

Abstract
Protein hydroxylation is a post-translational modification catalyzed by 2-oxoglutarate-dependent dioxygenases. The hydroxylation modification can take place on various amino acids, including but not limited to proline, lysine, asparagine, aspartate and histidine. A classical example of this modification is hypoxia inducible factor alpha (HIF-α) prolyl hydroxylation, which affects HIF-α protein stability via the Von-Hippel Lindau (VHL) tumor suppressor pathway, a Cullin 2-based E3 ligase adaptor protein frequently mutated in kidney cancer. In addition to protein stability regulation, protein hydroxylation may influence other post-translational modifications or the kinase activity of the modified protein (such as Akt and DYRK1A/B). In other cases, protein hydroxylation may alter protein-protein interaction and its downstream signaling events in vivo (such as OTUB1, MAPK6 and eEF2K). In this review, we highlight the recently identified protein hydroxylation targets and their pathophysiological roles, especially in cancer settings. Better understanding of protein hydroxylation will help identify novel therapeutic targets and their regulation mechanisms to foster development of more effective treatment strategies for various human cancers.
Keywords: hydroxylation, Hypoxia Inducible Factor alpha, Von-Hippel Lindau (VHL), Human Cancer
1. Introduction
1.1 2-oxoglutarate-dependent dioxygenases
2-oxoglutarate (2-OG)-dependent dioxygenases are iron-containing enzymes that pair substrate oxidation to the conversion of 2-OG to succinate and carbon dioxide [1]. The 2-OG-dependent oxygenase catalyzes the hydroxylation of proline and lysine residues, which was first identified in collagen biosynthesis [2]. Subsequently, it has been demonstrated that this type of post-translational modification also affects intracellular proteins. Specifically, the hypoxia-inducible factors (HIFs) have been found to be hydroxylated on both proline and asparagine residues by the prolyl hydroxylase domain proteins PHD1, PHD2, PHD3 (also called Egl nine homolog EglN2, EglN1, and EglN3) and factor inhibiting HIF (FIH), respectively [3]. Up to date, about seventy 2-OG dependent dioxygenases have been identified in the human genome [4]. According to their biological functions, it is possible to distinguish them into three major subclasses: histone demethylases, DNA/RNA demethylases/hydroxylases and protein hydroxylases [5].
Histone methylation is performed by the family of lysine and arginine methyltransferases, which can transfer up to three methyl groups to histone lysines and asparagines, subsequently triggering different activities according to the recruitment of specific effector proteins by the readers [6]. Importantly, the JumonjiC (JMJC) domain-containing histone demethylases, which can be further divided into seven subfamilies (KDM2-8), remove methyl groups from all three methyl lysine states, with concomitant production of succinate, carbon dioxide, and subsequent release of formaldehyde [7, 8].
Eukaryotic DNA and RNA methylation is primarily catalyzed by DNA and RNA methyltransferases, respectively, which epigenetically modulate cell fate without altering the nucleic acid sequence. DNA/RNA can be also methylated by endogenous and/or exogenous alkylating agents in a process referred to as methylation damage [9]. In humans, both regulatory and aberrant methylation can be oxidatively reversed by a non-heme iron-dependent dioxygenase superfamily composed of nine members (alkylation repair homologs ALKBH1-8, and fat-mass and obesity-associated FTO) [9]. Recently, the ten-eleven translocation (TET)/J-binding protein (JBP) family proteins (TET1, TET2, and TET3; JBP1 and JBP2), also belonging to the family of iron- and 2-OG-dependent dioxygenases, have been found to oxidize 5-methylcytosine [10].
In this review, we will mainly focus on protein hydroxylases. The primary residue to be hydroxylated in proteins is proline, and accordingly the hydroxylases that catalyze this reaction are called prolyl hydroxylases. In addition to proline, asparagine, aspartate, lysine and histidine have also been found to be hydroxylated in cells [11–14].
1.2 Prolyl Hydroxylases
The HIFs are transcription factors that coordinate cellular responses to low oxygen levels, aiming at increased oxygen delivery and reduced oxygen consumption [3]. Transcriptionally active HIFs are heterodimers composed of an alpha and ARNT (aryl hydrocarbon receptor nuclear translocator) subunits. In humans, the alpha subunit has three isoforms, namely HIF-1α, HIF-2α and HIF-3α. Different from the ARNT subunit that is constitutively expressed, the protein stabilization of the alpha subunit is oxygen-sensitive [3].
In normoxic conditions, HIFs have been found to be hydroxylated on one or two proline residues by the prolyl hydroxylase domain proteins PHD1/EglN2, PHD2/EglN1, and PHD3/EglN3 [15]. Specifically in regards to HIF-1α, its hydroxylation on proline 402 (Pro402) and 564 (Pro564) by PHDs triggers its recognition by a multimeric E3 ubiquitin ligase complex formed by the von Hippel-Lindau tumor suppressor protein (pVHL), elongin B and C, Cullin 2 (CUL2) and RING-box 1 (RBX1) proteins. Homologous to the Skp, Cullin, F-box containing (SCF) family of E3 ubiquitin ligase complexes, this complex targets HIF-1α for ubiquitin-mediated proteasomal degradation (Fig. 1A) [16]. It is important to point out that even though all three enzymes contribute to HIF regulation, PHD2/EglN1 is the major prolyl hydroxylase mediating HIF-1α hydroxylation in vivo [17–20].
Figure 1. HIF-1α regulation by hydroxylation.

(A) In the presence of oxygen, HIF-1α is hydroxylated by the prolyl hydroxylases PHD/EglN1, 2, 3 on prolines 402 and 564. This leads to HIF-1α recognition by the ubiquitin ligase complex composed of the von Hippel Lindau protein (cullin2) and RBX1 (RING-box1), which promotes the polyubiquitination of HIF-1α followed by its proteasomal degradation. (B) The lack of oxygen prevents the hydroxylation of HIF-1α by PHD, leading to its stabilization. HIF-1α can then migrate to the nucleus and associate with ARNT (aryl hydrocarbon receptor nuclear translocator) and the cofactor p300/CBP (cyclic AMP response element-binding protein). The HIF-1 complex binds to and induces the transcription of genes containing hypoxia-responsive elements (HRE) in their promoter region. (C) FIH (factor inhibiting HIF) can hydroxylate HIF on the asparagine 803 (Asn803) residue, which affects its binding with the transcriptional co-activator p300/CBP, thereby inhibiting HIF transcriptional activity.
Under hypoxic conditions, HIFs are not hydroxylated and, thus, can bind to the hypoxia-response elements (HRE) in the promoter of more than one hundred target genes involved in cell survival in low oxygen conditions [21]. These targeted genes include metabolic genes such as the glucose transporter 1 (GLUT1), the vascular endothelial growth factor (VEGF) that promotes angiogenesis, and erythropoietin (EPO) that controls erythropoiesis. Transforming growth factor alpha (TGFα), implicated in cell proliferation and survival, and C-X-C chemokine receptor type 4 (CXCR4), important for cell migration and invasion, are also characterized HIF targets (Fig. 1B) [22]. As all of these pathways promote the growth and progression of many tumors, HIFs are generally considered as attractive therapeutic targets in cancer.
Since the identification of HIF hydroxylation, much effort has been devoted to better understand whether and how prolyl hydroxylation regulates signaling pathways beyond the HIF-pathway. Over the course of the last decades, a handful of substrates have been identified as bona fide PHD substrates. Among them, the centrosomal protein of 192 kDa CEP192 and the inhibitor of nuclear factor kappa-B kinase subunit beta IKBKB are PHD1 substrates [23, 24], while the isoform 2 of pyruvate kinase PKM2 and the β-adrenergic receptor II are PHD3 substrates. Since these targets have been reviewed recently [5, 25], this review will largely focus on other most recently identified substrates.
1.3 Regulation of Prolyl Hydroxylases
PHD/EglN protein expression is tissue- and context-dependent. PHD2/EglN1 is ubiquitously expressed, whereas PHD1/EglN2 and PHD3/EglN3 are mainly observed in testis and heart, respectively [26]. PHDs are transcriptionally regulated by their downstream target HIF pathway [27, 28]. Of note, HIF-induced PHD3/EglN3 expression provides a negative feedback regulating HIFs in hypoxic conditions [29]. Furthermore, PHD2/EglN1 and PHD1/EglN2 can be transcriptionally. regulated by estrogen pathways in breast cancer cells [30–34]. At the protein level, the abundance of PHD3/EglN3, and to a lesser extent of PHD1/EglN2, is regulated by the E3 ubiquitin ligases SIAH1/2 in a proteasomal-dependent manner [35]. However, the manipulation of PHD/EglN catalytic activities has been a mystery for a long time. Previous research has reviewed extensively on how PHDs/EglNs serve as important nutrient sensors since their activity is regulated by the availability of oxygen, Fe2+, ascorbic acid and 2-OG [5]. Here we summarize the latest development on how PHD activity can be regulated.
Recent work from different groups reported that 2-hydroxyglutarate (2-HG), a small molecule produced by mutant isocitrate dehydrogenase 1 and 2 (IDH1/2) in glioblastoma, could manipulate dioxygenase catalytic activities, including PHDs/EglNs [36–40]. Mechanistically, 2-HG competes with 2-OG to bind with PHDs, leading to the accumulation of HIF-α [36]. However, some other findings demonstrated that the (R)-enantiomer of 2-HG, opposing to (S)-2HG, can largely stimulate PHD/EglN activities, diminish HIF-α levels, and contribute to the proliferation of human glioma and leukemia cells, therefore demonstrating that HIF-α functions as a tumor suppressor in the tissue-context of brain tumors or leukemia [38, 39].
More recently, L-cysteine has been reported to induce recombinant PHD2/EglN1 catalytic activity in an ascorbate-free condition in vitro and in cells [41]. Free cysteine was shown to protect PHDs/EglNs from self-catalyzed inactivation due to its oxidation [42–45]. Accordingly, the PHD/EglN proteins, previously recognized as oxygen sensors [3], now have also been defined as cysteine sensors. Physiologically, L-cysteine-mediated PHD/EglN protection can be antagonized by the presence of L-glutamate through inhibiting the glutamate-cysteine antiporter xCT [46, 47]. In certain cancers, such as triple-negative breast cancer (TNBC), the paracrine secretion of glutamate decreases the amount of free cysteine, leading to PHD/EglN inactivation and subsequent HIF accumulation [41], which has been shown to promote the growth, invasion and metastasis in both normoxic and hypoxic conditions [48, 49].
1.4 Other protein hydroxylases
In addition to prolyl hydroxylation, there have been reports showing that some proteins can be hydroxylated on asparagine, aspartate, lysine and histidine residues. For example, factor inhibiting HIF (FIH) can hydroxylate HIF on the asparagine 803 (Asn803) residue, which affects its binding with the transcriptional co-activator p300/cyclic AMP response element-binding protein (CBP), thereby inhibiting HIF transcriptional activity (Fig. 1C) [12]. It has been found that FIH can also hydroxylate aspartate residues in cytoskeletal ankyrinR and ankyrinB, leading to a decrease in their interaction with the cytoplasmic domain of band3 (CDB3) [13]. JMJD6, an enzyme reported to contain both arginine demethylase and lysine hydroxylase activity, has been shown to hydroxylate the splicing factor U2 small nuclear ribonucleoprotein auxiliary factor 65-kDa subunit (U2AF65) on lysine residues, leading to the alteration of some endogenous and reporter gene alternative RNA splicing [11]. Moreover, JMJD6 was shown to promote lysine hydroxylation on histones [50]. The dioxygenase NO66 can act as both a histone demethylase and a ribosomal histidine hydroxylase, catalyzing the hydroxylation of 60S ribosomal protein L8 on histidine 216 (His216), which raises the fascinating idea of a potential translational regulation by hydroxylases [14].
1.5 Mouse models of key proline or asparagine hydroxylases
A functional hypoxic response is essential for survival, as highlighted by the embryonic lethality of HIF-1α [51] and HIF-2α knockout mice [52]. Genetic inactivation of VHL in the mouse germ line also leads to embryonic death during mid-gestation, due to abnormal placental vascularization [53].
The prominent role of PHD2/EglN1 in oxygen sensing under basal physiological conditions is evidenced by the fact that PHD2/EglN1 ablation, like genetic inactivation of VHL or HIFs, results in severe placental and cardiac defects and, eventually, embryonic death between embryonic days 12.5 and 14.5, whereas PHD1/EglN2−/− and PHD3/EglN3−/− mice are viable and apparently normal [19, 54]. PHD1/EglN2−/− mouse shows the hypoxia tolerance due to its glucose metabolic switch from oxidative towards anaerobic ATP production [55]. Moreover, while the conditional deletion of PHD2/EglN1 in adult mice causes hyperactive angiogenesis and blood vessel dilation, the conditional knockout of PHD1/EglN2 or PHD3/EglN3 does not lead to angiogenic anomalies [19, 54] (Table 1). Adult mice lacking PHD2/EglN1 also display a severe erythrocytosis, due to the activation of the erythropoietin (EPO) pathway in the kidney. By contrast, PHD1/EglN2 or PHD3/EglN3 deficiency does not cause any apparent dysfunction. A moderate erythrocytosis occurs in PHD1/EglN2 and PHD3/EglN3 double knockout mouse, and this is associated with the activation of the hepatic HIF-2α/EPO pathway [56].
Table 1.
Mouse Models of Key Proline or Asparagine Hydroxylases
| Genotype | Type of knockout | Phenotype | Reference |
|---|---|---|---|
| PHD1/EglN2−/− |
|
|
|
| PHD2/EglN1−/− |
|
|
|
| PHD3/EglN3−/− |
|
|
|
|
PHD1/EglN2−/− PHD3/EglN3−/− |
|
|
|
| FIH−/− |
|
|
|
Different studies have investigated the role of PHD2/EglN1 in various tissues using tissue-specific knockout mice. Specifically, PHD2/EglN1 deletion in the epidermal or dermal tissues has been shown to improve wound healing and minimize ischemic injury by increasing HIF-1α cellular levels, therefore leading to the transcription of downstream angiogenic factors such as VEGF [57, 58]. Myeloid-specific PHD2/EglN1 knockout in mice promotes plaque progression by enhancing adhesion molecule expression on granulocytes and monocytes [59]. This is also achieved through HIF-1α pathway, as evidenced by the loss of the pro-atherogenic phenotype by PHD2/HIF-1α double knockout mice [60].
Distinctively, FIH deficient mice do not show apparent defects in the classical pathways controlled by HIF, such as angiogenesis, erythropoiesis, or development. Instead, FIH knockout mice demonstrate the features that highlight this dioxygenase as an important metabolic regulator [61]. Aside from the reduced body weight, these mice present an elevated metabolic rate, hyperventilation, improved glucose and lipid homeostasis. They are also resistant to high-fat-diet-induced weight gain and hepatic steatosis [61]. Interestingly, mice lacking FIH specifically in neurons display some of the main metabolic phenotypes of the global knockout animals, suggesting that FIH may regulate metabolism mainly through its function on the nervous system [61].
1.6 Therapeutic implications of PHD/EglN inhibitions in diseases
As discussed earlier, PHD2 conditional deletion leads to HIF stabilization, and consequently, increased levels of VEGF and EPO and enhanced angiogenesis [19, 58, 62]. These observations suggested that PHD inhibitors could be beneficial for patients affected by anemia and ischemia-related diseases. HIF stabilization may also be useful for the treatment of inflammatory diseases. PHD1 and FIH have been shown to enhance inflammation [63, 64], and PHD inhibitors such as dimethyloxalylglycine (DMOG) and FG-4497 significantly improve murine models of colitis [65, 66], suggesting that a hydroxylase inhibitor can be beneficial for blunting excessive inflammation. Over the last few years in-depth work has therefore been made to find PHD inhibitors by several means, such as in silico drug screening [67], combinatorial libraries [68] and high-throughput screening [69]. These efforts have led to the discovery of several compounds, four of which are currently being tested in human clinical trials [70].
Nevertheless, PHD inhibition can potentially cause deleterious effects that must be taken into account. HIF-α expression is frequently increased in numerous human diseases like tumors, erythrocytosis and pulmonary arterial hypertension, and some of its downstream targets can be inhibited for the treatment of these conditions [22]. However, it is important to point out that the role of HIFs in cancer is more complex than initially thought, as PHD inhibition has been found to impair tumor growth and invasiveness [71]. In another study, PHD2 inhibition in tumor cells has been shown to stimulate vessel formation yet resulting in tumor growth impairment. However, this effect was largely independent of HIF pathway, and was ascribed to the antiproliferative activity of the PHD2-driven TGFβ signaling pathway [72]. In order to minimize unwanted side effects, tissue-specific PHD inhibition should then be considered when treating specific pathological conditions. For instance, HIF can be selectively activated in alveolar epithelial cells when PHD inhibitors are inhaled [73]. Moreover, efforts should be made to seek isoform-selective PHD inhibitors, given the different expression and biological functions of individual PHDs [32].
1.7 Methods for the identification of novel potential protein hydroxylase substrates
Protein association detecting methods such as yeast two-hybrid screens and affinity purification (AP)-MS technology allowed to pinpoint certain IκB and Notch family members and ankyrin repeat and SOCS box protein 4 (ASB4) as additional FIH substrates [74–76], as well as the activating transcription factor 4 (ATF4) as a potential PHD3/EglN3 substrate [77].
An in silico screening for the consensus sequence LXXLAP (with X referring to any amino acid) allowed the finding of other PHD/EglN substrates involved in glucose metabolism and cell cycle regulation [23, 78]. Another approach based on a pharmacological substrate-trapping strategy linked to mass spectrometry has enabled the challenging detection of transient enzyme-substrate interactions, thus leading to the detection of several other FIH substrates [79]. It has thus become clear that hydroxylases have a large panel of downstream substrates, therefore regulating several cellular signaling pathways beyond the oxygen sensing pathway, largely through modulating HIF stability. However, since only a few experimental approaches proved efficient in detecting new substrates, little progress has been made so far in this field. As such, searching for new hydroxylation substrates becomes increasingly necessary in order to improve our understanding of how hydroxylation can modulate cellular responses to stimuli other than the hypoxic condition.
It is also important to point out that it is relatively difficult to differentiate between oxidation and hydroxylation from only mass spectrometry since both modifications can occur on a variety of amino acid side chains. Therefore, mass spectrometry fragmentation data need to be at high resolution and high coverage to confirm the assignment for the modification as previously shown [80]. Many hydroxylation events detected by mass spectrometry may not be mediated by prolyl hydroxylases. Therefore, it is critical to perform in vitro hydroxylation assays to validate whether these sites detected from mass spectrometry can be hydroxylated by various prolyl hydroxylases. Moreover, it is equally important to further investigate whether these sites are functionally important in a physiologically relevant system.
Here we give an overview of recently identified hydroxylation substrates (Table 2), primarily focusing on the biological readouts and the potential pathological role of this intriguing post-translational modification.
Table 2.
List of Recently Identified Hydroxylation Targets
| Protein | Potential hydroxylase | Hydroxylated residues | Effects of hydroxylation | Reference |
|---|---|---|---|---|
| FOXO3a | PHD1/EglN2 | Pro426, Pro437 | Dissociation from USP9x deubiquitinase, FOXO3a proteasomal degradation and subsequent cyclinD1 stabilization | Zheng et al. [87] |
| DYRK1A, DYRK1B | PHD1/EglN2 | Unknown | Increase of DYRK1A/B phosphorylation and kinase activity, disruption of phosphorylated ID2 interaction with pVHL. HIF2a ubiquitination and degradation | Lee et al. [90] |
| NDRG3 | PHD2/EglN1 | Pro294 | Increased association with pVHL, subsequent NDRG3 ubiquitination and degradation, decrease of NDRG3-regulated RAF-ERK1/2 kinase signaling | Lee et al. [94] |
| Akt | PHD2/EglN1 | Pro125, Pro313 | Increased interaction with pVHL and PP2a phosphatase, decrease Akt phosphorylation on Thr308 and Ser473 residues and therefore impaired Akt activity, subsequent decrease of Akt-mediated cell proliferation and tumorigenesis | Guo et al. [98] |
| EPOR | PHD3/EglN3 | Pro419, Pro426 | Increased binding with pVHL, EPOR proteasomal degradation, subsequent decrease of EPO-induced JAK-STAT signaling pathway and erythropoiesis | Heir et al. [103] |
| OTUB1 | FIH | Asn22 | Alteration of OTUB1 interaction with proteins important in cellular metabolism | Scholz et al. [104] |
| MAPK6 | PHD3/EglN3 | Pro25 | Dissociation from E3 ligase HUWE1 and subsequent MAPK6 protein stabilization | Rodriguez et al. [80] |
| RIPK4 | FIH | Asn | Modification of RIPK4 kinase activity, increased Wnt signaling pathway activity | Rodriguez et al. [80] |
| eEF2K | Unknown | Pro93 | Disruption of eEF2K-calmodulin complex, subsequent inhibition of eEF2K kinase activity and preservation of eEF2-dependent translation elongation and protein synthesis | Moore et al. [106] |
| β and γ actin | PHD3/EglN3 | Pro307, Pro322 | Impairment of actin polymerization | Luo et al. [109] |
| TR-α | PHD2/EglN1, PHD3/EglN3 | Pro160, Pro162 | Increased TR-α interaction with transcriptional co-repressor NCOR2, increased transcription of PLN | Xie et al. [112] |
| p53 | PHD1/EglN2 | Unknown | Binding with p33, phosphorylation on p53 Ser15 | Deschoemaeker et al. [113] |
| CERKL | PHD1/EglN2, PHD3/EglN3 | Unknown | Regulation of CERKL protein stability | Chen et al. [115] |
| FLNA | PHD2/EglN1 | Multiple Pro residues | Increased binding with pVHL E3 ligase and FLNA proteasomal degradation. | Segura et al. [116] |
| p53 | JMJD6 | Lys382 | Inhibition of p53 transcriptional activity | Wang et al. [121] |
2. Novel Hydroxylation Targets and Their Cellular Functions
2.1 FOXO3a prolyl hydroxylation
Forkhead box-O (FOXO) tumor suppressors function to suppress cell proliferation and cell survival by transcriptionally activating specific gene targets that are wired into diverse cancer regulatory pathways. Activation of the phosphatidylinositide 3-kinase (PI3K) by extracellular growth signals leads to FOXO phosphorylation at three conserved serine/threonine (Ser/Thr) sites by the serine/threonine kinase Akt, whereupon the FOXOs are translocated to the cytoplasm and degraded [81]. FOXO3a protein stability can also be regulated by the regulatory protein IkappaB or extracellular signal-regulated kinase (ERK)-mediated phosphorylation [82, 83].
Notably, the role of the FOXOs in cancer has recently received increasing support from genetic studies in mouse and human cancer. FOXOs are deleted or mutated in a variety of human cancers, such as leukemia and prostate cancer [84, 85]. Overexpression of FOXO3a inhibits breast tumor cell growth in vitro and tumor size in vivo, suggesting that FOXO3a acts largely as a tumor suppressor in breast cancer [82, 83]. Consistent with these observations from human cancer, the mouse genetic studies from Depinho group have established that broad somatic deletion of all three FOXOs (FOXO1, 3 and 4) engenders a cancer-prone condition, thus providing formal proof that FOXOs function as tumor suppressors in vivo [86].
Zheng et al. used an in vitro hydroxylation screening with recombinant PHD1/EglN2 and identified that FOXO3a is a bona fide PHD1/EglN2 substrate. Mechanistically, FOXO3a can be hydroxylated by PHD1/EglN2 on two different proline residues (Pro426 and 437), which triggers its dissociation from the ubiquitin specific peptidase 9, X-linked (USP9x) deubiquitinase (Fig. 2) [87]. As a result, FOXO3a undergoes proteasomal degradation. Since FOXO3a was reported to be a transcriptional repressor for cyclin D1, its destabilization leads to cyclin D1 accumulation. This study provides mechanistic insight into how PHD1/EglN2 transcriptionally regulates cyclin D1 in an enzymatic-dependent fashion, thereby contributing to breast tumorigenesis. In addition, it sheds light on the important role of FOXO3a and USP9x in breast cancer. Intriguingly, pVHL does not regulate FOXO3a protein stability in this setting (Zhang Q., unpublished finding). Thus, it remains to be determined which E3 ligase may mediate the hydroxylated FOXO3a for degradation.
Figure 2. FOXO3a regulation by hydroxylation.
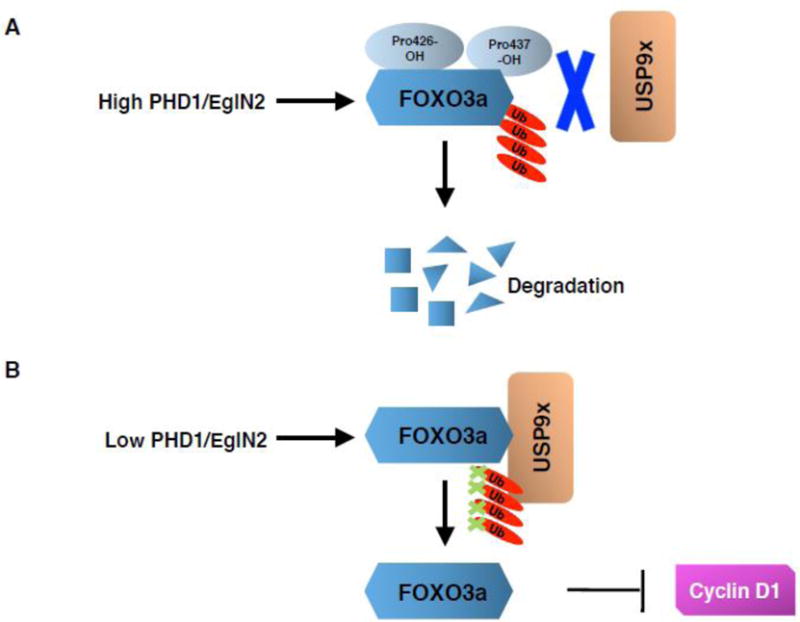
(A) The prolyl hydroxylase PHD1/EglN2 hydroxylates FOXO3a (Forkhead box-O3a) transcription factor on two proline residues (Pro426 and 437), thus preventing the binding with USP9x (ubiquitin specific peptidase 9, X-linked) deubiquitinase. This promotes FOXO3a ubiquitination and proteasomal degradation. (B) In the case of low activity levels of EglN2 (PHD1), e. g. under hypoxia, USP9x binds to FOXO3a thus preventing its ubiquitination and increasing its stability. FOXO3a being a transcriptional repressor for cyclin D1, its stabilization leads to a decreased cyclin D1 expression.
2.2 DYRK1A and DYRK1B prolyl hydroxylation
The inhibitor of differentiation (ID) protein family members (including ID1, 2, 3 and 4) are master regulators of stem cell function [88, 89]. During tumorigenesis, cancer cells hijacked their functions, therefore contributing to cancer stem cell function and tumorigenesis. However, the regulatory pathway of ID proteins remains largely undefined. By using glioblastoma cell as a model system, a recent paper by Lee et al. showed that dual-specificity tyrosine-(Y)-phosphorylation regulated kinases 1A and B (DYRK1A and DYRK1B) phosphorylate ID2 on threonine 27 (Thr27). Interestingly, PHD1/EglN2 hydroxylates DYRK1A and DYRK1B, thus promoting their phosphorylation and kinase activity in cells. As a result, ID2 is phosphorylated and prevented from interacting with pVHL, therefore preserving pVHL-dependent HIF-2α ubiquitination and degradation (Fig. 3A) [90].
Figure 3. DYRK1A/B regulation by hydroxylation.

(A) PHD1/EglN2 triggers DYRK1A and B (dual-specificity tyrosine-(Y)-phosphorylation regulated kinases 1A and B) hydroxylation, thus promoting their phosphorylation and kinase activity. Subsequently, ID2 (inhibitor of differentiation 2) is phosphorylated and prevented from interacting with pVHL ubiquitinase complex, therefore preserving pVHL-dependent HIF-2α ubiquitination and proteasomal degradation. (B) Under hypoxia or PHD1 depletion, DYRK1A and B cannot be efficiently hydroxylated or phosphorylated, thus preventing ID2 phosphorylation on Thr27. This promotes ID2 binding with pVHL that disturbs the scaffold protein CUL2 interaction with pVHL E3 ligase complex. As a consequence, pVHL cannot efficiently ubiquitinate HIF-2α. HIF-2α accumulation is then followed by transcriptional regulation of its downstream target gene expression.
On the other hand, in the presence of hypoxia or potentially PHD1 depletion, DYRK1A and DYRK1B cannot be efficiently hydroxylated or phosphorylated. Therefore, ID2 is not phosphorylated on Thr27, which subsequently promotes its binding with pVHL that displaces the scaffold protein CUL2 from its interaction with pVHL E3 ligase complex. Accordingly, pVHL cannot efficiently ubiquitinate HIF-2α, leading to HIF-2α accumulation followed by transcriptional regulation of its downstream target gene expression (Fig. 3B). This will contribute to glioma stemness, increased tumor growth and less favorable outcome in patients with glioblastoma. This elegant study reveals a novel regulatory pathway for ID2 function in glioblastoma affected by DYRK1A and DYRK1B prolyl hydroxylation, and indicates the potential role of PHD1/EglN2 as a tumor suppressor in glioblastoma by indirectly regulating HIF-2α levels and glioma stemness. However, it remains unclear whether PHD1/EglN2 directly hydroxylates DYRK1A/B on proline residues. In addition, it will be very interesting to further determine the molecular mechanism by which DYRK1A/B hydroxylation affects its phosphorylation and kinase activity, which will reveal the causal relationship between hydroxylation and phosphorylation events.
2.3 NDRG3 prolyl hydroxylation
N-Myc downstream-regulated gene (NDRG) contains 4 family members that are well conserved through evolution [91]. NDRG1 was first reported to be repressed by the N- and c-Myc proto-oncogenes [92, 93]. However, the regulation of other NDRG family members remains largely unknown. A recent paper by Lee et al. demonstrates that NDRG3 can be hydroxylated on the Pro294, potentially by PHD2/EglN1. Subsequently, NDRG3 hydroxylation triggers its binding with pVHL followed by NDRG3 ubiquitination and degradation (Fig. 4A) [94]. During tumor development, hypoxia provokes the accumulation of lactate that directly binds NDRG3, thus preventing its interaction with the pVHL complex. Therefore, NDRG3 can accumulate and contribute to downstream RAF-ERK1/2 kinase signaling (Fig. 4B).
Figure 4. NDRG3 regulation by hydroxylation.

(A) NDRG3 (N-Myc downstream-regulated gene) can be hydroxylated on proline 294, potentially by PHD2/EglN1. The hydroxylated NDRG3 binds to pVHL, which promotes NDRG3 ubiquitination and degradation. (B) Hypoxia can lead to the accumulation of lactate that directly binds NDRG3, thus preventing its interaction with the pVHL complex. Therefore, NDRG3 is stabilized and may contribute to downstream RAF-ERK1/2 kinase signaling.
There is a strong dependence of lactate-induced hypoxia for cell growth, angiogenesis and anti-apoptosis on NDRG3-mediated ERK1/2 activity [94]. There are also several interesting questions emerging from this elegant study. Firstly, does PHD2/EglN1 directly hydroxylate NDRG3 on Pro294 in vitro and in vivo? Secondly, it remains unclear how the interaction between NDRG3 and pVHL is decreased by hypoxia, and whether this could be due to the increased interaction between NDRG3 and lactate or to the decreased hydroxylation of NDRG3 under this metabolic condition. Thirdly, since hypoxia can induce PDK1 expression through HIF-1α, which will shift the pyruvate enzymatic reaction from producing acetyl-CoA to producing lactate important for the glycolytic pathway [95], it is likely that HIF-1α also contributes to lactate production level. If this is true, it is possible that the depletion of HIF pathway components will decrease lactate production and diminish NDRG3 accumulation under hypoxia. Therefore, it is likely that NDRG3 may be dependent on HIF signaling as a secondary effector that affects hypoxia responsive signaling in cancer.
2.4 Akt prolyl hydroxylation
VHL is a critical tumor suppressor gene that is inactivated by either mutation or hypermethylation in the majority of clear cell renal cell carcinoma (ccRCC), a lethal disease accounting for 85% of renal cancers and classically resistant to cytotoxic chemotherapy [96]. Preclinical studies demonstrated that Akt is frequently activated in ccRCC patients with pVHL loss [97], indicating the possible connection between pVHL loss and Akt hyper-activation.
The seminal findings from Guo et al. demonstrate that Akt can be hydroxylated on Pro125 and Pro313 residues by PHD2/EglN1 in vivo and in vitro [98]. pVHL binds directly with hydroxylated Akt and inhibits Akt activity, demonstrated by the decrease of Akt phosphorylation on threonine 308 (Thr308) and serine 473 (Ser473) [98]. Therefore, in cells under hypoxia or lacking functional pVHL, Akt activity is aberrantly elevated, therefore contributing to increased cell proliferation and tumorigenesis (Fig. 5A and 5B) [98]. Concerning the disease relevance, cancer-associated AKT mutations have been found to decrease Akt hydroxylation, therefore contributing to higher Akt phosphorylation.
Figure 5. Akt regulation by hydroxylation.

(A) Akt can be hydroxylated on Pro125 and Pro313 residues by PHD2/EglN1. pVHL and the phosphatase PP2A interact with hydroxylated Akt and inhibit Akt activity, as demonstrated by the decrease of Akt phosphorylation on threonine 308 (Thr308). (B) The lack of oxygen or functional pVHL prevents Akt hydroxylation and binding with pVHL and PP2A, therefore leading to the increase of Akt Thr308 phosphorylation and activity and contributing to increased cell proliferation and tumorigenesis.
By generating several Akt hydroxylation specific antibodies, this work shows the intimate connection between hydroxylation and kinase signaling pathways. Notably, in normoxic conditions, growth factor-induced Akt phosphorylationcould subsequently trigger the interaction of Akt with PHD2/EglN1, which in turn hydroxylates the activated Akt to promote pVHL-mediated suppression, presumably functioning as a negative feedback regulation to tightly regulate the kinase activity of Akt in vivo. Mechanistically, it also shows that Akt hydroxylation triggers a stronger interaction between Akt and the protein phosphatase 2A (PP2A) mediated by pVHL. (Fig. 5A). Interestingly enough, the Akt prolyl hydroxylation motif shares some similarities with the FOXO3a prolyl hydroxylation motif [87], raising the question of whether the “FOXO3alike” motif can be used to predict potential prolyl hydroxylation substrate pool in future research. This paper also suggests that not all of bona fide PHD/EglN substrates will carry the canonical LXXLAP HIF prolyl hydroxylation motif, which was identified in many proteins other than HIF such as IKKB, PKM2 and Cep192.
The recently identified GSPT or GPSS motif in FOXO3A and GSPS or GTPE motif in Akt, both of which can be proline-hydroxylated, might therefore be used to predict other putative hydroxylation sites. However, the limited number of published novel hydroxylation targets that can be used to predict these patterns limits the power of this in silico prediction. and awaits future research to solidify this motif.
In contrast to the finding that Akt hydroxylation compromises its phosphorylation, DYRK1A and DYRK1B hydroxylation positively regulates their phosphorylation. Therefore, it appears that there may be distinctive mechanisms that link hydroxylation and phosphorylation events, which also requires further detailed study in the near future.
2.5 EPOR prolyl hydroxylation
Erythropoietin (EPO) controls erythropoiesis by binding to its receptor (EPOR) on erythroid progenitor cells and subsequently activating JAK-STAT signaling pathways [99]. EPO has been well established to be a HIF-2α target gene [100, 101]. Mutation of PHD2/EglN1 was reported to induce EPO production largely in a HIF-2α-dependent manner [102]. However, the regulation of EPOR remains largely unknown.
Heir et al. [103] showed for the first time that EPOR protein levels are regulated by PHD3/EglN3 -mediated prolyl hydroxylation on Pro419 and Pro426 residues (Fig. 6A). Therefore, both hypoxia and prolyl hydroxylase inhibitors can induce EPOR protein stability. Mechanistically, EPOR protein stability regulation is through its binding with pVHL complex in a hydroxylation dependent manner and EPOR protein binds with the canonical pVHL substrate binding domain. Interestingly, while some of pVHL disease mutants still retain the ability to bind with hydroxylated HIF peptide, most of these mutants failed to bind with the EPOR hydroxylated peptide. This finding suggests that the EPO signaling is regulated by PHD3/EglN3 and the pVHL signaling pathway in human disease.
Figure 6. EPOR, MAPK6 and RIPK4 regulation by hydroxylation.

(A) PHD3/EglN3 -mediated prolyl hydroxylation on EPOR (erythropoietin receptor) Pro419 and Pro426 residues facilitates EPOR binding with pVHL complex, hereby causing its proteasomal degradation. PHD3/EglN3 depletion stabilizes EPOR and, therefore, stimulates JAK/STAT signaling pathway-driven erythropoiesis. (B) MAPK6 (Mitogen-activated protein kinase 6) can be hydroxylated by PHD3/EglN3 on proline 25 (Pro25), which potentially leads to its dissociation from HUWE1 (HECT, UBA and WWE domain containing 1) E3 ubiquitin protein ligase, thus protecting it from proteasomal degradation. (C) RIPK4 hydroxylation by FIH affects RIPK4 activity, therefore contributing to Wnt signaling pathway activity modulation.
2.6 OTUB1 asparagine hydroxylation
The ovarian tumor domain-containing ubiquitin aldehyde binding protein 1 (OTUB1) deubiquitinase was previously identified as a potential substrate for FIH hydroxylase [63]. OTUB1 was shown to be hydroxylated by FIH on the Asparagine 22 (Asn22) residue [104]. OTUB1 hydroxylation on Asn22 alters its binding behavior, especially with respect to proteins important in metabolic pathways. As a result, its hydroxylation changes the important pathways involved in cell metabolism. Scholz et al. [104] demonstrates a direct link between FIH-induced OTUB1 hydroxylation and cellular metabolism reprogramming, which may be important in human disease settings, especially in cancer where hypoxia serves as a hallmark and regulates cellular protein hydroxylation. Interestingly, OTUB1 Asn22 hydroxylation does not necessarily change its protein stability, which further indicates that not all hydroxylation modifications lead to changes in protein stability. As an alternative explanation, hydroxylation may change the binding complex repertoires, which may lead to profound changes important in various human diseases, such as cancer.
2.7 MAPK6 and RIPK4 prolyl and asparagine hydroxylation
Developing an enzyme trapping strategy by treating cells with the pan-prolyl hydroxylase inhibitor DMOG, combined with quantitative interaction proteomics, Rodriguez et al. [80] were able to identify some novel FIH and PHD substrates. As a proof of principle, they identified some of characterized PHD substrates from this proteomic approach, including CEP192 and FOXO3a [23, 80, 87]. Among potential PHD3/EglN3 binding partners/substrates, mitogen-activated protein kinase 6 (MAPK6) has been shown to be hydroxylated by PHD3/EglN3 on proline 25 (Pro25), which potentially leads to its dissociation from HECT, UBA and WWE domain containing 1, E3 ubiquitin protein ligase (HUWE1), thus protecting it from proteasomal degradation (Fig. 6B). Therefore, inhibition of MAPK6 hydroxylation by various prolyl hydroxylase inhibitors decreases MAPK6 protein stability in cells.
This regulation is distinctive of the canonical view that prolyl hydroxylation makes protein more susceptible for proteasomal degradation. This suggests that prolyl hydroxylation may affect protein stability via distinctive mechanisms depending on specific cellular contexts and prolyl hydroxylases. While PHD1/EglN2 and PHD2/EglN1 hydroxylation targets may be subjected to proteasomal degradation pathway, PHD3/EglN3 substrates may be protected from degradation upon being hydroxylated, while the underlying molecular mechanism warrants further in-depth investigation.
In addition, FIH was found to hydroxylate receptor-interacting serine-threonine kinase 4 (RIPK4) on asparagine residues, which did not mark RIPK4 for protein degradation. Interestingly, using TCF/LEF luciferase reporter to measure β-Catenin activity, RIPK4 hydroxylation was shown to increase Wnt signaling pathway activity (Fig. 6C). By defining the specific phosphorylation sites that are important for RIPK4 activity, Rodriguez and colleagues also pointed out that RIPK4 hydroxylation might affect RIPK4 kinase activity determined by the appearance of phosphorylation sites in mass spectrometry. This also suggests an enzymatic link between RIPK4 hydroxylation and phosphorylation. However, future research needs to be performed to examine the causal relationship and the molecular mechanism underlying how RIPK4 hydroxylation may affect its phosphorylation and activity in the cells. Although in this study a was used to identify potential prolyl or asparaginyl substrates, it still remains to be determined how these potential hydroxylation targets may affect cellular functions in a disease setting, such as cancer.
2.8 eEF2K prolyl hydroxylation
Translation elongation is mainly controlled by eukaryotic elongation factor 2 (eEF2) phosphorylation [105]. eEF2 kinase (eEF2K)-mediated eEF2 phosphorylation on the threonine 56 (Thr56) residue inhibits its ability to interact with ribosomes, therefore impairing translation elongation and protein synthesis. eEF2K is a calcium/calmodulin-dependent (CaM) α kinase. Hypoxia or prolyl hydroxylase inhibition was reported to activate eEF2K kinase activity, thereby contributing to increased eEF2 phosphorylation [106]. As a result, cells demonstrate decreased translation elongation and protein synthesis under hypoxia, which protects them from excessive usage of ATP. On the other hand, under normoxic condition, eEF2K can be hydroxylated on proline 98 residue (Pro98) that will disrupt its binding with calmodulin and inhibit its kinase activity (Fig. 7A). Therefore, eEF2 cannot be efficiently phosphorylated and protein synthesis can proceed.
Figure 7. eEF2K, β and γ-actin regulation by hydroxylation.

(A) Under normoxic condition, eEF2K (eukaryotic elongation factor 2 kinase) can be hydroxylated on proline 98 (Pro98), which disrupts its binding with calmodulin and inhibits its kinase activity. Therefore, eEF2 cannot be efficiently phosphorylated and protein synthesis proceeds. Hypoxic condition prevents eEF2K hydroxylation, allowing eEF2K interaction with calmodulin. Activated eEF2K phosphorylates eEF2 on threonine 56 (Thr56) therefore inhibiting its translation elongation activity. (B) β and γ-actin isoforms are hydroxylated by PHD3/EglN3 on Pro307 and Pro322 residues, which reduces actin polymerization. PHD3/EglN3 depletion leads to increased F-actin expression and cell motility in HeLa cells.
In summary, eEF2K serves as the protective mechanism for cells undergoing oxygen deprivation or ischemic condition. Consistent with this hypothesis, eEF2K knockout neurons showed increased usage of ATP and greater apoptosis when exposed to hypoxic condition [106]. This is the first report that eEF2K hydroxylation can affect its kinase activity and also provides some mechanistic insight on how cells rely on eEF2K for surviving under hypoxic conditions. In keeping with this notion, a previous study reported that eEF2K is protective for hypoxic cardiac muscle cells [107]. In a cancer setting that is characterized by hypoxia, eEF2K may also provide a protective mechanism to allow cancer cell survival. Therefore, developing a specific eEF2K inhibitor may be helpful for specifically killing these hypoxic cancer cells. However, it remains to be determined which prolyl hydroxylase may mediate the eEF2K hydroxylation and whether eEF2K hydroxylation would affect its binding with the pVHL E3 ligase complex.
2.9 Nonmuscle actin prolyl hydroxylation
Actin is very important for eukaryotic cell movement. Actin exists in three α isoforms and one γ isoform that are selectively expressed in certain cell types [108]. In addition, actin also constists of nonmuscle β and γ isoforms that are universally expressed in most cell types [108]. β and γ-actin isoforms were shown to be hydroxylated by PHD3/EglN3 on proline 307 (Pro307) and proline 322 (Pro322) residues, which impaired actin polymerization [109]. As a result, depletion of PHD3/EglN3 with multiple shRNAs led to increased F-actin expression and cell motility in HeLa cells. In addition, this regulation was largely dependent on PHD3/EglN3 activity (Fig. 7B). Therefore, this work solidifies the role of PHD3/EglN3 as a tumor suppressor in a cancer setting by inhibiting actin polymerization and cell motility [109]. Consistently, PHD3/EglN3 expression was shown to be downregulated in multiple cancers, including colon, melanoma and breast cancers [110, 111]. By regulating actin hydroxylation and cell motility, it is likely that PHD3/EglN3 may act as a tumor suppressor, inhibiting cell invasion and metastasis in these cancers. It will also be interesting to characterize additional PHD3/EglN3 substrates beside nonmuscle actin that may play an important role in cancer cell invasion and metastasis.
2.10 Thyroid hormone receptor alpha prolyl hydroxylation
By interacting with PHD2/EglN1 and PHD3/EglN3, thyroid hormone receptor α (TR-α) was reported to be hydroxylated at two proline residues, 160 and 162 (Pro160 and Pro162) [112]. Inhibition of TR-α hydroxylation (by hypoxia, for example) can increase its binding with the transcriptional nuclear receptor corepressor 2 (NCOR2). As a result, it leads to decreased transcription of phospholamban (PLN). In ischemic condition, decreased PLN in the heart leads to abnormal CaMKII activation, which can potentiate cardiomyocyte apoptosis and hypertrophy. In a physiologically relevant condition, PHD2/EglN1 and PHD3/EglN3 knockout mice showed an elevation in PLN expression in the heart and displayed a cardiac hypertrophy phenotype. It still remains to be determined whether PHD2/EglN1 or PHD3/EglN3 can directly hydroxylate TR-α on these proline residues. In addition, TR-α proline hydroxylation does not change its protein level or stability. Rather, it changes the interaction with the nuclear receptor corepressor 2 (NCOR2) and affects PLN transcription, which provides a new mechanistic insight on how prolyl hydroxylation can affect a range of biological pathways, from canonical protein stability to protein-protein binding complexes.
2.11 p53 prolyl hydroxylation
The tumor protein p53 gene (Tp53) is one of the most important tumor suppressors that is mutated in more than 50% of tumors. PHD1/EglN2 depletion was shown to sensitize colon cancer cells to chemotherapy drug treatment such as fluoracil (5-FU), characterized by enhanced apoptosis [113]. Interestingly, this enhanced response is dependent on intact p53 and p53 phosphorylation on the serine 15 (Ser15) residue, which was normally increased by chemotherapy drug treatment. Upon PHD1/EglN2 depletion, p53 Ser15 phosphorylation is decreased following 5-FU treatment when compared to control [113]. Similar results were obtained when treating cells with the pan-hydroxylase inhibitor DMOG, suggesting that Ser15 phosphorylation was regulated by prolyl hydroxylase activity [113].
Mechanistically, Deschoemaeker et al. [113] showed that reduced p53 hydroxylation impaired p53 phosphorylation induced by the proline-directed Ser/Thr MAP kinase p38α. p53 hydroxylation is essential for p38 binding with p53 and induces p53 phosphorylation on Ser15. However, it remains unclear whether p53 can serve as a direct PHD1/EglN2 substrate. In essence, this paper shows a causal link between PHD1/EglN2 silencing and reduced p53-mediated DNA repair following chemotherapy. Therefore, PHD1/EglN2 inhibition can be used as a therapeutic intervention strategy in colon cancer in combination with chemotherapy. Together with previous report that PHD1/EglN2 regulates cyclin D1 in breast tumorigenesis in an enzymatic-dependent manner [33], these findings strongly support the development of specific PHD1/EglN2 inhibitors in cancer therapy.
2.12 CERKL prolyl hydroxylation
Ceramid kinase-like (CERKL) is an understudied protein involved in oxidative stress and mitochondrial signaling [114]. For example, CERKL interacts with the mitochondrial thioredoxin (TRX2) and activates its important functions in the mitochondria. Accordingly, depletion of CERKL leads to retinal degeneration due to oxidative damage in both human and mouse models. However, the regulation of CERKL remains largely unknown. Chen et al. [115] found that CERKL was ubiquitinated, which was mediated by the pVHL E3 ligase complex. Depletion of VHL leads to increased CERKL protein levels. In addition, some preliminary data suggested that PHD1/EglN2 and PHD3/EglN3 might be important for regulating CERKL protein stability, possibly through hydroxylation [115]. It still remains to be determined whether PHD1/EglN2- and PHD3/EglN3-regulated CERKL hydroxylation may mediate its protein stability controlled by pVHL. In addition, the physiological relevance of CERKL in human diseases, such as cancer, remains to be further determined.
2.13 Filamin A prolyl hydroxylation
Hypoxia was reported to induce reversible spine regression, an effort by neurons to avoid the energy crisis by decreasing synaptic transmission [116]. Filamin A (FLNA) is an important actin cross-linker. The FLNA/F-actin ratios promote orthogonal networks or parallel actin bundles [117]. As such, FLNA regulates many aspects of neuronal pathways, including dendritic morphogenesis, neuronal migration and axonal growth cone [118–120]. The role of FLNA in spine regression remains largely unclear.
Segura et al. [116] showed that under normoxic conditions, PHD2/EglN1 promotes FLNA hydroxylation on multiple proline residues, including Pro2309, Pro2316 and possibly Pro2312, which will lead to FLNA binding with pVHL E3 ligase complex and degradation by the proteasome. Inhibition of PHD2/EglN1 activity by genetic approach (PHD2/EglN1 loss), pharmacological approach (such as DMOG) or physiological relevant condition (such as hypoxia) can lead to FLNA stabilization and immature filopodium-like dendritic protrusion, therefore inducing spine regression and reduced synaptic density [116]. Since FLNA has to be precisely regulated in order to prevent neurological defects, PHD2/EglN1 -mediated FLNA hydroxylation and its subsequent regulation by pVHL provides a molecular mechanism for its tight regulation in neurological context in vivo [116].
2.14 p53 lysine hydroxylation
Wang et al. [121] recently reported that the tumor suppressor protein p53 physically associates with the Jumonji C domain-containing protein JMJD6. As a result, JMJD6 hydroxylates p53 on the lysine 382 (Lys382) residue, leading to inhibition of its transcriptional activity [121]. JMJD6 depletion induces cell apoptosis and G1-phase cell cycle arrest, increases the sensitivity to cell death induced by DNA damaging agents and inhibits p53-dependent colon cell proliferation [121].
Of note, several types of human cancers, especially colon cancer, show an upregulation of JMJD6 expression, and elevated JMJD6 nuclear localization positively correlates with the aggressiveness of colon adenocarcinomas [121]. This finding suggests that JMJD6 could be considered as a therapeutic target in colon cancer, but more in-depth investigations are warranted to pinpoint the critical role of JMJD6 in tumorigenesis.
3. Conclusion and future perspectives
Until recently, protein hydroxylation was largely considered as a relatively specialized post-translational modification of the extracellular collagens and proteins with collagen-like sequences. The finding that the HIFs can be hydroxylated and, more importantly, that such modification strongly affects their signaling, has led to the hypothesis that post-translational hydroxylation may play a more prominent role than once we thought. Over the last two decades, the combination of in silico genomic analysis, protein association methods and pharmacological substrate-trapping followed by mass spectrometry analysis has allowed the discovery of several novel hydroxylation substrates, confirming the notion that protein hydroxylation extensively influences signaling pathways. In most cases, hydroxylation has been shown to modify protein stability, either altering the interaction with protein deubiquitinases (e.g. FOXO3a) or ubiquitin ligases (e.g. NDRG3, EPOR, MAPK6), or indirectly preserving the composition of ubiquitin ligase complexes (e.g. DYRK1A/B). However, hydroxylation can also affect the enzymatic activity of certain proteins, perturbing their interaction with direct activators (e.g. eEF2K) or influencing the occurrence of other post-translational modifications that in turn affect their activity (e.g. Akt, TR-α and p53). Moreover, protein binding pattern or homeostasis can be altered by hydroxylation, as evidenced by the example of OTUB1.
In general, proline hydroxylation leads to altered protein stability (such as for HIF-αs and FOXO3a) and asparagine hydroxylation contributes to altered protein-protein interactions (such as HIF-αs and OTUB1). However, the recent reports summarized above suggest that proline hydroxylation can also influence protein-protein interactions and other post-translational modifications (as illustrated by the examples of Akt, TR-α and eEF2K). These non-canonical functions of proline (or asparagine) hydroxylation remain to be further explored in future in-depth studies. The technical improvement of the last few years should enable a large-scale discovery of novel substrates, thus allowing a better understanding of how protein hydroxylation affects cell signaling and, in a larger perspective, cell pathophysiology.
Highlights.
Protein hydroxylation is catalyzed by 2-oxoglutarate-dependent dioxygenases.
A canonical example is HIFα proline hydroxylation by PHD/EglNs.
PHD/EglN inhibitors have been shown to exhibit therapeutic potential.
Protein hydroxylation can affect protein stability and protein-protein interaction.
Targeting protein hydroxylation could be a strategy for the treatment of cancers.
Acknowledgments
The authors sincerely apologize to all those colleagues whose important work was not cited in this paper owing to space limitations. They thank the members of Wei and Zhang laboratory for critical reading and discussion of the manuscript. W.W. is a Leukemia & Lymphoma Society (LLS) research scholar. Q.Z is a Kimmel Scholar and V Scholar. J.G. is an NRSA T32 trainee and supported by 5T32HL007893-17. This work was supported in part by US National Institutes of Health (NIH) grants to W.W. (GM094777 and CA177910), K99/R00 (CA160351) award from NIH (QZ), University Cancer Research Fund Tier 2 Innovator Award (QZ) from University of North Carolina at Chapel Hill, Kimmel Scholar Award (QZ), The V Foundation Scholar Award (QZ), DOD Career Development Award (QZ) and Susan G. Komen Career Catalyst Award (QZ).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
References
- 1.Loenarz C, Schofield CJ. Expanding chemical biology of 2-oxoglutarate oxygenases. Nature chemical biology. 2008;4:152–156. doi: 10.1038/nchembio0308-152. [DOI] [PubMed] [Google Scholar]
- 2.Hutton JJ, Jr, Kaplan A, Udenfriend S. Conversion of the amino acid sequence gly-pro-pro in protein to gly-pro-hyp by collagen proline hydroxylase. Archives of biochemistry and biophysics. 1967;121:384–391. doi: 10.1016/0003-9861(67)90091-4. [DOI] [PubMed] [Google Scholar]
- 3.Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Molecular cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 4.Losman JA, Kaelin WG., Jr What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes & development. 2013;27:836–852. doi: 10.1101/gad.217406.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ploumakis A, Coleman ML. OH, the Places You’ll Go! Hydroxylation, Gene Expression, and Cancer. Molecular cell. 2015;58:729–741. doi: 10.1016/j.molcel.2015.05.026. [DOI] [PubMed] [Google Scholar]
- 6.Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13:343–357. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klose RJ, Kallin EM, Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet. 2006;7:715–727. doi: 10.1038/nrg1945. [DOI] [PubMed] [Google Scholar]
- 8.Hojfeldt JW, Agger K, Helin K. Histone lysine demethylases as targets for anticancer therapy, Nature reviews. Drug discovery. 2013;12:917–930. doi: 10.1038/nrd4154. [DOI] [PubMed] [Google Scholar]
- 9.Shen L, Song CX, He C, Zhang Y. Mechanism and function of oxidative reversal of DNA and RNA methylation. Annual review of biochemistry. 2014;83:585–614. doi: 10.1146/annurev-biochem-060713-035513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Webby CJ, Wolf A, Gromak N, Dreger M, Kramer H, Kessler B, Nielsen ML, Schmitz C, Butler DS, Yates JR, 3rd, Delahunty CM, Hahn P, Lengeling A, Mann M, Proudfoot NJ, Schofield CJ, Bottger A. Jmjd6 catalyses lysyl-hydroxylation of U2AF65, a protein associated with RNA splicing. Science. 2009;325:90–93. doi: 10.1126/science.1175865. [DOI] [PubMed] [Google Scholar]
- 12.Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes & development. 2002;16:1466–1471. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang M, Ge W, Chowdhury R, Claridge TD, Kramer HB, Schmierer B, McDonough MA, Gong L, Kessler BM, Ratcliffe PJ, Coleman ML, Schofield CJ. Asparagine and aspartate hydroxylation of the cytoskeletal ankyrin family is catalyzed by factor-inhibiting hypoxia-inducible factor. The Journal of biological chemistry. 2011;286:7648–7660. doi: 10.1074/jbc.M110.193540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ge W, Wolf A, Feng T, Ho CH, Sekirnik R, Zayer A, Granatino N, Cockman ME, Loenarz C, Loik ND, Hardy AP, Claridge TD, Hamed RB, Chowdhury R, Gong L, Robinson CV, Trudgian DC, Jiang M, Mackeen MM, McCullagh JS, Gordiyenko Y, Thalhammer A, Yamamoto A, Yang M, Liu-Yi P, Zhang Z, Schmidt-Zachmann M, Kessler BM, Ratcliffe PJ, Preston GM, Coleman ML, Schofield CJ. Oxygenase-catalyzed ribosome hydroxylation occurs in prokaryotes and humans. Nature chemical biology. 2012;8:960–962. doi: 10.1038/nchembio.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 16.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 17.Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. The EMBO journal. 2003;22:4082–4090. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.To KK, Huang LE. Suppression of hypoxia-inducible factor 1alpha (HIF-1alpha) transcriptional activity by the HIF prolyl hydroxylase EGLN1. The Journal of biological chemistry. 2005;280:38102–38107. doi: 10.1074/jbc.M504342200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takeda K, Ho VC, Takeda H, Duan LJ, Nagy A, Fong GH. Placental but not heart defects are associated with elevated hypoxia-inducible factor alpha levels in mice lacking prolyl hydroxylase domain protein 2. Molecular and cellular biology. 2006;26:8336–8346. doi: 10.1128/MCB.00425-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Minamishima YA, Moslehi J, Bardeesy N, Cullen D, Bronson RT, Kaelin WG., Jr Somatic inactivation of the PHD2 prolyl hydroxylase causes polycythemia and congestive heart failure. Blood. 2008;111:3236–3244. doi: 10.1182/blood-2007-10-117812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weidemann A, Johnson RS. Biology of HIF-1alpha. Cell death and differentiation. 2008;15:621–627. doi: 10.1038/cdd.2008.12. [DOI] [PubMed] [Google Scholar]
- 22.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moser SC, Bensaddek D, Ortmann B, Maure JF, Mudie S, Blow JJ, Lamond AI, Swedlow JR, Rocha S. PHD1 links cell-cycle progression to oxygen sensing through hydroxylation of the centrosomal protein Cep192. Developmental cell. 2013;26:381–392. doi: 10.1016/j.devcel.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F, Godson C, Nielsen JE, Moynagh P, Pouyssegur J, Taylor CT. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:18154–18159. doi: 10.1073/pnas.0602235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Markolovic S, Wilkins SE, Schofield CJ. Protein Hydroxylation Catalyzed by 2-Oxoglutarate-dependent Oxygenases. The Journal of biological chemistry. 2015;290:20712–20722. doi: 10.1074/jbc.R115.662627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oehme F, Ellinghaus P, Kolkhof P, Smith TJ, Ramakrishnan S, Hutter J, Schramm M, Flamme I. Overexpression of PH-4, a novel putative proline 4-hydroxylase, modulates activity of hypoxia-inducible transcription factors. Biochemical and biophysical research communications. 2002;296:343–349. doi: 10.1016/s0006-291x(02)00862-8. [DOI] [PubMed] [Google Scholar]
- 27.del Peso L, Castellanos MC, Temes E, Martin-Puig S, Cuevas Y, Olmos G, Landazuri MO. The von Hippel Lindau/hypoxia-inducible factor (HIF) pathway regulates the transcription of the HIF-proline hydroxylase genes in response to low oxygen. The Journal of biological chemistry. 2003;278:48690–48695. doi: 10.1074/jbc.M308862200. [DOI] [PubMed] [Google Scholar]
- 28.Metzen E, Stiehl DP, Doege K, Marxsen JH, Hellwig-Burgel T, Jelkmann W. Regulation of the prolyl hydroxylase domain protein 2 (phd2/egln-1) gene: identification of a functional hypoxia-responsive element. The Biochemical journal. 2005;387:711–717. doi: 10.1042/BJ20041736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Henze AT, Riedel J, Diem T, Wenner J, Flamme I, Pouyseggur J, Plate KH, Acker T. Prolyl hydroxylases 2 and 3 act in gliomas as protective negative feedback regulators of hypoxia-inducible factors. Cancer research. 2010;70:357–366. doi: 10.1158/0008-5472.CAN-09-1876. [DOI] [PubMed] [Google Scholar]
- 30.Mak P, Chang C, Pursell B, Mercurio AM. Estrogen receptor beta sustains epithelial differentiation by regulating prolyl hydroxylase 2 transcription. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:4708–4713. doi: 10.1073/pnas.1221654110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Porter DA, Krop IE, Nasser S, Sgroi D, Kaelin CM, Marks JR, Riggins G, Polyak K. A SAGE (serial analysis of gene expression) view of breast tumor progression. Cancer research. 2001;61:5697–5702. [PubMed] [Google Scholar]
- 32.Appelhoff RJ, Tian YM, Raval RR, Turley H, Harris AL, Pugh CW, Ratcliffe PJ, Gleadle JM. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. The Journal of biological chemistry. 2004;279:38458–38465. doi: 10.1074/jbc.M406026200. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Q, Gu J, Li L, Liu J, Luo B, Cheung HW, Boehm JS, Ni M, Geisen C, Root DE, Polyak K, Brown M, Richardson AL, Hahn WC, Kaelin WG, Jr, Bommi-Reddy A. Control of cyclin D1 and breast tumorigenesis by the EglN2 prolyl hydroxylase. Cancer cell. 2009;16:413–424. doi: 10.1016/j.ccr.2009.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seth P, Krop I, Porter D, Polyak K. Novel estrogen and tamoxifen induced genes identified by SAGE (Serial Analysis of Gene Expression) Oncogene. 2002;21:836–843. doi: 10.1038/sj.onc.1205113. [DOI] [PubMed] [Google Scholar]
- 35.Nakayama K, Frew IJ, Hagensen M, Skals M, Habelhah H, Bhoumik A, Kadoya T, Erdjument-Bromage H, Tempst P, Frappell PB, Bowtell DD, Ronai Z. Siah2 regulates stability of prolyl-hydroxylases, controls HIF1alpha abundance, and modulates physiological responses to hypoxia. Cell. 2004;117:941–952. doi: 10.1016/j.cell.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 36.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, Leung IK, Li XS, Woon EC, Yang M, McDonough MA, King ON, Clifton IJ, Klose RJ, Claridge TD, Ratcliffe PJ, Schofield CJ, Kawamura A. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO reports. 2011;12:463–469. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, Travins J, Weiss S, Looper R, Ligon KL, Verhaak RG, Yan H, Kaelin WG., Jr Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483:484–488. doi: 10.1038/nature10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, Cowley GS, Root DE, Ebert BL, Kaelin WG., Jr (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339:1621–1625. doi: 10.1126/science.1231677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, Wellen KE, O’Rourke DM, Berger SL, Chan TA, Levine RL, Mellinghoff IK, Thompson CB. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Briggs KJ, Koivunen P, Cao S, Backus KM, Olenchock BA, Patel H, Zhang Q, Signoretti S, Gerfen GJ, Richardson AL, Witkiewicz AK, Cravatt BF, Clardy J, Kaelin WG., Jr Paracrine Induction of HIF by Glutamate in Breast Cancer: EglN1 Senses Cysteine. Cell. 2016;166:126–139. doi: 10.1016/j.cell.2016.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Flashman E, Hoffart LM, Hamed RB, Bollinger JM, Jr, Krebs C, Schofield CJ. Evidence for the slow reaction of hypoxia-inducible factor prolyl hydroxylase 2 with oxygen. The FEBS journal. 2010;277:4089–4099. doi: 10.1111/j.1742-4658.2010.07804.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hirsila M, Koivunen P, Xu L, Seeley T, Kivirikko KI, Myllyharju J. Effect of desferrioxamine and metals on the hydroxylases in the oxygen sensing pathway. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2005;19:1308–1310. doi: 10.1096/fj.04-3399fje. [DOI] [PubMed] [Google Scholar]
- 44.Chowdhury R, Flashman E, Mecinovic J, Kramer HB, Kessler BM, Frapart YM, Boucher JL, Clifton IJ, McDonough MA, Schofield CJ. Studies on the reaction of nitric oxide with the hypoxia-inducible factor prolyl hydroxylase domain 2 (EGLN1) Journal of molecular biology. 2011;410:268–279. doi: 10.1016/j.jmb.2011.04.075. [DOI] [PubMed] [Google Scholar]
- 45.Mecinovic J, Chowdhury R, Flashman E, Schofield CJ. Use of mass spectrometry to probe the nucleophilicity of cysteinyl residues of prolyl hydroxylase domain 2. Analytical biochemistry. 2009;393:215–221. doi: 10.1016/j.ab.2009.06.029. [DOI] [PubMed] [Google Scholar]
- 46.Narang VS, Pauletti GM, Gout PW, Buckley DJ, Buckley AR. Suppression of cystine uptake by sulfasalazine inhibits proliferation of human mammary carcinoma cells. Anticancer Res. 2003;23:4571–4579. [PubMed] [Google Scholar]
- 47.Narang VS, Pauletti GM, Gout PW, Buckley DJ, Buckley AR. Sulfasalazine-induced reduction of glutathione levels in breast cancer cells: enhancement of growth-inhibitory activity of Doxorubicin. Chemotherapy. 2007;53:210–217. doi: 10.1159/000100812. [DOI] [PubMed] [Google Scholar]
- 48.Wong CC, Zhang H, Gilkes DM, Chen J, Wei H, Chaturvedi P, Hubbi ME, Semenza GL. Inhibitors of hypoxia-inducible factor 1 block breast cancer metastatic niche formation and lung metastasis. Journal of molecular medicine. 2012;90:803–815. doi: 10.1007/s00109-011-0855-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang H, Wong CC, Wei H, Gilkes DM, Korangath P, Chaturvedi P, Schito L, Chen J, Krishnamachary B, Winnard PT, Jr, Raman V, Zhen L, Mitzner WA, Sukumar S, Semenza GL. HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene. 2012;31:1757–1770. doi: 10.1038/onc.2011.365. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 50.Unoki M, Masuda A, Dohmae N, Arita K, Yoshimatsu M, Iwai Y, Fukui Y, Ueda K, Hamamoto R, Shirakawa M, Sasaki H, Nakamura Y. Lysyl 5-hydroxylation, a novel histone modification, by Jumonji domain containing 6 (JMJD6) The Journal of biological chemistry. 2013;288:6053–6062. doi: 10.1074/jbc.M112.433284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kotch LE, Iyer NV, Laughner E, Semenza GL. Defective vascularization of HIF-1alpha-null embryos is not associated with VEGF deficiency but with mesenchymal cell death. Dev Biol. 1999;209:254–267. doi: 10.1006/dbio.1999.9253. [DOI] [PubMed] [Google Scholar]
- 52.Patel SA, Simon MC. Biology of hypoxia-inducible factor-2alpha in development and disease. Cell death and differentiation. 2008;15:628–634. doi: 10.1038/cdd.2008.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gnarra JR, Ward JM, Porter FD, Wagner JR, Devor DE, Grinberg A, Emmert-Buck MR, Westphal H, Klausner RD, Linehan WM. Defective placental vasculogenesis causes embryonic lethality in VHL-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:9102–9107. doi: 10.1073/pnas.94.17.9102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takeda K, Cowan A, Fong GH. Essential role for prolyl hydroxylase domain protein 2 in oxygen homeostasis of the adult vascular system. Circulation. 2007;116:774–781. doi: 10.1161/CIRCULATIONAHA.107.701516. [DOI] [PubMed] [Google Scholar]
- 55.Aragones J, Schneider M, Van Geyte K, Fraisl P, Dresselaers T, Mazzone M, Dirkx R, Zacchigna S, Lemieux H, Jeoung NH, Lambrechts D, Bishop T, Lafuste P, Diez-Juan A, Harten SK, Van Noten P, De Bock K, Willam C, Tjwa M, Grosfeld A, Navet R, Moons L, Vandendriessche T, Deroose C, Wijeyekoon B, Nuyts J, Jordan B, Silasi-Mansat R, Lupu F, Dewerchin M, Pugh C, Salmon P, Mortelmans L, Gallez B, Gorus F, Buyse J, Sluse F, Harris RA, Gnaiger E, Hespel P, Van Hecke P, Schuit F, Van Veldhoven P, Ratcliffe P, Baes M, Maxwell P, Carmeliet P. Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nature genetics. 2008;40:170–180. doi: 10.1038/ng.2007.62. [DOI] [PubMed] [Google Scholar]
- 56.Takeda K, Aguila HL, Parikh NS, Li X, Lamothe K, Duan LJ, Takeda H, Lee FS, Fong GH. Regulation of adult erythropoiesis by prolyl hydroxylase domain proteins. Blood. 2008;111:3229–3235. doi: 10.1182/blood-2007-09-114561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kalucka J, Ettinger A, Franke K, Mamlouk S, Singh RP, Farhat K, Muschter A, Olbrich S, Breier G, Katschinski DM, Huttner W, Weidemann A, Wielockx B. Loss of epithelial hypoxia-inducible factor prolyl hydroxylase 2 accelerates skin wound healing in mice. Molecular and cellular biology. 2013;33:3426–3438. doi: 10.1128/MCB.00609-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zimmermann AS, Morrison SD, Hu MS, Li S, Nauta A, Sorkin M, Meyer NP, Walmsley GG, Maan ZN, Chan DA, Gurtner GC, Giaccia AJ, Longaker MT. Epidermal or dermal specific knockout of PHD-2 enhances wound healing and minimizes ischemic injury. PloS one. 2014;9:e93373. doi: 10.1371/journal.pone.0093373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ikeda J, Ichiki T, Matsuura H, Inoue E, Kishimoto J, Watanabe A, Sankoda C, Kitamoto S, Tokunou T, Takeda K, Fong GH, Sunagawa K. Deletion of phd2 in myeloid lineage attenuates hypertensive cardiovascular remodeling. Journal of the American Heart Association. 2013;2:e000178. doi: 10.1161/JAHA.113.000178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pfluecke C, Christoph M, Ibrahim K, Mensch M, Barthel P, Franke K, Wielockx B, Strasser RH, Wunderlich C, Poitz DM. Phd-2 knockout promotes plaque progression via hif1 alpha and increases the expression of mac-1, psgl-1 and vla-4 on monocytes and granulocytes in mice. J Am Coll Cardiol. 2013;61:E1829–E1829. [Google Scholar]
- 61.Zhang N, Fu Z, Linke S, Chicher J, Gorman JJ, Visk D, Haddad GG, Poellinger L, Peet DJ, Powell F, Johnson RS. The asparaginyl hydroxylase factor inhibiting HIF-1alpha is an essential regulator of metabolism. Cell metabolism. 2010;11:364–378. doi: 10.1016/j.cmet.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takeda K, Fong GH. Prolyl hydroxylase domain 2 protein suppresses hypoxia-induced endothelial cell proliferation. Hypertension. 2007;49:178–184. doi: 10.1161/01.HYP.0000251360.40838.0f. [DOI] [PubMed] [Google Scholar]
- 63.Scholz CC, Cavadas MA, Tambuwala MM, Hams E, Rodriguez J, von Kriegsheim A, Cotter P, Bruning U, Fallon PG, Cheong A, Cummins EP, Taylor CT. Regulation of IL-1beta-induced NF-kappaB by hydroxylases links key hypoxic and inflammatory signaling pathways. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:18490–18495. doi: 10.1073/pnas.1309718110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Scholz CC, Taylor CT. Targeting the HIF pathway in inflammation and immunity. Curr Opin Pharmacol. 2013;13:646–653. doi: 10.1016/j.coph.2013.04.009. [DOI] [PubMed] [Google Scholar]
- 65.Cummins EP, Seeballuck F, Keely SJ, Mangan NE, Callanan JJ, Fallon PG, Taylor CT. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology. 2008;134:156–165. doi: 10.1053/j.gastro.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 66.Robinson A, Keely S, Karhausen J, Gerich ME, Furuta GT, Colgan SP. Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology. 2008;134:145–155. doi: 10.1053/j.gastro.2007.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Teli MK, Rajanikant GK. Identification of novel potential HIF-prolyl hydroxylase inhibitors by in silico screening. Mol Divers. 2012;16:193–202. doi: 10.1007/s11030-011-9338-x. [DOI] [PubMed] [Google Scholar]
- 68.Vachal P, Miao S, Pierce JM, Guiadeen D, Colandrea VJ, Wyvratt MJ, Salowe SP, Sonatore LM, Milligan JA, Hajdu R, Gollapudi A, Keohane CA, Lingham RB, Mandala SM, DeMartino JA, Tong X, Wolff M, Steinhuebel D, Kieczykowski GR, Fleitz FJ, Chapman K, Athanasopoulos J, Adam G, Akyuz CD, Jena DK, Lusen JW, Meng J, Stein BD, Xia L, Sherer EC, Hale JJ. 1,3,8-Triazaspiro[4.5]decane-2,4-diones as efficacious pan-inhibitors of hypoxia-inducible factor prolyl hydroxylase 1–3 (HIF PHD1-3) for the treatment of anemia. Journal of medicinal chemistry. 2012;55:2945–2959. doi: 10.1021/jm201542d. [DOI] [PubMed] [Google Scholar]
- 69.Rabinowitz MH. Inhibition of hypoxia-inducible factor prolyl hydroxylase domain oxygen sensors: tricking the body into mounting orchestrated survival and repair responses. Journal of medicinal chemistry. 2013;56:9369–9402. doi: 10.1021/jm400386j. [DOI] [PubMed] [Google Scholar]
- 70.Kim SY, Yang EG. Recent Advances in Developing Inhibitors for Hypoxia-Inducible Factor Prolyl Hydroxylases and Their Therapeutic Implications. Molecules. 2015;20:20551–20568. doi: 10.3390/molecules201119717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mazzone M, Dettori D, Oliveira RLeitede, Loges S, Schmidt T, Jonckx B, Tian YM, Lanahan AA, Pollard P, Ruiz de Almodovar C, De Smet F, Vinckier S, Aragones J, Debackere K, Luttun A, Wyns S, Jordan B, Pisacane A, Gallez B, Lampugnani MG, Dejana E, Simons M, Ratcliffe P, Maxwell P, Carmeliet P. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell. 2009;136:839–851. doi: 10.1016/j.cell.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Klotzsche-von Ameln A, Muschter A, Mamlouk S, Kalucka J, Prade I, Franke K, Rezaei M, Poitz DM, Breier G, Wielockx B. Inhibition of HIF prolyl hydroxylase-2 blocks tumor growth in mice through the antiproliferative activity of TGFbeta. Cancer research. 2011;71:3306–3316. doi: 10.1158/0008-5472.CAN-10-3838. [DOI] [PubMed] [Google Scholar]
- 73.Eckle T, Brodsky K, Bonney M, Packard T, Han J, Borchers CH, Mariani TJ, Kominsky DJ, Mittelbronn M, Eltzschig HK. HIF1A reduces acute lung injury by optimizing carbohydrate metabolism in the alveolar epithelium. PLoS biology. 2013;11:e1001665. doi: 10.1371/journal.pbio.1001665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cockman ME, Lancaster DE, Stolze IP, Hewitson KS, McDonough MA, Coleman ML, Coles CH, Yu X, Hay RT, Ley SC, Pugh CW, Oldham NJ, Masson N, Schofield CJ, Ratcliffe PJ. Posttranslational hydroxylation of ankyrin repeats in IkappaB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH) Proceedings of the National Academy of Sciences of the United States of America. 2006;103:14767–14772. doi: 10.1073/pnas.0606877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Coleman ML, McDonough MA, Hewitson KS, Coles C, Mecinovic J, Edelmann M, Cook KM, Cockman ME, Lancaster DE, Kessler BM, Oldham NJ, Ratcliffe PJ, Schofield CJ. Asparaginyl hydroxylation of the Notch ankyrin repeat domain by factor inhibiting hypoxia-inducible factor. The Journal of biological chemistry. 2007;282:24027–24038. doi: 10.1074/jbc.M704102200. [DOI] [PubMed] [Google Scholar]
- 76.Ferguson JE, 3rd, Wu Y, Smith K, Charles P, Powers K, Wang H, Patterson C. ASB4 is a hydroxylation substrate of FIH and promotes vascular differentiation via an oxygen-dependent mechanism. Molecular and cellular biology. 2007;27:6407–6419. doi: 10.1128/MCB.00511-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Koditz J, Nesper J, Wottawa M, Stiehl DP, Camenisch G, Franke C, Myllyharju J, Wenger RH, Katschinski DM. Oxygen-dependent ATF-4 stability is mediated by the PHD3 oxygen sensor. Blood. 2007;110:3610–3617. doi: 10.1182/blood-2007-06-094441. [DOI] [PubMed] [Google Scholar]
- 78.Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, Cole RN, Pandey A, Semenza GL. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145:732–744. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cockman ME, Webb JD, Kramer HB, Kessler BM, Ratcliffe PJ. Proteomics-based identification of novel factor inhibiting hypoxia-inducible factor (FIH) substrates indicates widespread asparaginyl hydroxylation of ankyrin repeat domain-containing proteins. Mol Cell Proteomics. 2009;8:535–546. doi: 10.1074/mcp.M800340-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rodriguez J, Pilkington R, Garcia Munoz A, Nguyen LK, Rauch N, Kennedy S, Monsefi N, Herrero A, Taylor CT, von Kriegsheim A. Substrate-Trapped Interactors of PHD3 and FIH Cluster in Distinct Signaling Pathways. Cell reports. 2016;14:2745–2760. doi: 10.1016/j.celrep.2016.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24:7410–7425. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
- 82.Hu MC, Lee DF, Xia W, Golfman LS, Ou-Yang F, Yang JY, Zou Y, Bao S, Hanada N, Saso H, Kobayashi R, Hung MC. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004;117:225–237. doi: 10.1016/s0092-8674(04)00302-2. [DOI] [PubMed] [Google Scholar]
- 83.Yang JY, Zong CS, Xia W, Yamaguchi H, Ding Q, Xie X, Lang JY, Lai CC, Chang CJ, Huang WC, Huang H, Kuo HP, Lee DF, Li LY, Lien HC, Cheng X, Chang KJ, Hsiao CD, Tsai FJ, Tsai CH, Sahin AA, Muller WJ, Mills GB, Yu D, Hortobagyi GN, Hung MC. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nature cell biology. 2008;10:138–148. doi: 10.1038/ncb1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Borkhardt A, Repp R, Haas OA, Leis T, Harbott J, Kreuder J, Hammermann J, Henn T, Lampert F. Cloning and characterization of AFX, the gene that fuses to MLL in acute leukemias with a t(X;11)(q13;q23) Oncogene. 1997;14:195–202. doi: 10.1038/sj.onc.1200814. [DOI] [PubMed] [Google Scholar]
- 85.Hillion J, Le Coniat M, Jonveaux P, Berger R, Bernard OA. AF6q21, a novel partner of the MLL gene in t(6;11)(q21;q23), defines a forkhead transcriptional factor subfamily. Blood. 1997;90:3714–3719. [PubMed] [Google Scholar]
- 86.Paik JH, Kollipara R, Chu G, Ji H, Xiao Y, Ding Z, Miao L, Tothova Z, Horner JW, Carrasco DR, Jiang S, Gilliland DG, Chin L, Wong WH, Castrillon DH, DePinho RA. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell. 2007;128:309–323. doi: 10.1016/j.cell.2006.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zheng X, Zhai B, Koivunen P, Shin SJ, Lu G, Liu J, Geisen C, Chakraborty AA, Moslehi JJ, Smalley DM, Wei X, Chen X, Chen Z, Beres JM, Zhang J, Tsao JL, Brenner MC, Zhang Y, Fan C, DePinho RA, Paik J, Gygi SP, Kaelin WG, Jr, Zhang Q. Prolyl hydroxylation by EglN2 destabilizes FOXO3a by blocking its interaction with the USP9x deubiquitinase. Genes & development. 2014;28:1429–1444. doi: 10.1101/gad.242131.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lasorella A, Benezra R, Iavarone A. The ID proteins: master regulators of cancer stem cells and tumour aggressiveness, Nature reviews. Cancer. 2014;14:77–91. doi: 10.1038/nrc3638. [DOI] [PubMed] [Google Scholar]
- 89.Perk J, Iavarone A, Benezra R. Id family of helix-loop-helix proteins in cancer, Nature reviews. Cancer. 2005;5:603–614. doi: 10.1038/nrc1673. [DOI] [PubMed] [Google Scholar]
- 90.Lee SB, Frattini V, Bansal M, Castano AM, Sherman D, Hutchinson K, Bruce JN, Califano A, Liu G, Cardozo T, Iavarone A, Lasorella A. An ID2-dependent mechanism for VHL inactivation in cancer. Nature. 2016;529:172–177. doi: 10.1038/nature16475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Melotte V, Qu X, Ongenaert M, van Criekinge W, de Bruine AP, Baldwin HS, van Engeland M. The N-myc downstream regulated gene (NDRG) family: diverse functions, multiple applications. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2010;24:4153–4166. doi: 10.1096/fj.09-151464. [DOI] [PubMed] [Google Scholar]
- 92.Li J, Kretzner L. The growth-inhibitory Ndrg1 gene is a Myc negative target in human neuroblastomas and other cell types with overexpressed N- or c-myc. Mol Cell Biochem. 2003;250:91–105. doi: 10.1023/a:1024918328162. [DOI] [PubMed] [Google Scholar]
- 93.Zhang J, Chen S, Zhang W, Zhang J, Liu X, Shi H, Che H, Wang W, Li F, Yao L. Human differentiation-related gene NDRG1 is a Myc downstream-regulated gene that is repressed by Myc on the core promoter region. Gene. 2008;417:5–12. doi: 10.1016/j.gene.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 94.Lee DC, Sohn HA, Park ZY, Oh S, Kang YK, Lee KM, Kang M, Jang YJ, Yang SJ, Hong YK, Noh H, Kim JA, Kim DJ, Bae KH, Kim DM, Chung SJ, Yoo HS, Yu DY, Park KC, Yeom YI. A lactate-induced response to hypoxia. Cell. 2015;161:595–609. doi: 10.1016/j.cell.2015.03.011. [DOI] [PubMed] [Google Scholar]
- 95.Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell metabolism. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 96.Kaelin WG., Jr Molecular basis of the VHL hereditary cancer syndrome, Nature reviews. Cancer. 2002;2:673–682. doi: 10.1038/nrc885. [DOI] [PubMed] [Google Scholar]
- 97.Hager M, Haufe H, Kemmerling R, Hitzl W, Mikuz G, Moser PL, Kolbitsch C. Increased activated Akt expression in renal cell carcinomas and prognosis. J Cell Mol Med. 2009;13:2181–2188. doi: 10.1111/j.1582-4934.2008.00488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Guo J, Chakraborty AA, Liu P, Gan W, Zheng X, Inuzuka H, Wang B, Zhang J, Zhang L, Yuan M, Novak J, Cheng JQ, Toker A, Signoretti S, Zhang Q, Asara J, Kaelin W, Wei W. pVHL suppresses kinase activity of Akt in a proline-hydroxylation-dependent manner. Science. 2016;353:929–932. doi: 10.1126/science.aad5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Watowich SS. The erythropoietin receptor: molecular structure and hematopoietic signaling pathways. J Investig Med. 2011;59:1067–1072. doi: 10.231/JIM.0b013e31820fb28c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ratcliffe PJ. HIF-1 and HIF-2: working alone or together in hypoxia? The Journal of clinical investigation. 2007;117:862–865. doi: 10.1172/JCI31750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rankin EB, Biju MP, Liu Q, Unger TL, Rha J, Johnson RS, Simon MC, Keith B, Haase VH. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. The Journal of clinical investigation. 2007;117:1068–1077. doi: 10.1172/JCI30117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Haase VH. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013;27:41–53. doi: 10.1016/j.blre.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Heir P, Srikumar T, Bikopoulos G, Bunda S, Poon BP, Lee JE, Raught B, Ohh M. Oxygen-dependent Regulation of Erythropoietin Receptor Turnover and Signaling. The Journal of biological chemistry. 2016;291:7357–7372. doi: 10.1074/jbc.M115.694562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Scholz CC, Rodriguez J, Pickel C, Burr S, Fabrizio JA, Nolan KA, Spielmann P, Cavadas MA, Crifo B, Halligan DN, Nathan JA, Peet DJ, Wenger RH, Von Kriegsheim A, Cummins EP, Taylor CT. FIH Regulates Cellular Metabolism through Hydroxylation of the Deubiquitinase OTUB1. PLoS biology. 2016;14:e1002347. doi: 10.1371/journal.pbio.1002347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kaul G, Pattan G, Rafeequi T. Eukaryotic elongation factor-2 (eEF2): its regulation and peptide chain elongation. Cell Biochem Funct. 2011;29:227–234. doi: 10.1002/cbf.1740. [DOI] [PubMed] [Google Scholar]
- 106.Moore CE, Mikolajek H, Regufeda Mota S, Wang X, Kenney JW, Werner JM, Proud CG. Elongation Factor 2 Kinase Is Regulated by Proline Hydroxylation and Protects Cells during Hypoxia. Molecular and cellular biology. 2015;35:1788–1804. doi: 10.1128/MCB.01457-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Terai K, Hiramoto Y, Masaki M, Sugiyama S, Kuroda T, Hori M, Kawase I, Hirota H. AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Molecular and cellular biology. 2005;25:9554–9575. doi: 10.1128/MCB.25.21.9554-9575.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Perrin BJ, Ervasti JM. The actin gene family: function follows isoform. Cytoskeleton (Hoboken) 2010;67:630–634. doi: 10.1002/cm.20475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Luo W, Lin B, Wang Y, Zhong J, O’Meally R, Cole RN, Pandey A, Levchenko A, Semenza GL. PHD3-mediated prolyl hydroxylation of nonmuscle actin impairs polymerization and cell motility. Molecular biology of the cell. 2014;25:2788–2796. doi: 10.1091/mbc.E14-02-0775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Xue J, Li X, Jiao S, Wei Y, Wu G, Fang J. Prolyl hydroxylase-3 is down-regulated in colorectal cancer cells and inhibits IKKbeta independent of hydroxylase activity. Gastroenterology. 2010;138:606–615. doi: 10.1053/j.gastro.2009.09.049. [DOI] [PubMed] [Google Scholar]
- 111.Place TL, Fitzgerald MP, Venkataraman S, Vorrink SU, Case AJ, Teoh ML, Domann FE. Aberrant promoter CpG methylation is a mechanism for impaired PHD3 expression in a diverse set of malignant cells. PloS one. 2011;6:e14617. doi: 10.1371/journal.pone.0014617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Xie L, Pi X, Townley-Tilson WH, Li N, Wehrens XH, Entman ML, Taffet GE, Mishra A, Peng J, Schisler JC, Meissner G, Patterson C. PHD2/3-dependent hydroxylation tunes cardiac response to beta-adrenergic stress via phospholamban. The Journal of clinical investigation. 2015;125:2759–2771. doi: 10.1172/JCI80369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Deschoemaeker S, Di Conza G, Lilla S, Martin-Perez R, Mennerich D, Boon L, Hendrikx S, Maddocks OD, Marx C, Radhakrishnan P, Prenen H, Schneider M, Myllyharju J, Kietzmann T, Vousden KH, Zanivan S, Mazzone M. PHD1 regulates p53-mediated colorectal cancer chemoresistance. EMBO Mol Med. 2015;7:1350–1365. doi: 10.15252/emmm.201505492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Li C, Wang L, Zhang J, Huang M, Wong F, Liu X, Liu F, Cui X, Yang G, Chen J, Liu Y, Wang J, Liao S, Gao M, Hu X, Shu X, Wang Q, Yin Z, Tang Z, Liu M. CERKL interacts with mitochondrial TRX2 and protects retinal cells from oxidative stress-induced apoptosis. Biochimica et biophysica acta. 2014;1842:1121–1129. doi: 10.1016/j.bbadis.2014.04.009. [DOI] [PubMed] [Google Scholar]
- 115.Chen J, Liu F, Li H, Archacki S, Gao M, Liu Y, Liao S, Huang M, Wang J, Yu S, Li C, Tang Z, Liu M. pVHL interacts with Ceramide kinase like (CERKL) protein and ubiquitinates it for oxygen dependent proteasomal degradation. Cell Signal. 2015;27:2314–2323. doi: 10.1016/j.cellsig.2015.08.011. [DOI] [PubMed] [Google Scholar]
- 116.Segura I, Lange C, Knevels E, Moskalyuk A, Pulizzi R, Eelen G, Chaze T, Tudor C, Boulegue C, Holt M, Daelemans D, Matondo M, Ghesquiere B, Giugliano M, Ruizde Almodovar C, Dewerchin M, Carmeliet P. The Oxygen Sensor PHD2 Controls Dendritic Spines and Synapses via Modification of Filamin A. Cell reports. 2016;14:2653–2667. doi: 10.1016/j.celrep.2016.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.van der Flier A, Sonnenberg A. Function and interactions of integrins. Cell Tissue Res. 2001;305:285–298. doi: 10.1007/s004410100417. [DOI] [PubMed] [Google Scholar]
- 118.Zhang L, Bartley CM, Gong X, Hsieh LS, Lin TV, Feliciano DM, Bordey A. MEK-ERK1/2-dependent FLNA overexpression promotes abnormal dendritic patterning in tuberous sclerosis independent of mTOR. Neuron. 2014;84:78–91. doi: 10.1016/j.neuron.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sarkisian MR, Bartley CM, Chi H, Nakamura F, Hashimoto-Torii K, Torii M, Flavell RA, Rakic P. MEKK4 signaling regulates filamin expression and neuronal migration. Neuron. 2006;52:789–801. doi: 10.1016/j.neuron.2006.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zheng L, Michelson Y, Freger V, Avraham Z, Venken KJ, Bellen HJ, Justice MJ, Wides R. Drosophila Ten-m and filamin affect motor neuron growth cone guidance. PloS one. 2011;6:e22956. doi: 10.1371/journal.pone.0022956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang F, He L, Huangyang P, Liang J, Si W, Yan R, Han X, Liu S, Gui B, Li W, Miao D, Jing C, Liu Z, Pei F, Sun L, Shang Y. JMJD6 promotes colon carcinogenesis through negative regulation of p53 by hydroxylation. PLoS biology. 2014;12:e1001819. doi: 10.1371/journal.pbio.1001819. [DOI] [PMC free article] [PubMed] [Google Scholar]