxorubicin. NMR Biomed. 2015;28(3):281–90. doi: 10.1002/nbm.3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chu GL, Dewey WC. The role of low intracellular or extracellular pH in sensitization to hyperthermic radiosensitization. Radiat Res. 1988;115(3):576–85. [PubMed] [Google Scholar]
- 10.Chu GL, Wang ZH, Hyun WC, Pershadsingh HA, Fulwyler MJ, Dewey WC. The role of intracellular pH and its variance in low pH sensitization of killing by hyperthermia. Radiat Res. 1990;122(3):288–93. [PubMed] [Google Scholar]
- 11.Lyons JC, Kim GE, Song CW. Modification of intracellular pH and thermosensitivity. Radiat Res. 1992;129(1):79–87. [PubMed] [Google Scholar]
- 12.Kim JH, Alfieri A, Kim SH, Young CW, Silvestrini B. Radiosensitization of meth-a fibro-sarcoma in mice by lonidamine. Oncology. 1984;41:36–38. doi: 10.1159/000225883. [DOI] [PubMed] [Google Scholar]
- 13.Kim JH, Alfieri AA, Kim SH, Young CW. Potentiation of radiation effects on 2 murine tumors by lonidamine. Cancer Research. 1986;46(3):1120–1123. [PubMed] [Google Scholar]
- 14.Golding JP, Wardhaugh T, Patrick L, Turner M, Phillips JB, Bruce JI, et al. Targeting tumour energy metabolism potentiates the cytotoxicity of 5-aminolevulinic acid photodynamic therapy. Br J Cancer. 2013;109(4):976–82. doi: 10.1038/bjc.2013.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Price GS, Page RL, Riviere JE, Cline JM, Thrall DE. Effect of whole-body hyperthermia on the pharmacokinetics and toxicity of lonidamine in dogs. International Journal of Hyperthermia. 1995;11(4):531–544. doi: 10.3109/02656739509022488. [DOI] [PubMed] [Google Scholar]
- 16.Price GS, Page RL, Riviere JE, Cline JM, Thrall DE. Pharmacokinetics and toxicity of oral and intravenous lonidamine in dogs. Cancer Chemotherapy and Pharmacology. 1996;38(2):129–135. doi: 10.1007/s002800050460. [DOI] [PubMed] [Google Scholar]
- 17.Cervantes-Madrid D, Duenas-Gonzalez A. Antitumor effects of a drug combination targeting glycolysis, glutaminolysis and de novo synthesis of fatty acids. Oncol Rep. 2015;34(3):1533–42. doi: 10.3892/or.2015.4077. [DOI] [PubMed] [Google Scholar]
- 18.Sadeghi RN, Karami-Tehrani F, Salami S. Targeting prostate cancer cell metabolism: impact of hexokinase and CPT-1 enzymes. Tumour Biol. 2015;36(4):2893–905. doi: 10.1007/s13277-014-2919-4. [DOI] [PubMed] [Google Scholar]
- 19.Floridi A, Paggi MG, D’Atri S, De Martino C, Marcante ML, Silvestrini B, et al. Effect of lonidamine on the energy metabolism of Ehrlich ascites tumor cells. Cancer Res. 1981;41(11 Pt 1):4661–6. [PubMed] [Google Scholar]
- 20.Floridi A, Paggi MG, Marcante ML, Silvestrini B, Caputo A, De Martino C. Lonidamine, a selective inhibitor of aerobic glycolysis of murine tumor cells. J Natl Cancer Inst. 1981;66(3):497–9. [PubMed] [Google Scholar]
- 21.Halestrap AP. Monocarboxylic Acid Transport. Comprehensive Physiology. 2013;3:1611–1643. doi: 10.1002/cphy.c130008. [DOI] [PubMed] [Google Scholar]
- 22.Floridi A, Bellocci M, Paggi MG, Marcante ML, De Martino C. Changes of energy metabolism in the germ cells and Ehrlich ascites tumor cells. Chemotherapy. 1981;27(Suppl 2):50–60. doi: 10.1159/000238045. [DOI] [PubMed] [Google Scholar]
- 23.Mathupala SP, Ko YH, Pedersen PL. Hexokinase-2 bound to mitochondria: cancer’s stygian link to the “Warburg Effect” and a pivotal target for effective therapy. Semin Cancer Biol. 2009;19(1):17–24. doi: 10.1016/j.semcancer.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Floridi ADAS, Paggi MG, Marcante ML, Martino DC, Silvestrini B, Caputo A, Lehninger A. Binding of lonidamine to ehrlich ascites tumor and liver mitochondria. In: TG, et al., editors. Membranes in Tumor Growth. Amsterdam: Elsevier Biomedical Press; 1982. pp. 559–565. [Google Scholar]
- 25.Floridi A, Lehninger AL. Action of the antitumor and antispermatogenic agent lonidamine on electron transport in Ehrlich ascites tumor mitochondria. Arch Biochem Biophys. 1983;226(1):73–83. doi: 10.1016/0003-9861(83)90272-2. [DOI] [PubMed] [Google Scholar]
- 26.Ben-Horin H, Tassini M, Vivi A, Navon G, Kaplan O. Mechanism of action of the antineoplastic drug lonidamine: 31P and 13C nuclear magnetic resonance studies. Cancer Res. 1995;55(13):2814–21. [PubMed] [Google Scholar]
- 27.Mardor Y, Kaplan O, Sterin M, Ruiz-Cabello J, Ash E, Roth Y, et al. Noninvasive real-time monitoring of intracellular cancer cell metabolism and response to lonidamine treatment using diffusion weighted proton magnetic resonance spectroscopy. Cancer Res. 2000;60(18):5179–86. [PubMed] [Google Scholar]
- 28.Ben-Yoseph O, Lyons JC, Song CW, Ross BD. Mechanism of action of lonidamine in the 9L brain tumor model involves inhibition of lactate efflux and intracellular acidification. J Neurooncol. 1998;36(2):149–57. doi: 10.1023/a:1005819604858. [DOI] [PubMed] [Google Scholar]
- 29.Zhou R, Bansal N, Leeper DB, Glickson JD. Intracellular acidification of human melanoma xenografts by the respiratory inhibitor m-iodobenzylguanidine plus hyperglycemia: a 31P magnetic resonance spectroscopy study. Cancer Res. 2000;60(13):3532–6. [PubMed] [Google Scholar]
- 30.Zhou R, Bansal N, Leeper DB, Pickup S, Glickson JD. Enhancement of hyperglycemia-induced acidification of human melanoma xenografts with inhibitors of respiration and ion transport. Acad Radiol. 2001;8(7):571–82. doi: 10.1016/S1076-6332(03)80681-5. [DOI] [PubMed] [Google Scholar]
- 31.Halestrap AP. Mitochondrial Pyruvate Carrier - Kinetics and Specificity for Substrates and Inhibitors. Biochemical Journal. 1975;148(1):85–96. doi: 10.1042/bj1480085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shestov AA, Mancuso A, Lee SC, Guo L, Nelson DS, Roman JC, et al. Bonded Cumomer Analysis of Human Melanoma Metabolism Monitored by 13C NMR Spectroscopy of Perfused Tumor Cells. J Biol Chem. 2016;291(10):5157–71. doi: 10.1074/jbc.M115.701862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nath K, Nelson DS, Putt ME, Leeper DB, Garman B, Nathanson KL, et al. Comparison of the Lonidamine Potentiated Effect of Nitrogen Mustard Alkylating Agents on the Systemic Treatment of DB-1 Human Melanoma Xenografts in Mice. PLoS One. 2016 Jun 10;11(6):e0157125. doi: 10.1371/journal.pone.0157125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loeber R, Michaelson E, Fang Q, Campbell C, Pegg AE, Tretyakova N. Cross-linking of the DNA repair protein Omicron6-alkylguanine DNA alkyltransferase to DNA in the presence of antitumor nitrogen mustards. Chem Res Toxicol. 2008;21(4):787–95. doi: 10.1021/tx7004508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skarsgard LD, Skwarchuk MW, Vinczan A, Kristl J, Chaplin DJ. The cytotoxicity of melphalan and its relationship to pH, hypoxia and drug uptake. Anticancer Res. 1995;15(1):219–23. [PubMed] [Google Scholar]
- 36.Wong P, Lee C, Tannock IF. Reduction of intracellular pH as a strategy to enhance the pH-dependent cytotoxic effects of melphalan for human breast cancer cells. Clin Cancer Res. 2005;11(9):3553–7. doi: 10.1158/1078-0432.CCR-04-2472. [DOI] [PubMed] [Google Scholar]
- 37.McCoy CL, Parkins CS, Chaplin DJ, Griffiths JR, Rodrigues LM, Stubbs M. The effect of blood flow modification on intra- and extracellular pH measured by 31P magnetic resonance spectroscopy in murine tumours. Br J Cancer. 1995;72(4):905–11. doi: 10.1038/bjc.1995.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raghunand N, Altbach MI, van Sluis R, Baggett B, Taylor CW, Bhujwalla ZM, et al. Plasmalemmal pH-gradients in drug-sensitive and drug-resistant MCF-7 human breast carcinoma xenografts measured by 31P magnetic resonance spectroscopy. Biochem Pharmacol. 1999;57(3):309–12. doi: 10.1016/s0006-2952(98)00306-2. [DOI] [PubMed] [Google Scholar]
- 39.Gerweck LE, Seetharaman K. Cellular pH gradient in tumor versus normal tissue: potential exploitation for the treatment of cancer. Cancer Res. 1996;56(6):1194–8. [PubMed] [Google Scholar]
- 40.Floridi A, Bruno T, Miccadei S, Fanciulli M, Federico A, Paggi MG. Enhancement of doxorubicin content by the antitumor drug lonidamine in resistant Ehrlich ascites tumor cells through modulation of energy metabolism. Biochem Pharmacol. 1998;56(7):841–9. doi: 10.1016/s0006-2952(98)00054-9. [DOI] [PubMed] [Google Scholar]
- 41.Kuin A, Aalders M, Lamfers M, van Zuidam DJ, Essers M, Beijnen JH, et al. Potentiation of anti-cancer drug activity at low intratumoral pH induced by the mitochondrial inhibitor m-iodobenzylguanidine (MIBG) and its analogue benzylguanidine (BG) Br J Cancer. 1999;79(5–6):793–801. doi: 10.1038/sj.bjc.6690127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burd R, Lavorgna SN, Daskalakis C, Wachsberger PR, Wahl ML, Biaglow JE, et al. Tumor oxygenation and acidification are increased in melanoma xenografts after exposure to hyperglycemia and meta-iodo-benzylguanidine. Radiat Res. 2003;159(3):328–35. doi: 10.1667/0033-7587(2003)159[0328:toaaai]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 43.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330(7503):1304–5. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Franciosi M, Lucisano G, Lapice E, Strippoli GF, Pellegrini F, Nicolucci A. Metformin therapy and risk of cancer in patients with type 2 diabetes: systematic review. PLoS One. 2013;8(8):e71583. doi: 10.1371/journal.pone.0071583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Z, Ren L, Liu C, Xia T, Zha X, Wang S. Phenformin Induces Cell Cycle Change, Apoptosis, and Mesenchymal-Epithelial Transition and Regulates the AMPK/mTOR/p70s6k and MAPK/ERK Pathways in Breast Cancer Cells. PLoS One. 2015;10(6):e0131207. doi: 10.1371/journal.pone.0131207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Madiraju AK, Erion DM, Rahimi Y, Zhang XM, Braddock DT, Albright RA, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510(7506):542–6. doi: 10.1038/nature13270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marchiq I, Le Floch R, Roux D, Simon MP, Pouyssegur J. Genetic disruption of lactate/H+ symporters (MCTs) and their subunit CD147/BASIGIN sensitizes glycolytic tumor cells to phenformin. Cancer Res. 2015;75(1):171–80. doi: 10.1158/0008-5472.CAN-14-2260. [DOI] [PubMed] [Google Scholar]
- 48.Orecchioni S, Reggiani F, Talarico G, Mancuso P, Calleri A, Gregato G, et al. The biguanides metformin and phenformin inhibit angiogenesis, local and metastatic growth of breast cancer by targeting both neoplastic and microenvironment cells. Int J Cancer. 2015;136(6):E534–44. doi: 10.1002/ijc.29193. [DOI] [PubMed] [Google Scholar]
- 49.Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife. 2014;3:e02242. doi: 10.7554/eLife.02242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nancolas B, Guo L, Zhou R, Nath K, Nelson DS, Leeper DB, et al. The anti-tumour agent lonidamine is a potent inhibitor of the mitochondrial pyruvate carrier and plasma membrane monocarboxylate transporters. Biochem J. 2016;473(7):929–36. doi: 10.1042/BJ20151120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Halestrap AP, Price NT. The proton-linked monocarboxylate transporter (MCT) family: structure, function and regulation. Biochem J. 1999;343(Pt 2):281–99. [PMC free article] [PubMed] [Google Scholar]
- 52.Guo L, Shestov AA, Worth AJ, Nath K, Nelson DS, Leeper DB, et al. Inhibition of mitochondrial complex II by the anti-cancer agent lonidamine. J Biol Chem. 2016;291(1):42–57. doi: 10.1074/jbc.M115.697516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Drose S. Differential effects of complex II on mitochondrial ROS production and their relation to cardioprotective pre- and postconditioning. Biochim Biophys Acta. 2013;1827(5):578–87. doi: 10.1016/j.bbabio.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 54.Yankovskaya V, Horsefield R, Tornroth S, Luna-Chavez C, Miyoshi H, Leger C, et al. Architecture of succinate dehydrogenase and reactive oxygen species generation. Science. 2003;299(5607):700–4. doi: 10.1126/science.1079605. [DOI] [PubMed] [Google Scholar]
- 55.Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci. 2014;39(8):347–54. doi: 10.1016/j.tibs.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blair IA. Endogenous glutathione adducts. Curr Drug Metab. 2006;7(8):853–72. doi: 10.2174/138920006779010601. [DOI] [PubMed] [Google Scholar]
- 57.Shestov AA, Valette J, Deelchand DK, Ugurbil K, Henry PG. Metabolic modeling of dynamic brain 13C NMR multiplet data: concepts and simulations with a two-compartment neuronal-glial model. Neurochem Res. 2012;37(11):2388–401. doi: 10.1007/s11064-012-0782-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shestov AA, Lee SC, Nath K, Guo L, Nelson DS, Roman JC, Leeper DB, Wasik MA, Blair IA, Glickson JD. 13C MRS and LC-MS Flux Analysis of Tumor Intermediary Metabolism. Front Oncol. 2016 Jun 15;6:135. doi: 10.3389/fonc.2016.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shestov AA, Valette J, Ugurbil K, Henry PG. On the reliability of 13C metabolic modeling with two-compartment neuronal-glial models. J Neurosci Res. 2007;85(15):3294–303. doi: 10.1002/jnr.21269. [DOI] [PubMed] [Google Scholar]
- 60.Lee SC, Marzec M, Liu X, Wehrli S, Kantekure K, Ragunath PN, et al. Decreased lactate concentration and glycolytic enzyme expression reflect inhibition of mTOR signal transduction pathway in B-cell lymphoma. NMR Biomed. 2013;26(1):106–14. doi: 10.1002/nbm.2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mendes-Mourao J, Halestrap AP, Crisp DM, Pogson CI. The involvement of mitochondrial pyruvate transport in the pathways of gluconeogenesis from serine and alanine in isolated rat and mouse liver cells. FEBS Lett. 1975;53(1):29–32. doi: 10.1016/0014-5793(75)80674-0. [DOI] [PubMed] [Google Scholar]
- 62.Di Cosimo S, Ferretti G, Papaldo P, Carlini P, Fabi A, Cognetti F. Lonidamine: Efficacy and safety in clinical trials for the treatment of solid tumors. Drugs of Today. 2003;39(3):157–173. doi: 10.1358/dot.2003.39.3.799451. [DOI] [PubMed] [Google Scholar]
- 63.Mansi JL, Degraeff A, Newell DR, Glaholm J, Button D, Leach MO, et al. A Phase-II clinical and pharmacokinetic study of lonidamine in patients with advanced breast-cancer. British Journal of Cancer. 1991;64(3):593–597. doi: 10.1038/bjc.1991.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roehrborn CG. The development of lonidamine for benign prostatic hyperplasia and other indications. Rev Urol. 2005;7(Suppl 7):S12–20. [PMC free article] [PubMed] [Google Scholar]
- 65.Robustelli della Cuna G, Pedrazzoli P. Toxicity and clinical tolerance of lonidamine. Semin Oncol. 1991;18(2 Suppl 4):18–22. [PubMed] [Google Scholar]
- 66.Silvestrini B. Lonidamine: an overview. Semin Oncol. 1991;18(2 Suppl 4):2–6. [PubMed] [Google Scholar]
- 67.Floridi A, Paggi MG, Fanciulli M. Modulation of glycolysis in neuroepithelial tumors. J Neurosurg Sci. 1989;33(1):55–64. [PubMed] [Google Scholar]
- 68.Mansi JL, de Graeff A, Newell DR, Glaholm J, Button D, Leach MO, et al. A phase II clinical and pharmacokinetic study of Lonidamine in patients with advanced breast cancer. Br J Cancer. 1991;64(3):593–7. doi: 10.1038/bjc.1991.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gatzemeier U, Toomes H, Picollo R, Christoffel V, Lucker PW, Ulmer J. Single- and multiple dose pharmacokinetics of lonidamine in patients suffering from non-small-cell lung cancer. Arzneimittelforschung. 1991;41(4):436–9. [PubMed] [Google Scholar]
- 70.Dogliotti L, Danese S, Berruti A, Zola P, Buniva T, Bottini A, et al. Cisplatin, epirubicin, and lonidamine combination regimen as first-line chemotherapy for metastatic breast cancer: a pilot study. Cancer Chemother Pharmacol. 1998;41(4):333–8. doi: 10.1007/s002800050747. [DOI] [PubMed] [Google Scholar]
- 71.Gebbia V, Borsellino N, Testa A, Latteri MA, Milia V, Valdesi M, et al. Cisplatin and epirubicin plus oral lonidamine as first-line treatment for metastatic breast cancer: a phase II study of the Southern Italy Oncology Group (GOIM) Anticancer Drugs. 1997;8(10):943–8. doi: 10.1097/00001813-199711000-00005. [DOI] [PubMed] [Google Scholar]
- 72.Lopez M, Vici P, Di Lauro L, Paoletti G, Gionfra T, Conti F, et al. Intrapatient comparison of single-agent epirubicin with or without lonidamine in metastatic breast cancer. Eur J Cancer. 1995;31A(10):1611–4. doi: 10.1016/0959-8049(95)00200-3. [DOI] [PubMed] [Google Scholar]
- 73.Nistico C, Garufi C, Milella M, D’Ottavio AM, Vaccaro A, Fabi A, et al. Weekly epirubicin plus lonidamine in advanced breast carcinoma. Breast Cancer Res Treat. 1999;56(3):233–7. doi: 10.1023/a:1006213815195. [DOI] [PubMed] [Google Scholar]
- 74.Comella P, Frasci G, Panza N, Manzione L, Lorusso V, Di Rienzo G, et al. Cisplatin, gemcitabine, and vinorelbine combination therapy in advanced non-small-cell lung cancer: a phase II randomized study of the Southern Italy Cooperative Oncology Group. J Clin Oncol. 1999;17(5):1526–34. doi: 10.1200/JCO.1999.17.5.1526. [DOI] [PubMed] [Google Scholar]
- 75.Contu A, Olmeo NA, Pani P, Deriu A, Ortu S, Paga C. Lonidamine in non-small-cell lung cancer: a phase II study. Tumori. 1991;77(1):52–5. doi: 10.1177/030089169107700113. [DOI] [PubMed] [Google Scholar]
- 76.Portalone L, Lombardi A, Antilli A, Cruciani AR, Magliacani V, Mugnaini L, et al. Treatment of inoperable non-small cell lung carcinoma stage IIIb and IV with cisplatin, epidoxorubicin, vindesine and lonidamine: a phase II study. Tumori. 1999;85(4):239–42. doi: 10.1177/030089169908500405. [DOI] [PubMed] [Google Scholar]
- 77.Bottalico C, Lorusso V, Brandi M, Micelli G, Rella CA, Coviello M, et al. Correlation between HPLC-determined lonidamine serum levels and clinical response in patients with advanced ovarian cancer. Anticancer Res. 1996;16(6B):3865–9. [PubMed] [Google Scholar]
- 78.De Lena M, Lorusso V, Bottalico C, Brandi M, De Mitrio A, Catino A, et al. Revertant and potentiating activity of lonidamine in patients with ovarian cancer previously treated with platinum. J Clin Oncol. 1997;15(10):3208–13. doi: 10.1200/JCO.1997.15.10.3208. [DOI] [PubMed] [Google Scholar]
- 79.De Lena M, Lorusso V, Latorre A, Fanizza G, Gargano G, Caporusso L, et al. Paclitaxel, cisplatin and lonidamine in advanced ovarian cancer. A phase II study. Eur J Cancer. 2001;37(3):364–8. doi: 10.1016/s0959-8049(00)00400-7. [DOI] [PubMed] [Google Scholar]
- 80.Gadducci A, Brunetti I, Muttini MP, Fanucchi A, Dargenio F, Giannessi PG, et al. Epidoxorubicin and lonidamine in refractory or recurrent epithelial ovarian cancer. Eur J Cancer. 1994;30A(10):1432–5. doi: 10.1016/0959-8049(94)00231-s. [DOI] [PubMed] [Google Scholar]
- 81.Colella E, Merlano M, Blengio F, Angelini F, Ausili Cefaro GP, Scasso F, et al. Randomised phase II study of methotrexate (MTX) versus methotrexate plus lonidamine (MTX + LND) in recurrent and/or metastatic carcinoma of the head and neck. Eur J Cancer. 1994;30A(7):928–30. doi: 10.1016/0959-8049(94)90116-3. [DOI] [PubMed] [Google Scholar]
- 82.Magno L, Terraneo F, Bertoni F, Tordiglione M, Bardelli D, Rosignoli MT, et al. Double-blind randomized study of lonidamine and radiotherapy in head and neck cancer. Int J Radiat Oncol Biol Phys. 1994;29(1):45–55. doi: 10.1016/0360-3016(94)90225-9. [DOI] [PubMed] [Google Scholar]
- 83.Magno L, Terraneo F, Ciottoli GB. Lonidamine and radiotherapy in head and neck cancers. A pilot study. Oncology. 1984;41(Suppl 1):113–5. doi: 10.1159/000225898. [DOI] [PubMed] [Google Scholar]
- 84.Amadori D, Frassineti GL, De Matteis A, Mustacchi G, Santoro A, Cariello S, et al. Modulating effect of lonidamine on response to doxorubicin in metastatic breast cancer patients: results from a multicenter prospective randomized trial. Breast Cancer Res Treat. 1998;49(3):209–17. doi: 10.1023/a:1006063412726. [DOI] [PubMed] [Google Scholar]
- 85.Berruti A, Bitossi R, Gorzegno G, Bottini A, Alquati P, De Matteis A, et al. Time to progression in metastatic breast cancer patients treated with epirubicin is not improved by the addition of either cisplatin or lonidamine: final results of a phase III study with a factorial design. J Clin Oncol. 2002;20(20):4150–9. doi: 10.1200/JCO.2002.08.012. [DOI] [PubMed] [Google Scholar]
- 86.Calabresi F, Di Lauro L, Marolla P, Curcio CG, Paoletti G, Calabro A, et al. Fluorouracil, doxorubicin, and cyclophosphamide versus fluorouracil, doxorubicin, and cyclophosphamide plus lonidamine for the treatment of advanced breast cancer: a multicentric randomized clinical study. Semin Oncol. 1991;18(2 Suppl 4):66–72. [PubMed] [Google Scholar]
- 87.Dogliotti L, Berruti A, Buniva T, Zola P, Bau MG, Farris A, et al. Lonidamine significantly increases the activity of epirubicin in patients with advanced breast cancer: results from a multicenter prospective randomized trial. J Clin Oncol. 1996;14(4):1165–72. doi: 10.1200/JCO.1996.14.4.1165. [DOI] [PubMed] [Google Scholar]
- 88.Pacini P, Rinaldini M, Algeri R, Guarneri A, Tucci E, Barsanti G, et al. FEC (5-fluorouracil, epidoxorubicin and cyclophosphamide) versus EM (epidoxorubicin and mitomycin-C) with or without lonidamine as first-line treatment for advanced breast cancer. A multicentric randomised study. Final results. Eur J Cancer. 2000;36(8):966–75. doi: 10.1016/s0959-8049(00)00068-x. [DOI] [PubMed] [Google Scholar]
- 89.Buccheri G, Ferrigno D. A randomised trial of MACC chemotherapy with or without lonidamine in advanced non-small cell lung cancer. Cuneo Lung Cancer Study Group (CuLCaSG) Eur J Cancer. 1994;30A(10):1424–31. doi: 10.1016/0959-8049(94)00286-e. [DOI] [PubMed] [Google Scholar]
- 90.De Marinis F, Rinaldi M, Ardizzoni A, Bruzzi P, Pennucci MC, Portalone L, et al. The role of vindesine and lonidamine in the treatment of elderly patients with advanced non-small cell lung cancer: a phase III randomized FONICAP trial. Italian Lung Cancer Task Force Tumori. 1999;85(3):177–82. doi: 10.1177/030089169908500306. [DOI] [PubMed] [Google Scholar]
- 91.Gatzemeier U, Cavalli F, Haussinger K, Kaukel E, Koschel G, Martinelli G, et al. Phase III trial with and without lonidamine in non-small cell lung cancer. Semin Oncol. 1991;18(2 Suppl 4):42–8. [PubMed] [Google Scholar]
- 92.Ianniello GP, De Cataldis G, Comella P, Scarpati MD, Maiorino A, Brancaccio L, et al. Cisplatin, epirubicin, and vindesine with or without lonidamine in the treatment of inoperable nonsmall cell lung carcinoma: a multicenter randomized clinical trial. Cancer. 1996;78(1):63–9. doi: 10.1002/(SICI)1097-0142(19960701)78:1<63::AID-CNCR11>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 93.Scarantino CW, McCunniff AJ, Evans G, Young CW, Paggiarino DA. A prospective randomized comparison of radiation therapy plus lonidamine versus radiation therapy plus placebo as initial treatment of clinically localized but nonresectable nonsmall cell lung cancer. Int J Radiat Oncol Biol Phys. 1994;29(5):999–1004. doi: 10.1016/0360-3016(94)90394-8. [DOI] [PubMed] [Google Scholar]
- 94.Di Cosimo S, Ferretti G, Papaldo P, Carlini P, Fabi A, Cognetti F. Lonidamine: efficacy and safety in clinical trials for the treatment of solid tumors. Drugs Today (Barc) 2003;39(3):157–74. doi: 10.1358/dot.2003.39.3.799451. [DOI] [PubMed] [Google Scholar]
- 95.Robins HI, Longo WL, Lagoni RK, Neville AJ, Hugander A, Schmitt CL, et al. Phase I trial of lonidamine with whole body hyperthermia in advanced cancer. Cancer Res. 1988;48(22):6587–92. [PubMed] [Google Scholar]