

ABSTRACT
In antitumor immunity, dendritic cells (DCs) capture, process, and present tumor antigens to T cells, initiating a tumoricidal response. However, DCs are often dysfunctional due to their exposure to the tumor microenvironment (TME), leading to tumor escape from immune surveillance. Here, a vital role of microRNA-155 (miR-155) in regulating the function of DCs in breast cancer is reported. Host miR-155 deficiency enhanced breast cancer growth in mice, accompanied by reduced DCs in the tumors and draining lymph nodes. miR-155 deficiency in DCs impaired their maturation, migration ability, cytokine production, and the ability to activate T cells. We demonstrate that miR-155 regulates DC migration through epigenetic modulation of CCR7 expression. Moreover, IL-6 and IL-10, two cytokines abundant in the TME, are found to impair DC maturation by suppressing miR-155 expression. Furthermore, animal studies show that a lack of miR-155 diminishes the effectiveness of DC-based immunotherapy for breast cancer. In conclusion, these findings suggest that miR-155 is a master regulator of DC function in breast cancer, including maturation, cytokine secretion, migration toward lymph nodes, and activation of T-cells. These results suggest that boosting the expression of a single microRNA, miR-155, may significantly improve the efficacy of DC-based immunotherapies for breast cancer.
KEYWORDS: Breast cancer, CCR7, dendritic cell, epigenetic modulation, Immunotherapy, microRNA-155, tumor microenvironment
Abbreviations
- BMDC
bone marrow-derived dendritic cell
- CCL
C-C motif chemokine ligand
- CCR
C-C chemokine receptor type
- ChIP
Chromatin immunoprecipitation
- CSFE
Carboxyfluorescein succinimidyl ester
- DC
dendritic cell
- GM-CSF
granulocyte macrophage colony-stimulating factor
- IL
interleukin
- Jarid2
Jumonji, AT Rich Interactive Domain 2
- MDSC
myeloid-derived suppressor cell
- PRC2
Polycomb Repressive Complex 2
- SOCS-1
suppressor of cytokine signaling 1
- Suz12
Suppressor of Zeste 12
- TME
tumor microenvironment
Introduction
Immunotherapies are becoming mainstay therapies for several types of cancer.1 Although passive immunotherapies, best represented by T-cell immunomodulatory monoclonal antibodies (checkpoint inhibitors) and adoptive T cell transfer, are making their way into clinics,2 the development of active immunotherapies using tumor antigen-loaded dendritic cells (DCs) has been relatively stagnant.3 In antitumor immune responses, DCs capture tumor antigens, migrate to the draining lymph nodes, and present antigens to naive T cells, resulting in the generation of tumor-specific effector T cells.4-7 However, in tumor-bearing hosts, DCs display a relatively immature and dysfunctional phenotype due to weak tumor immunogenicity and immunosuppressive factors secreted by tumor cells or tumor-associated immunosuppressive cells,4,8,9 resulting in tumor escape from immune surveillance.10 These same caveats also diminish the efficacy of DC-based vaccines for cancers.3 To reinvigorate DC-based immunotherapies, in addition to identifying more potent tumor-specific antigens, new strategies to enhance DC functions through manipulating their intrinsic regulatory machineries and their interaction with the immunosuppressive microenvironment are of tremendous clinical value.
microRNA-155 (miR-155) has been identified as an oncogene in several hematological and solid tumors,11-15 and thus miR-155 inhibition has been suggested as an antitumor strategy.16-19 However, one elegant study suggested that upregulation of miR-155 in breast cancer cells may render these cells more sensitive to radiotherapy.20 Moreover, the pivotal role of miR-155 in host immunity makes the miR-155 inhibition approach for cancer therapy questionable.21-24 We and others recently demonstrated a vital role of miR-155 in defending against infectious diseases and cancer by regulating functions of multiple types of immune cells.25-29 Initial evidence has also shown the importance of miR-155 in DC function in various in vitro settings.22,30,31 However, systemic studies using animal models to examine if miR-155 affects DC functions in tumors are lacking. Here, we reveal a critical role of miR-155 in driving an effective antitumor response in breast cancer via regulation of DC maturation, migration, and T cell activation, and suggest that boosting the expression of miR-155 may significantly improve the efficacy of DC-based immunotherapies for breast cancer.
Results
Host miR-155 deficiency enhances breast cancer growth and metastasis
To examine if host miR-155 plays a role in breast cancer, an orthotopic breast cancer mouse model was used. WT and miR-155−/− C57BL/6 mice were inoculated with EO771 cells in the fourth mammary glands, and tumor growth was monitored. The results showed that host miR-155 deficiency drastically enhanced EO771 tumor growth and metastasis (Fig. 1A–C; Fig. S2A); the effects were much more robust than those previously observed in melanoma and lung cancer models.27,29
Figure 1.

Enhanced breast cancer progression and perturbed leukocyte profile in miR-155−/− mice. (A) Growth curve of EO771 tumors in WT (n = 14) and miR-155−/− mice (n = 20). Tumor volume is shown as mm3. Twenty-five days post-tumor cell inoculation, tumors and draining lymph nodes were removed and analyzed. (B) Average tumor weight in WT and miR-155−/− mice (left); representative tumors are shown (right). (C) Quantification of tumor nodules per lung in WT (n = 7) and miR-155−/− mice (n = 10). (D) and (E) Representative flow cytometry graphs and percentage (D) and absolute cell number (E) of tumor-infiltrating DCs per gram tumor tissue of WT and miR-155−/− mice (n = 5/group) are shown. (F) Average weight of inguinal lymph nodes (left) and representative tumor-draining lymph nodes are shown (right). (G) Absolute cell number of indicated leukocytes within the lymph nodes of WT and miR-155−/− mice (n = 5/group). *p < 0.05; **p < 0.01; ***p < 0.001 by Student's t test.
We previously found that miR-155 plays pivotal roles in regulating the dynamics and functions of myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages in the tumor microenvironment (TME) in melanoma and lung cancer.27,29 To investigate if host miR-155 deficiency influences immune responses in the breast cancer model, flow cytometry was performed to determine the leukocyte profile in the spleen, lymph nodes, and tumor tissue. We found that in the spleens of miR-155−/− breast tumor-bearing mice, there were significantly increased MDSCs (CD11b+ Gr1+) and decreased T cells (CD3+) (Fig. S2B and C) compared to those in WT mice. Interestingly, DCs (CD11c+) were remarkably decreased in the tumor tissue of miR-155−/− mice relative to WT counterparts (Fig. 1D and E), while were comparable in spleens (Fig. S2B and C). We further found that tumor-bearing miR-155−/− mice had much smaller draining lymph nodes with fewer total cells than WT mice (Fig. 1F; Fig. S2D). Flow cytometry analysis showed that lymph nodes of miR-155−/− mice contained much fewer DCs, B cells (CD19+), and T cells compared to those of WT mice (Fig. 1G), whereas the percentages of these cells showed no difference between miR-155−/− and WT mice (Fig. S2E). Furthermore, we observed a remarkable reduction in the classical CD8α+ sub-population of DCs in both the spleen and lymph nodes of tumor-bearing miR-155−/− mice relative to WT mice (Fig. S2F and G). These cells are critical to cross-presenting tumor antigens to CD8+ T cells. Meanwhile, another DC sub-population, plasmacytoid DCs (pDC, CD11c+/B220+) were also decreased in the lymph nodes of tumor-bearing miR-155−/− mice (Fig. S2H).
miR-155 is critical for DC maturation in breast cancer
In cancer immune surveillance, immature DCs capture tumor antigens and undergo maturation, accompanied by the upregulation of MHC-II and co-stimulatory molecules as well as the secretion of cytokines.6,32 DC maturation is a prerequisite for antigen presentation and T cell activation. It was reported that miR-155 is required for toll-like receptor ligand-induced DC maturation.30 To examine if miR-155 regulates DC maturation in breast cancer, we measured MHC-II and costimulatory molecule expression on DCs of multiple organs from WT and miR-155−/− mice carrying EO771 tumors. We found an overall defective pattern of expression of MHC-II and costimulatory markers on splenic DCs (Fig. 2A), tumor-infiltrating DCs (Fig. 2B), and lymph node DCs (Fig. 2C) from miR-155−/− mice compared to WT counterparts. To evaluate the effects of miR-155 deficiency on DC maturation in vitro, we generated bone marrow-derived dendritic cells (BMDCs) and pulsed them with EO771 cell lysate and EO771-conditioned medium (ECM). We used the combination of tumor cell lysate and ECM because we found the combination is more effective than either tumor cell lysate or ECM alone in promoting DC maturation (Fig. S1). Apoptotic tumor cells lysate contains the entire antigen repertoire for DCs to elicit a broad polyclonal tumor-specific response. Meanwhile, soluble mediators released by dying cells such as high mobility group box 1 (HMGB1), heat shock proteins (HSP) may serve as toll-like receptor ligands and effective vaccine adjuvants for DC activation.7 In agreement with the in vivo data, the pulsed miR-155−/− BMDCs exhibited significantly lower expression of maturation markers compared to WT ones (Fig. 2D).
Figure 2.
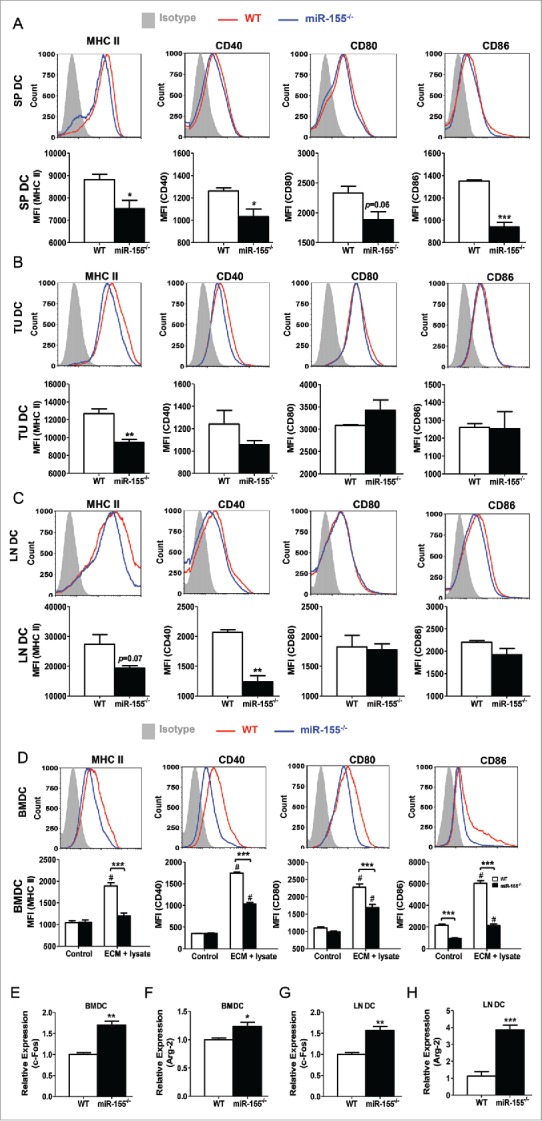
miR-155 deficiency impairs DC maturation. Tumor-bearing mice were sacrificed 25 d post-inoculation, and single cell suspensions were acquired from spleen, tumor tissue, and lymph nodes. (A)–(C) Expression of MHC II, CD40, CD80, and CD86 on DCs was analyzed by flow cytometry. Representative flow cytometry graphs (upper) and column summary of the data (lower) are shown. Spleen DCs (SP DC) (A) and lymph node DCs (LN DC) (C) were gated from CD11c+ population, whereas tumor-infiltrating DCs (TU DC) (B) were gated from CD45+ CD11c+ population. Mean fluorescence intensity (MFI) of cell markers from four experiments were quantified. (D) BMDCs were treated with tumor cell lysate plus ECM in vitro for 48 h and the expression of MHC II, CD40, CD80, and CD86 was determined by flow cytometry (n = 3). Representative flow cytometry graphs of ECM+cell lysate-treated samples are shown (upper) and MFI of cell markers were quantified (lower). (E) and (F) mRNA levels of c-Fos (E) and Arg-2 (F) in BMDCs treated with tumor cell lysate plus ECM were determined by RT-PCR (n = 3). (G) and (H) mRNA levels of c-Fos (G) and Arg-2 (H) in DCs isolated from tumor-draining lymph nodes were determined by RT-PCR (n = 3). (A)–(C), (E)–(H), Student's t test; D, two-way ANOVA followed by the Tukey multiple comparison test. #p < 0.05 versus control group; *p < 0.05; **p < 0.01; ***p < 0.001 vs. WT group.
Recent studies have shown that repression of c-Fos and Arginase-2, both verified as miR-155 targets, was critical for DC maturation and function in various contexts.30,31 To determine if miR-155 affects the expression of these genes in DCs loaded with tumor-associated antigens, we treated BMDCs in vitro with tumor cell lysate and ECM, and found that miR-155−/− BMDCs expressed substantially higher levels of c-Fos and Arg-2 compared to WT ones (Fig. 2E and F). Consistent with these findings, both c-Fos and Arg-2 expression levels were significantly enhanced in lymph node DCs of tumor-bearing miR-155−/− mice relative to WT mice (Fig. 2G and H). Taken together, these results indicate a requirement of miR-155 expression for efficient maturation of DCs in breast cancer.
miR-155−/− DCs are defective in stimulating T cell activation and proliferation
After maturation, DCs are poised to present antigens to and activate T cells. To examine if miR-155 deficiency in DCs affects their ability to stimulate T cell activation and proliferation, we co-cultured naive splenic T cells from healthy WT mice with WT or miR-155−/− BMDCs pulsed with tumor cell lysate and ECM. We found that T cell activation induced by pulsed miR-155−/− BMDCs was significantly impaired characterized by decreased expression of CD25 and CD69 (Fig. 3A and B). T cell proliferation induced by pulsed miR-155−/− BMDCs was significantly inhibited compared to that induced by WT BMDCs (Fig. 3C). Furthermore, when co-cultured with miR-155−/− BMDCs pulsed by tumor cell lysate and ECM, T cells displayed a significant decrease in IFNγ production (Fig. 3D).
Figure 3.
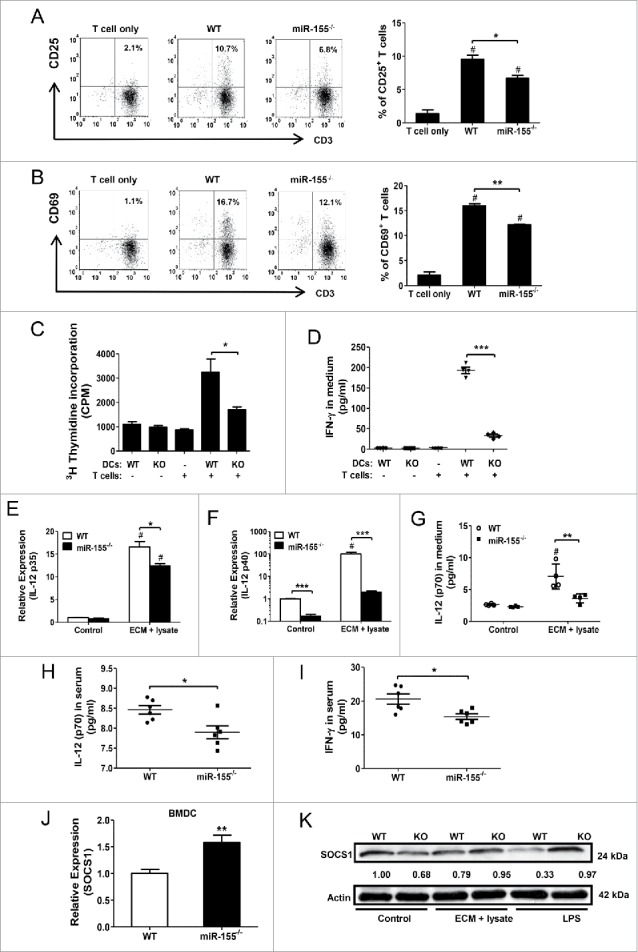
miR-155 is critical for DC-mediated T cell activation. Naive T cells isolated from spleens of healthy WT mice were co-cultured with tumor cell lysate and ECM pulsed WT or miR-155−/− BMDCs (T cells:DCs = 10:1). Twenty-four hours after co-culture, T cell activation was determined by examining CD25 and CD69 expression on CD3+ cells by flow cytometry. T cell only group served as control. (A) Representative scatter plots (left) and percentage of CD3+ CD25+ cells (right) are shown (n = 3). (B) Representative scatter plots (left) and percentage of CD3+ CD69+ cells (right) are shown (n = 3). (C) Five days after co-culture, T cell proliferation was measured by [3H] thymidine incorporation assay (n = 4). (D) Co-culture media was collected on day 3 and IFNγ concentrations were measured by ELISA; BMDC and T cell only group were used as controls (n = 4). (E) and (F) mRNA levels of IL-12 p35 (E) and IL-12 p40 (F) were determined by RT-PCR in WT and miR-155−/− BMDCs with or without treatment (n = 3). (G) Forty-eight hours post-treatment, IL-12 p70 concentrations in BMDC culture media were determined by ELISA; BMDCs without tumor lysate and ECM treatment were used as controls (n = 4). (H) and (I) IL-12 p70 (H) and IFNγ (I) concentrations in sera of WT and miR-155−/− tumor-bearing mice at the end point (Day 25) were determined by ELISA (n = 6). (J) and (K) mRNA (J) and protein levels (K) of SOCS1 in BMDCs treated with tumor lysate and ECM were determined by RT-PCR and western blot, respectively. LPS stimulated cells were used as positive control. (A)–(D) one-way ANOVA followed by the Tukey multiple comparison test. #p < 0.05 versus T cell only group. (E)–(G) two-way ANOVA following by the Tukey multiple comparison test. #p < 0.05 versus control group. (H)–(J) Student's t test. *p < 0.05; **p < 0.01; ***p < 0.001 versus WT group.
In order for mature DCs to effectively stimulate T cells, not only is antigen-presentation through MHC-antigen complex and co-stimulatory molecules required, but also additional signals such as IL-12 are necessary.33 IL-12 augments IFNγ production by CD4+ T cells, NK cells, and CD8+ T cells, and promotes longer conjugation events between CD8+ T cells and DCs.34 Since miR-155 was reported to regulate IL-12 production by targeting SOCS1 in DCs,22 we determined IL-12 production in DCs in the context of breast cancer. We found that upon tumor cell lysate and ECM treatment, miR-155−/− BMDCs displayed impaired expression of IL-12 mRNAs (both p35 and p40 sub-units) (Fig. 3E and F) and reduced secretion of IL-12p70 (Fig. 3G) compared to WT BMDCs. In agreement with the in vitro data, we found a reduction of both IL-12p70 and IFNγ concentrations in miR-155−/− mouse sera (Fig. 3H and I). As expected, both mRNA and protein levels of SOCS1 were enhanced in miR-155−/−-deficient BMDCs treated with tumor cell lysate and ECM, relative to WT BMDCs (Fig. 3J and K), suggesting that miR-155 positively regulates IL-12 production in DCs through inhibition of SOCS-1 expression in breast cancer.
miR-155 deficiency impairs dendritic cell migration by suppressing CCR7 expression
To present tumor antigens to and activate T cells, DCs need to migrate to the draining lymph nodes where naive T cells reside in the deep cortex. To examine if miR-155 deficiency restricts the migratory capacity of DCs to nearby lymph nodes, we performed in vivo DC migration experiments by inoculating CFSE-labeled WT or miR-155−/− DCs pulsed with tumor cell lysate and ECM into the groins of tumor-bearing WT mice. Forty-eight hours later, the percentage of CSFE positive cells in the draining lymph nodes of WT BMDC recipients was 0.32%, whereas it was only 0.18% in miR-155−/− BMDC recipients (Fig. 4A). Similarly, in an in vitro migration assay, upon tumor cell lysate and ECM treatment, miR-155−/− BMDCs displayed a defective migratory capacity toward CCL19 (Fig. 4B).
Figure 4.

miR-155 affects DC migration by epigenetically regulating CCR7 expression. (A) In vivo migration of CFSE-labeled DCs toward draining lymph nodes was measured by flow cytometry. WT or miR-155−/− BMDCs were pulsed with tumor cell lysate and ECM, labeled with CFSE, and implanted into the groins of tumor-bearing WT mice. Typical scatter plots (left) and percentages (right) of CFSE+ population are shown (n = 4). (B) In vitro migration of WT or miR-155−/− BMDCs pulsed with tumor cell lysate and ECM was determined by trans-well migration assay. Immature BMDCs maintained in DC medium were used as controls. Representative fluorescence images are shown (left). Cells were counted in 10 random fields per sample at 200 × magnification and quantified (right) (n = 3). (C) CCR7 mRNA level in DCs isolated from spleen, tumor, and lymph node of WT or miR-155−/− mice carrying EO771 tumors was determined by RT-PCR (n = 3). (D) CCR7 mRNA level in BMDCs treated with tumor lysate and ECM in vitro was determined by RT-PCR; immature BMDCs served as controls (n = 3). (E) and (F) Cell surface CCR7 expression on DCs isolated from spleen (E) or lymph node (F) of tumor-bearing WT or miR-155−/− mice was determined by flow cytometry. MFIs of CCR7 from three independent experiments are shown. (G) Quantified MFI of CCR7 on BMDCs matured by tumor cell lysate and ECM in vitro was shown. (H) Enrichment of H3K27me3 at Ccr7 promoter (left) and first intron (right) in WT and miR-155−/− BMDCs treated with tumor lysate and ECM was determined by qPCR. (I and J) Jarid2 protein levels in DCs isolated from lymph nodes and spleens of WT and miR-155−/− tumor-bearing mice (I), and in tumor-associated antigen-treated BMDCs (J) were detected by western blot; relative intensities of Jarid2 were labeled under the bands. (K) ChIP-qPCR was performed to detect the recruitment of Suz12 at the Ccr7 promoter (left) and first intron (right) in WT and miR-155−/− BMDCs. (A), (C), (E) and (F), (H), and (K) Student's t test. (B), (D), and (G) two-way ANOVA followed by the Tukey multiple comparison test. #p < 0.05 versus control group; *p < 0.05; **p < 0.01; ***p < 0.001 versus WT group.
CCR7 is the driving force for DCs to migrate following the CCL19/CCL21 gradient in lymph nodes.35 We measured CCR7 mRNA levels in DCs isolated from tumor-bearing mice and observed lower CCR7 mRNA expression in spleen, tumor, and lymph node DCs from miR-155−/− mice compared to their WT counterparts (Fig. 4C). Moreover, miR-155−/− BMDCs, when pulsed with tumor cell lysate and ECM, also exhibited a reduced CCR7 level compared with WT ones (Fig. 4D). We further determined cell surface CCR7 expression on DCs by flow cytometry, and significantly observed lower CCR7 MFI on DCs in the spleen (Fig. 4E; Fig. S3A) and lymph nodes (Fig. 4F; Fig. S3B) of tumor-bearing miR-155−/− mice; in vitro matured miR-155−/− BMDCs also exhibited a lower level of CCR7 than WT ones (Fig. 4G; Fig. S3C). Taken together, these results indicate that, in breast cancer, reduced CCR7 expression in miR-155−/− DCs may restrain their migration toward lymph nodes.
miR-155 epigenetically regulates CCR7 expression in DCs
It was recently reported that histone 3 lysine 27 trimethylation (H3K27me3) modulates CCR7 expression in DCs.36 To investigate if miR-155 regulates CCR7 expression in DCs by affecting H3K27me3, a chromatin immunoprecipitation (ChIP) assay was performed. MiR-155−/− BMDCs pulsed with tumor cell lysate and ECM were found to contain significantly more H3K27me3 at the CCR7 promoter and first intron than WT BMDCs (Fig. 4H). Jumonji, AT Rich Interactive Domain 2 (Jarid2), a direct miR-155 target, recruits Polycomb Repressive Complex 2 (PRC2) to specific sites of the genome and represses target gene expression through H3K27me3.37-39 We analyzed Jarid2 expression in DCs isolated from spleens and lymph nodes of tumor-bearing mice and observed an increased expression of Jarid2 in miR-155−/− DCs relative to WT ones (Fig. 4I). A similar result was obtained in miR-155−/− BMDCs when treated with tumor cell lysate and ECM (Fig. 4J). To determine whether increased Jarid2 in miR-155−/− BMDCs represses CCR7 expression by recruiting PRC2, another ChIP assay was performed to detect a core PRC2 component: Suppressor of Zeste (Suz12); and an increased Suz12 occupancy at the CCR7 promoter was observed in miR-155−/− BMDCs (Fig. 4K). Taken together, these data suggest that miR155 directly represses the expression of the DNA binding protein Jarid2, thus indirectly diminishes the recruitment of PRC2 complex to the promoter region of CCR7. Reduced PRC2 complex further decreases H3K27me3 presence at CCR7 locus, leading to enhanced expression of CCR7 in DCs.
Tumor cell-derived IL-6 and IL-10 inhibit DC function via repressing miR-155
Above data suggest that miR-155 is essential for DC maturation and function in the antitumor response to breast cancer. We speculate that miR-155 upregulation may be defective in DCs in breast cancer, and thus fewer DCs are sufficiently mature to migrate to lymph nodes and trigger effective antitumor immunity, whereas relatively immature DCs are retained at the tumor site or in circulation. We compared miR-155 expression in DCs isolated from lymph nodes, spleen, and tumor tissues of the same tumor-bearing WT mice. We observed a significantly lower miR-155 level in splenic and tumor-infiltrating DCs compared to that of lymph node DCs (Fig. 5A). In line with miR-155 expression, the expression of DC maturation markers CD40, CD86, and CCR7 was significantly lower in splenic and tumor-infiltrating DCs relative to lymph node DCs (Fig. 5B–D; Fig. S4).
Figure 5.
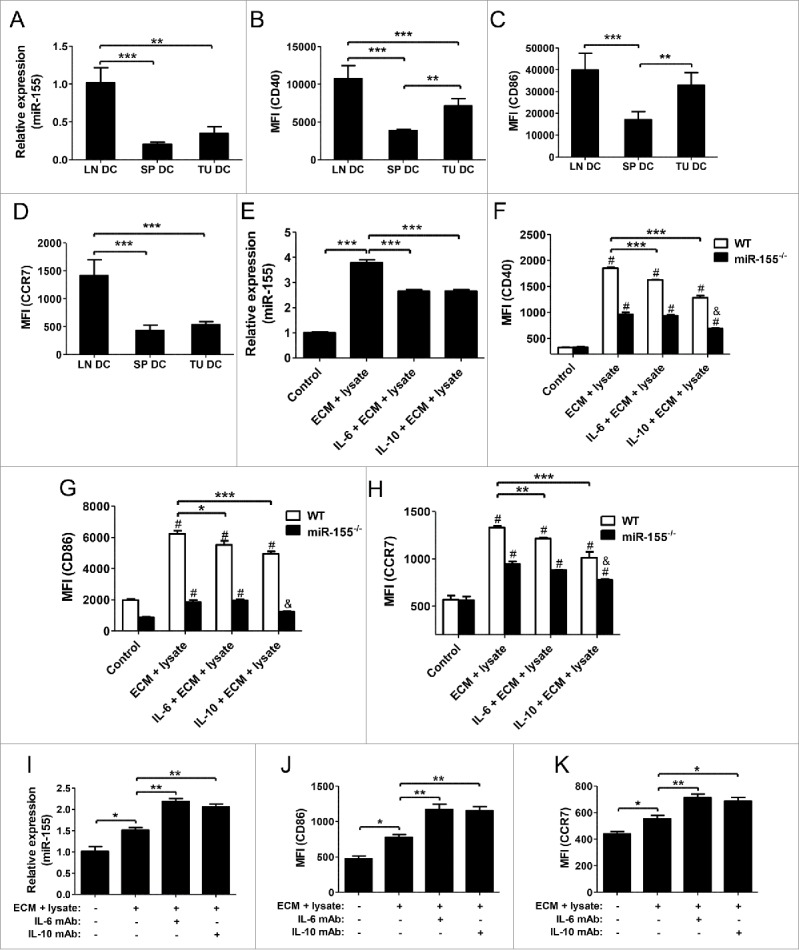
IL-6 and IL-10 inhibit DC maturation through suppressing miR-155. (A) miR-155 expression in lymph node DCs, splenic DCs, and tumor-infiltrating DCs of tumor-bearing WT mice was detected by RT-PCR (n = 3). (B)–(D) CD40 (B), CD86 (C), and CCR7 (D) expression levels on above DCs were determined by flow cytometry under the same voltage and compensation conditions (n = 3). (E)–(H) WT BMDCs were pretreated with IL-6 (100 ng/mL) or IL-10 (50 ng/mL) for 24 h prior to the additional treatment with EO771 tumor cell lysate and ECM; cells without ECM plus cell lysate treatment were used as negative control (n = 3). Expression levels of miR-155 (E) and CD40 (F), CD86 (G), and CCR7 (H) were determined by RT-PCR and flow cytometry, respectively. (I)–(K) To neutralize IL-6 and IL-10 in ECM, IL-6 mAb (5 μg/mL), or IL-10 mAb (1 μg/mL) was added into the treatment medium and incubated for 2 h at 37°C before BMDC stimulation (n = 3). Expression of miR-155 (I), CD86 (J), and CCR7 (K) were determined by RT-PCR and flow cytometry, respectively. (A)–(E) and (I)–(K) one-way ANOVA followed by the Tukey multiple comparison test; (F)–(H) two-way ANOVA followed by the Tukey multiple comparison test. #p < 0.05 versus their respective control group; &p < 0.05 relative to positive control; *p < 0.05; **p < 0.01; ***p < 0.001.
It has been shown that DCs in the TME possess a relatively immature phenotype,40 and the dysfunction of DCs in tumors may be a consequence of their exposure to soluble factors, such as IL-10 and IL-6, in the TME.41,42 To elucidate if these soluble factors inhibit DC maturation via suppressing miR-155 expression, we conducted in vitro experiments by pretreating WT and miR-155−/− BMDCs with IL-6 or IL-10 before pulsing them with maturation stimuli. We found that both IL-6 and IL-10 pretreatment significantly inhibited miR-155 expression (Fig. 5E) and maturation of WT BMDCs (Fig. 5F–H). Conversely, depletion of IL-6 or IL-10 from tumor cell-conditioned medium using neutralizing antibodies significantly elevated the expression of miR-155 (Fig. 5I) and DC maturation markers (Fig. 5J and K; Fig. S5). Taken together, these results demonstrate that some soluble factors in the TME, such as IL-6 and IL-10, impair DC maturation through diminishing miR-155 upregulation. Therefore, targeted removal of these inhibitory soluble factors in TME may unleash the full potential of DCs to trigger antitumor immunity in breast cancer.
miR-155 deficiency diminishes the efficacy of DC-based immunotherapy for breast cancer
To confirm the contribution of DC miR-155 to the antitumor immune response to breast cancer, mice carrying orthotopic EO771 breast tumors were adoptively transferred with WT or miR-155−/− BMDCs pulsed with tumor cell lysate and ECM. A set of mice were sacrificed 48 h after the first DC vaccine administration, and enlargement of both draining lymph nodes and spleens were observed in WT but not in miR-155−/− BMDC-treated mice (Fig. 6A and C). Also, an augmentation of activated T cells was only detected in the draining lymph nodes (Fig 6B; Fig. S6A), but not in the spleens (Fig. 6D; Fig. S6B) of WT BMDC-transferred mice. When the mice were sacrificed 5 d post-DC transfer, significantly smaller tumors were observed in mice that received WT BMDCs but not in those received miR-155−/− cells, compared to mice without receiving DCs (Fig. 6E). To monitor tumor growth and survival rate, additional DC vaccine was injected twice a week for three consecutive weeks in another set of mice. Although tumor growth rates in both groups were restricted compared to control mice, miR-155−/−-deficient DCs showed less beneficial effects (Fig. 6F). Moreover, WT DC-treated mice survived much longer than those treated with miR-155−/− DCs (Fig. 6G).
Figure 6.
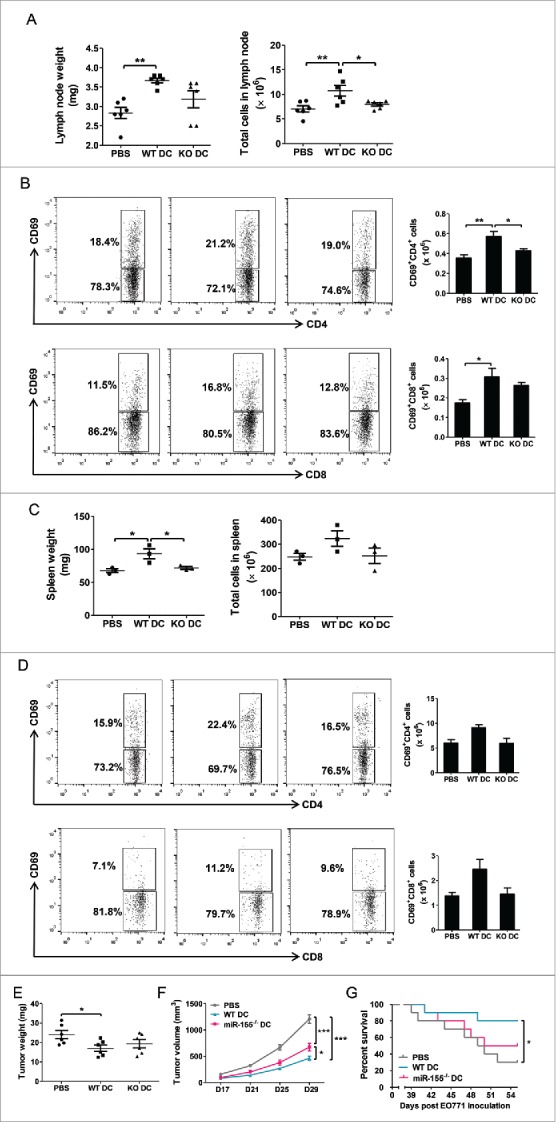
miR-155 deficiency diminishes the efficacy of DC-based immunotherapy for breast cancer. WT C57Bl/6 mice were implanted with EO771 tumor cells; 24 h later, WT BMDCs (WT DC) or miR-155−/− BMDCs (KO DC) matured by tumor cell lysate and ECM in vitro were injected s.c. as vaccines. (A)–(D) Forty-eight hours post DC vaccination, lymph nodes and spleens were removed. Mice injected with PBS served as control. Lymph nodes were weighed (left) and absolute cell numbers in lymph nodes were counted (right) (A). Activated CD4+ T cells (upper) and CD8+ T cells (lower) in lymph nodes were measured by flow cytometry. Representative flow cytometry graphs (left) and column summary of activated T cell number (right) are shown (B). Spleens were weighed (left) and absolute cell numbers in spleens were counted (right) (C). Activated CD4+ T cells (left) and CD8+ T cells in spleens were measured by flow cytometry. Representative flow cytometry graphs (left) and column summary of activated T cell number (right) are shown (D). (E) Five days post DC vaccination, some mice were sacrificed and tumors were weighed. (F) and (G) DC vaccine was injected twice a week for consecutive three weeks in some mice; growth kinetics of EO771 tumors was followed until Day 29 after tumor inoculation (F), and survival rates of tumor-bearing mice were monitored until Day 55 after tumor inoculation (G). N = 10 in each group. (A)–(F), one-way ANOVA followed by the Tukey multiple comparison test. (G) Survival data were analyzed with the Mantel-Cox log-rank test. *p < 0.05; **p < 0.01; ***p < 0.001.
Discussion
By applying orthotopic breast cancer models and using an in vitro cell culture system mimicking the TME, we have demonstrated in this study that miR-155 is required for DCs to exert effective functions in the antitumor response, including maturation, cytokine secretion, migration toward lymph nodes, and activation of T-cells, and that tumor-derived IL-6 and IL-10 can disrupt DC dynamics and function by compromising miR-155 induction in TME (Fig. 7). We further showed that miR-155 deficiency diminished the therapeutic potency of DC-based vaccine for breast cancer.
Figure 7.
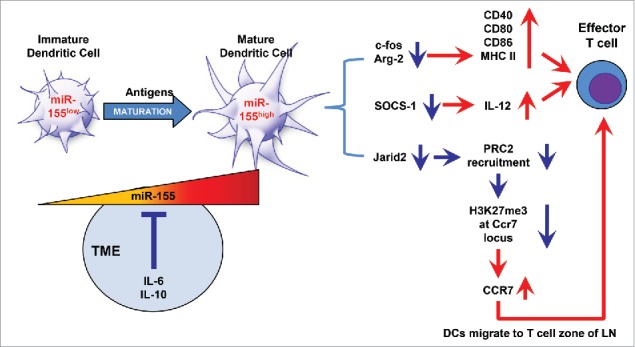
Schematic graph depicting the molecular mechanisms underlying miR-155-mediated antitumor immune response in DCs. During early tumor development, immature DCs recognize and take up tumor antigens, resulting in elevated miR-155 expression, which affects the following three functions of DCs: (1) DC maturation: Upregulation of MHC II and costimulatory molecules (CD40, CD80, and CD86) by miR-155-mediated suppression of c-Fos and Arg-2 contributes to DCs maturation; (2) T cell activation: Enhanced IL-12 p70 production by miR-155 via inhibiting SOCS1 is essential to elicit T cell activation; and (3) DC migration: miR-155 reduces H3K27me3 enrichment at the Ccr7 locus through repressing Jarid2 expression and PRC2 recruitment. Elevated CCR7 expression drives DCs migration toward the T cell zone of draining lymph nodes, where DCs activate effector T cells and initiate a tumor-specific immune response. However, with the development of breast cancer, miR-155 expression is repressed by gradually elevated soluble factors (such as IL-6 and IL-10), mainly secreted by tumor cells and immunosuppressive cells in the TME, leading to DC dysfunction and ultimately tumor escape from immune surveillance.
It has been reported that during DC maturation by TLR ligands in various settings, suppression of miR-155 targets, such as SOCS1, c-Fos, and Arg-2, is required for MHC II and costimulatory molecule expression as well as IL-12 production.22,30,31 Our current study demonstrated for the first time in the setting of breast cancer that miR-155−/− DCs displayed a profound defect in the ability to process and present tumor antigens to T cells accompanied by an accumulation of the aforementioned miR-155 targets. Upregulation of CCR7 expression is critical for driving mature DCs to migrate toward the T cell zone of draining lymph nodes.35,43 One of the major obstacles DC-based immunotherapy faces is that the migration of ex vivo pulsed DCs to lymph nodes is defective.44 Correlation between CCR7 and miR-155 expression in DCs has been found in previous gene screening,45 but whether miR-155 regulates CCR7 expression on DCs in the context of cancer has not been reported. Our results revealed that miR-155 can manipulate H3K27me3 enrichment at the CCR7 locus by targeting Jarid2, a direct miR-155 target, thus epigenetically upregulate CCR7 expression.
In many tumors, DCs remain immature, and thus are ineffective in triggering antitumor immunity. Tumor-derived soluble factors such as IL-6 and IL-10 are believed to impair DC functions in the TME,41,42 In our study, we found that both IL-6 and IL-10 inhibited BMDCs maturation through repression of miR-155 upregulation, and their neutralizing antibodies de-repressed miR-155 upregulation and consequently promoted DC maturation. The underlying molecular mechanisms, however, need further elucidation.
Importantly, when we applied DC vaccines in orthotopic breast cancer models and revealed a diminished tumor-eliminating effect of miR-155−/− DC vaccine. It was recently reported that miR-155 upregulation in DCs is sufficient to break immune tolerance through targeting SHIP1 in the context of auto-immunity.46 Our data suggest that ex vivo achieved miR-155 overexpression in DCs may substantially improve the antitumor efficacy of a DC-vaccine for breast cancer.
In summary, our results revealed a crucial role of miR-155 for DCs in initiating an effective antitumor immune response and diminished miR-155 upregulation in DCs in the TME may be one of the mechanisms by which tumor cells escape immune surveillance. Our study suggested that boosting the expression of a single microRNA, miR-155, may significantly improve the efficacy of DC-based immunotherapies for breast cancer and possibly other solid tumors. In addition, an ex vivo DC engineering strategy to generate miR-155-overexpressing DC-based anticancer therapies can avoid the potential oncogenic side-effects of systemic miR-155 delivery.
Materials and methods
Mice
All animal experiments were approved by the Institutional Animal Care and Use Committee at the University of South Carolina. Wild type (WT) and miR-155−/− C57BL/6 mice, as well as Balb/c mice at the age of 8–10 weeks were obtained from Jackson Laboratories. Mice were maintained in pathogen-free conditions at the University of South Carolina according to National Institutes of Health guidelines.
Cell culture, tumor-conditioned medium, and tumor cell lysate
The 4T1 cells were obtained directly from the American Type Culture Collection (ATCC) in 2013. EO771 cells, developed from an ER+ spontaneous mammary adenocarcinoma,47,48 were obtained in 2012 and maintained in culture as previously described.49 These two cell lines were authenticated in 2016 by IDEXX Laboratories (IDEXX BioResearch Case #7479-2016). The samples were confirmed to be of mouse origin and no mammalian interspecies contamination was detected. A genetic profile was generated for each sample using a panel of microsatellite markers for genotyping. The 4T1 cells were confirmed to match identically to the genetic profile established for this cell line. The genetic profile for EO771 cell line is more consistent with having been derived from a mouse with a mixed/stock genetic background. A very similar profile has been seen in other sources of EO771 cell line.50
Cells were maintained in high-glucose Dulbecco's modified Eagle's medium (DMEM, Gibco, 11995065) supplemented with 10% fetal bovine serum (FBS,Gibco, 10437028) and a combination of penicillin/streptomycin (Gibco, 15140122) at 37°C in a humidified 5% CO2 atmosphere.
To obtain tumor-conditioned medium, EO771 or 4T1 cells were seeded at 5 × 106 cells per 75 cm2 bottle and cultured to 70% confluence. The medium was then replaced with serum-free DMEM. After 48 h, the culture medium was collected, filtered through 0.45 μm filters, and further concentrated 20-fold using Centrifugal Filters with a 3K molecular weight cutoff (Millipore, UFC900324).
For preparation of tumor cell lysate, tumor cells were cultured for 48 h in serum-free DMEM and then disrupted by four freeze-thaw cycles in liquid nitrogen and a 37°C water bath. The solution was centrifuged at 1,000 × g for 10 min to remove insoluble cell fragments, and the supernatant was referred to as cell lysate and used as a source of tumor-associated antigen.
Orthotopic breast cancer model
A mouse orthotopic breast cancer model was established as previously described.49 Briefly, 2 × 105 EO771 or 4T1 cells in 20 μL of PBS were injected on both sides of the fourth pair of mammary fat pads of WT and miR-155−/− mice. The tumor size was measured using a caliper on indicated days. Tumor volume was determined by the formula: length × width2/2. At the experimental end point, mice were sacrificed; lymph nodes, lungs, tumors, and spleens were removed, weighed, and processed for FACS, immunohistochemistry (IHC) analysis, and other analyses.
Cell isolation
Cells from lymph nodes and spleens were isolated by mechanical disruption. Tumors were weighed, cut into small fragments (< 3 mm), and digested in 5 mL of dissociation solution (RPMI 1640 medium (Gibco, 11875-093) supplemented with 10% FBS, Collagenase type I (200 U/mL, Worthington-biochem, LS004197) and DNase I (100 μg/mL, Roche, 10104159001)) for 1 h at 37°C. Erythrocytes were lysed by red blood cell lysing buffer (Sigma, 11814389001). Cell suspensions were passed through 70-μm cell strainers, and then washed and resuspended in staining buffer (PBS with 2% FBS).
For DC and T cell purification, 1 × 108 cells isolated as described above were sequentially incubated with 20 μL PE-conjugated CD11c (Biolegend, 117307) or CD3 (Biolegend, 100308) antibody, 100 μL PE selection cocktail, and 50 μL magnetic nanoparticles (EasySep™ Mouse PE Positive Selection Kit, 18554), and then were separated using the magnet according to the manufacturer's instructions. In all samples, a purity of >95% was achieved as determined by flow cytometry.
Flow cytometry
Flow cytometry analysis was performed as previously described.27 Briefly, RBC depleted cells were stained with fluorescein conjugated antibodies (The antibodies were listed in Table S1) in staining buffer for 30 min on ice, in the dark. Samples were washed twice with staining buffer; cells were acquired using a BD FACS Aria II flow cytometer and data were analyzed by FACSDIVA software. In most cases, 20,000 live events were collected per sample.
Generation of bone marrow-derived DCs (BMDCs) and tumor antigen pulsing
Bone marrow cells were flushed from mouse femurs and tibias and erythrocytes were depleted by red blood cell lysing buffer. The resulting cells were cultured at a density of 1 × 106 cells/mL in DC medium (RPMI 1640 medium supplemented with 10% FBS and a combination of penicillin/streptomycin, 50 μM β-mercaptoethanol, 10 ng/mL recombinant granulocyte macrophage colony-stimulating factor (rGM-CSF, BioAbChem, 42-GMCSF), and 10 ng/mL rIL-4 (BioAbChem, 42-IL4). Fresh medium was added on Day 3. After 7 d of culture, loosely adherent cells were harvested by gentle pipetting (each preparation was confirmed >90% positive for CD11c (Fig. S1A)) and resuspended in DC medium or treatment medium (DC medium with the addition of 20% (v/v) of concentrated ECM and 100 μg/mL of tumor lysate) to a final density of 0.5 × 106 cells/mL and then cultured for 48 h (the combination of tumor lysate and tumor-conditioned medium was superior to either lysate or ECM alone in initiating DC maturation (Fig. S1B)). After pulsation, DCs were collected by non-enzymatic cell dissociation solution (Sigma, C5914) for various purposes.
T cell activation and proliferation assays
BMDCs (1 × 105 cells in 1 mL of DC medium or treatment medium) were placed in 24-well plates for 48 h. Then, the supernatant was removed and RPMI 1640 complete medium containing 1 × 106 purified CD3+ T cells was added into each well. For T cell activation assays, T cells were harvested after 24 h of co-culture, and cell surface expression of CD25 or CD69 was assessed with flow cytometry. For T cell proliferation assays, cells were co-cultured for 5 d, with 1 µCi [3H] thymidine (Amersham Pharmacia Biotech, NET027E) added to each well during the final 18 h. An equivalent amount of fresh medium was replaced on Day 3. Soluble rIL-2 (20 U/mL, Biolegend, 714604) was applied to support the proliferation of purified T cells. Cells were harvested and the incorporated radioactivity was measured in a β-scintillation counter (Microbeta 1450). Proliferation of T cells or BMDCs alone was examined in parallel as controls.
Enzyme-linked immunosorbent assay (ELISA)
Cell-free supernatant from BMDC cultures or BMDC/T-cell co-cultures was harvested at the indicated time points. Concentration of IL-12/p70 or IFNγ was measured. To measure cytokine concentrations in sera, blood samples were collected from WT and miR-155−/− mice bearing breast tumors and allowed to clot for 30 min at room temperature; the samples were then centrifuged at 3,000 × g for 10 min; the serum layer was removed and diluted 1:5. Cytokine concentrations were determined by Mouse IL-12 (p70) ELISA MAX™ Deluxe kit (Biolegend, 433604) and Mouse IFNγ ELISA MAX™ Deluxe kit (Biolegend, 430804) according to the manufacturer's instructions. All samples were tested in triplicate.
Quantitative real-time PCR (qPCR) for mRNA expression
Total RNA was extracted using QIAzol Lysis Reagent (Qiagen, 79306). One microgram of RNA from each sample was reverse-transcribed using iScript™ cDNA Synthesis Kit (Bio-Rad, 1708891). qPCR was performed on a Bio-Rad CFX96 system using iQ™ SYBR® Green Supermix (Bio-Rad, 1708882). All primers used for qPCR analysis were synthesized by Integrated DNA Technologies. The primer sequences were listed in Table S2. All assays were performed following the manufacturer's instructions. The relative amount of target mRNA was determined using the comparative threshold (Ct) method by normalizing target mRNA Ct values to those of 18S RNA. PCR thermal cycling conditions were 3 min at 95°C, 40 cycles of 15 s at 95°C, and 58 s at 60°C. Samples were run in triplicate.
miR-155 expression quantification
miR-155 expression was measured according to the manufacturer's instructions using the miScript PCR System (QIAGEN) which is comprised of the miScript II RT Kit (218161), miScript SYBR Green PCR Kit (218073), and miScript Primer Assay (See Table S2).
Western blot analysis
Cells were lysed in RIPA buffer (Thermo Scientific, 89900) supplemented with protease inhibitor cocktail (Sigma, 20-201). Total cellular extracts (30 μg) were separated in 4%–20% SDS-PAGE precast gels (Bio-rad, 4561095) and transferred onto nitrocellulose membranes. Membranes were first probed with anti-SOCS1 (1:1000, abcam, ab48137), anti-Jarid2 (1: 1000, Genetex, GTX100657) or anti-β-actin (1:1000, Sigma,A2206) antibodies, followed by goat anti-rabbit secondary antibody conjugated with HRP (1:5000, Millipore,AP132P). Protein detection was performed using Pierce ECL Western Blotting Substrate (Thermo Scientific, 32106).
In vivo DC migration
Following treatment with tumor lysate and tumor-conditioned medium for 48 h, 1 × 106 WT, or miR-155−/− BMDCs were labeled with Carboxyfluorescein succinimidyl ester (CSFE,Biolegend, 4223801) according to the manufacturer's protocol and then injected subcutaneously (s.c.) into the groins of the WT mice implanted with EO771 cells 1 d earlier. Lymph nodes were harvested 48 h after BMDC injection, and CFSE-labeled cells were determined by flow cytometry.
In vitro DC migration
24-well plates were pre-equilibrated by adding 0.5 mL of serum-free RPMI 1640 medium (SFM) containing 100 ng/mL CCL19 (Sigma, SRP4495); then 2 × 105 BMDCs in 0.3 mL of RPMI 1640 were seeded into the upper chamber of trans-well inserts with 8 μm pore size (Corning, 3422) and allowed to migrate for 3 h at 37°C. The upper surfaces of the trans-well inserts were swabbed using cotton buds. Cells that migrated to the lower surfaces were fixed with 4% formaldehyde and stained with 4',6-diamidino-2-phenylindole (DAPI, 1 μg/mL, Sigma, 102362) for 1 min. The inserts were then cut out, mounted onto slides, and imaged under a Nikon ECLIPSE E600 fluorescence microscope at 200 × magnification (ten fields per membrane, triplicate for each experimental group). DAPI stained cells were quantified using Image-Pro Plus analysis software (Media Cybernetics).
ChIP assay
BMDCs (6 × 106) seeded in 100-mm dishes were treated with tumor lysate and tumor-conditioned medium for 48 h. ChIP assay was performed following standard protocol. In brief, cultures were cross-linked with 1% paraformaldehyde and chromatin was sheared to 200 base pairs. Chromatin (20 µg/IP) was immunoprecipitated with antibodies against histone H3K27me3 or SUZ12, or with a negative control IgG. ChIP-derived DNA was recovered and quantified by qPCR. Data reflect percent input of each qPCR reaction with the indicated primer mixes. Antibodies and validation primers were listed in Tables S1 and 2.
Immunization
Twenty-four hours post-tumor inoculation in WT mice, 0.5 × 106 tumor-associated antigen pulsed BMDCs were injected s.c. into the groins of the mice once, or twice a week for three consecutive weeks for the following experiments: mice injected once with the DC vaccine were sacrificed 48 h later for in vivo T cell activation analysis; mice that received repeated DC immunizations were used for monitoring tumor growth, survival rate and pulmonary metastasis.
Statistical analysis
All statistical analysis was performed using the GraphPad Prism software 5.0. The data were presented as the mean ± SEM. When applicable, unpaired student's t-test, one-way or two-way ANOVA followed by Tukey multiple comparison test were used to determine significance. Survival data were analyzed with the Mantel-Cox log-rank test. p < 0.05 was considered to be statistically significant.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by NIH R01HL116626 and AT003961-8455 (to DF), and National Natural Science Foundation of China 3097112 (to SH).
Author contributions
J.W., F.Y. and D.F. designed the experiments and wrote the manuscript. J.W. carried out most of the study and performed the statistical analyses. S.I. assisted in performing ChIP assays and flow cytometry. X.J., S.L., Y.W. and W.L. assisted in sacrificing mice and collecting samples for the animal studies. Y.W. performed immunohistochemistry analysis. S.H. and W.A. helped in the design of the study, assisted in interpreting the data, and revised the manuscript. All authors have agreed to the submission of this manuscript for publication.
References
- 1.Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2014; 342:1432-3; PMID:24357284; http://dx.doi.org/18799145 10.1126/science.342.6165.1432 [DOI] [PubMed] [Google Scholar]
- 2.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2012; 480:480-9; PMID:22193102; http://dx.doi.org/18799145 10.1038/nature10673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu H, Cao X. Dendritic cell vaccines in cancer immunotherapy: from biology to translational medicine. Front Med 2012; 5:323-32; PMID:22198743; http://dx.doi.org/18799145 10.1007/s11684-011-0172-4 [DOI] [PubMed] [Google Scholar]
- 4.Melief CJ. Cancer immunotherapy by dendritic cells. Immunity 2008; 29:372-83; PMID:18799145; http://dx.doi.org/ 10.1016/j.immuni.2008.08.004 [DOI] [PubMed] [Google Scholar]
- 5.Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature 2007; 449:419-26; PMID:17898760; http://dx.doi.org/ 10.1038/nature06175 [DOI] [PubMed] [Google Scholar]
- 6.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer 2012; 12:265-77; PMID:22437871; http://dx.doi.org/ 10.1038/nrc3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dhodapkar MV, Dhodapkar KM, Palucka AK. Interactions of tumor cells with dendritic cells: balancing immunity and tolerance. Cell Death Differ 2008; 15:39-50; PMID:17948027; http://dx.doi.org/ 10.1038/sj.cdd.4402247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest 2007; 117:1155-66; PMID:17476345; http://dx.doi.org/ 10.1172/JCI31422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marigo I, Dolcetti L, Serafini P, Zanovello P, Bronte V. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev 2008; 222:162-79; PMID:18364001; http://dx.doi.org/ 10.1111/j.1600-065X.2008.00602.x [DOI] [PubMed] [Google Scholar]
- 10.Pinzon-Charry A, Maxwell T, Lopez JA. Dendritic cell dysfunction in cancer: a mechanism for immunosuppression. Immunol Cell Biol 2005; 83:451-61; PMID:16174093; http://dx.doi.org/ 10.1111/j.1440-1711.2005.01371.x [DOI] [PubMed] [Google Scholar]
- 11.Jiang S, Zhang HW, Lu MH, He XH, Li Y, Gu H, Liu MF, Wang ED. MicroRNA-155 functions as an OncomiR in breast cancer by targeting the suppressor of cytokine signaling 1 gene. Cancer Res 2010; 70:3119-27; PMID:20354188; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-4250 [DOI] [PubMed] [Google Scholar]
- 12.Kluiver J, Poppema S, de Jong D, Blokzijl T, Harms G, Jacobs S, Kroesen BJ, van den Berg A. BIC and miR-155 are highly expressed in Hodgkin, primary mediastinal and diffuse large B cell lymphomas. J Pathol 2005; 207:243-9; PMID:16041695; http://dx.doi.org/ 10.1002/path.1825 [DOI] [PubMed] [Google Scholar]
- 13.Wang B, Majumder S, Nuovo G, Kutay H, Volinia S, Patel T, Schmittgen TD, Croce C, Ghoshal K, Jacob ST. Role of microRNA-155 at early stages of hepatocarcinogenesis induced by choline-deficient and amino acid-defined diet in C57BL/6 mice. Hepatology 2009; 50:1152-61; PMID:19711427; http://dx.doi.org/ 10.1002/hep.23100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M et al.. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A 2006; 103:2257-61; PMID:16461460; http://dx.doi.org/ 10.1073/pnas.0510565103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baffa R, Fassan M, Volinia S, O'Hara B, Liu CG, Palazzo JP, Gardiman M, Rugge M, Gomella LG, Croce CM et al.. MicroRNA expression profiling of human metastatic cancers identifies cancer gene targets. J Pathol 2009; 219:214-21; PMID:19593777; http://dx.doi.org/ 10.1002/path.2586 [DOI] [PubMed] [Google Scholar]
- 16.Iorio MV, Croce CM. MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol Med 2012; 4:143-59; PMID:22351564; http://dx.doi.org/ 10.1002/emmm.201100209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Roccaro AM, Rombaoa C, Flores L, Obad S, Fernandes SM, Sacco A, Liu Y, Ngo H, Quang P et al.. LNA-mediated anti-miR-155 silencing in low-grade B-cell lymphomas. Blood 2012; 120:1678-86; PMID:22797699; http://dx.doi.org/ 10.1182/blood-2012-02-410647 [DOI] [PubMed] [Google Scholar]
- 18.Zhang M, Zhou X, Wang B, Yung BC, Lee LJ, Ghoshal K, Lee RJ. Lactosylated gramicidin-based lipid nanoparticles (Lac-GLN) for targeted delivery of anti-miR-155 to hepatocellular carcinoma. J Control Release 2013; 168:251-61; PMID:23567045; http://dx.doi.org/ 10.1016/j.jconrel.2013.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gambari R, Fabbri E, Borgatti M, Lampronti I, Finotti A, Brognara E, Bianchi N, Manicardi A, Marchelli R, Corradini R. Targeting microRNAs involved in human diseases: a novel approach for modification of gene expression and drug development. Biochem Pharmacol 2011; 82:1416-29; PMID:21864506; http://dx.doi.org/ 10.1016/j.bcp.2011.08.007 [DOI] [PubMed] [Google Scholar]
- 20.Gasparini P, Lovat F, Fassan M, Casadei L, Cascione L, Jacob NK, Carasi S, Palmieri D, Costinean S, Shapiro CL et al.. Protective role of miR-155 in breast cancer through RAD51 targeting impairs homologous recombination after irradiation. Proc Natl Acad Sci USA 2014; 111:4536-41; PMID:24616504; http://dx.doi.org/ 10.1073/pnas.1402604111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O'Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci USA 2007; 104:1604-9; PMID:17242365; http://dx.doi.org/ 10.1073/pnas.0610731104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu C, Huang X, Zhang X, Roensch K, Cao Q, Nakayama KI, Blazar BR, Zeng Y, Zhou X. miR-221 and miR-155 regulate human dendritic cell development, apoptosis, and IL-12 production through targeting of p27kip1, KPC1, and SOCS-1. Blood 2011; 117:4293-303; PMID:21355095; http://dx.doi.org/ 10.1182/blood-2010-12-322503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vigorito E, Kohlhaas S, Lu D, Leyland R. miR-155: an ancient regulator of the immune system. Immunol Rev 2013; 253:146-57; PMID:23550644; http://dx.doi.org/ 10.1111/imr.12057 [DOI] [PubMed] [Google Scholar]
- 24.O'Connell RM, Kahn D, Gibson WS, Round JL, Scholz RL, Chaudhuri AA, Kahn ME, Rao DS, Baltimore D. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity 2010; 33:607-19; PMID:20888269; http://dx.doi.org/ 10.1016/j.immuni.2010.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vigorito E, Perks KL, Abreu-Goodger C, Bunting S, Xiang Z, Kohlhaas S, Das PP, Miska EA, Rodriguez A, Bradley A et al.. microRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity 2007; 27:847-59; PMID:18055230; http://dx.doi.org/ 10.1016/j.immuni.2007.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, Robbins J, Martinez J, Tabas I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab 2012; 15:545-53; PMID:22445600; http://dx.doi.org/ 10.1016/j.cmet.2012.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, Yu F, Jia X, Iwanowycz S, Wang Y, Huang S, Ai W, Fan D. MicroRNA-155 deficiency enhances the recruitment and functions of myeloid-derived suppressor cells in tumor microenvironment and promotes solid tumor growth. Int J Cancer 2015; 136:E602-13; PMID:25143000; http://dx.doi.org/ 10.1002/ijc.29151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ji Y, Wrzesinski C, Yu Z, Hu J, Gautam S, Hawk NV, Telford WG, Palmer DC, Franco Z, Sukumar M et al.. miR-155 augments CD8+ T-cell antitumor activity in lymphoreplete hosts by enhancing responsiveness to homeostatic gammac cytokines. Proc Natl Acad Sci USA 2015; 112:476-81; PMID:25548153; http://dx.doi.org/ 10.1073/pnas.1422916112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu F, Jia X, Du F, Wang J, Wang Y, Ai W, Fan D. miR-155-deficient bone marrow promotes tumor metastasis. Mol Cancer Res 2013; 11:923-36; PMID:23666369; http://dx.doi.org/ 10.1158/1541-7786.MCR-12-0686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dunand-Sauthier I, Santiago-Raber ML, Capponi L, Vejnar CE, Schaad O, Irla M, Seguin-Estevez Q, Descombes P, Zdobnov EM, Acha-Orbea H et al.. Silencing of c-Fos expression by microRNA-155 is critical for dendritic cell maturation and function. Blood 2011; 117:4490-500; PMID:21385848; http://dx.doi.org/ 10.1182/blood-2010-09-308064 [DOI] [PubMed] [Google Scholar]
- 31.Dunand-Sauthier I, Irla M, Carnesecchi S, Seguin-Estevez Q, Vejnar CE, Zdobnov EM, Santiago-Raber ML, Reith W. Repression of arginase-2 expression in dendritic cells by microRNA-155 is critical for promoting T cell proliferation. J Immunol 2014; 193:1690-700; PMID:25009204; http://dx.doi.org/ 10.4049/jimmunol.1301913 [DOI] [PubMed] [Google Scholar]
- 32.Radford KJ, Tullett KM, Lahoud MH. Dendritic cells and cancer immunotherapy. Curr Opin Immunol 2014; 27:26-32; PMID:24513968; http://dx.doi.org/ 10.1016/j.coi.2014.01.005 [DOI] [PubMed] [Google Scholar]
- 33.Del Vecchio M, Bajetta E, Canova S, Lotze MT, Wesa A, Parmiani G, Anichini A. Interleukin-12: biological properties and clinical application. Clin Cancer Res 2007; 13:4677-85; PMID:17699845; http://dx.doi.org/ 10.1158/1078-0432.CCR-07-0776 [DOI] [PubMed] [Google Scholar]
- 34.Henry CJ, Ornelles DA, Mitchell LM, Brzoza-Lewis KL, Hiltbold EM. IL-12 produced by dendritic cells augments CD8+ T cell activation through the production of the chemokines CCL1 and CCL17. J Immunol 2008; 181:8576-84; PMID:19050277; http://dx.doi.org/ 10.4049/jimmunol.181.12.8576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Forster R, Davalos-Misslitz AC, Rot A. CCR7 and its ligands: balancing immunity and tolerance. Nat Rev Immunol 2008; 8:362-71; PMID:18379575; http://dx.doi.org/ 10.1038/nri2297 [DOI] [PubMed] [Google Scholar]
- 36.Moran TP, Nakano H, Kondilis-Mangum HD, Wade PA, Cook DN. Epigenetic control of Ccr7 expression in distinct lineages of lung dendritic cells. J Immunol 2014; 193:4904-13; PMID:25297875; http://dx.doi.org/ 10.4049/jimmunol.1401104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Landeira D, Sauer S, Poot R, Dvorkina M, Mazzarella L, Jorgensen HF, Pereira CF, Leleu M, Piccolo FM, Spivakov M et al.. Jarid2 is a PRC2 component in embryonic stem cells required for multi-lineage differentiation and recruitment of PRC1 and RNA Polymerase II to developmental regulators. Nat Cell Biol 2010; 12:618-24; PMID:20473294; http://dx.doi.org/ 10.1038/ncb2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bolisetty MT, Dy G, Tam W, Beemon KL. Reticuloendotheliosis virus strain T induces miR-155, which targets JARID2 and promotes cell survival. J Virol 2009; 83:12009-17; PMID:19759154; http://dx.doi.org/ 10.1128/JVI.01182-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Escobar TM, Kanellopoulou C, Kugler DG, Kilaru G, Nguyen CK, Nagarajan V, Bhairavabhotla RK, Northrup D, Zahr R, Burr P et al.. miR-155 activates cytokine gene expression in Th17 cells by regulating the DNA-binding protein Jarid2 to relieve polycomb-mediated repression. Immunity 2014; 40:865-79; PMID:24856900; http://dx.doi.org/ 10.1016/j.immuni.2014.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bell D, Chomarat P, Broyles D, Netto G, Harb GM, Lebecque S, Valladeau J, Davoust J, Palucka KA, Banchereau J. In breast carcinoma tissue, immature dendritic cells reside within the tumor, whereas mature dendritic cells are located in peritumoral areas. J Exp Med 1999; 190:1417-26; PMID:10562317; http://dx.doi.org/ 10.1084/jem.190.10.1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steinbrink K, Jonuleit H, Muller G, Schuler G, Knop J, Enk AH. Interleukin-10-treated human dendritic cells induce a melanoma-antigen-specific anergy in CD8(+) T cells resulting in a failure to lyse tumor cells. Blood 1999; 93:1634-42; PMID:10029592 [PubMed] [Google Scholar]
- 42.Park SJ, Nakagawa T, Kitamura H, Atsumi T, Kamon H, Sawa S, Kamimura D, Ueda N, Iwakura Y, Ishihara K et al.. IL-6 regulates in vivo dendritic cell differentiation through STAT3 activation. J Immunol 2004; 173:3844-54; PMID:15356132; http://dx.doi.org/ 10.4049/jimmunol.173.6.3844 [DOI] [PubMed] [Google Scholar]
- 43.Rot A, von Andrian UH. Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol 2004; 22:891-928; PMID:15032599; http://dx.doi.org/ 10.1146/annurev.immunol.22.012703.104543 [DOI] [PubMed] [Google Scholar]
- 44.De Vries IJ, Krooshoop DJ, Scharenborg NM, Lesterhuis WJ, Diepstra JH, Van Muijen GN, Strijk SP, Ruers TJ, Boerman OC, Oyen WJ et al.. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res 2003; 63:12-7; PMID:12517769 [PubMed] [Google Scholar]
- 45.Holmstrom K, Pedersen AW, Claesson MH, Zocca MB, Jensen SS. Identification of a microRNA signature in dendritic cell vaccines for cancer immunotherapy. Hum Immunol 2010; 71:67-73; PMID:19819280; http://dx.doi.org/ 10.1016/j.humimm.2009.10.001 [DOI] [PubMed] [Google Scholar]
- 46.Lind EF, Millar DG, Dissanayake D, Savage JC, Grimshaw NK, Kerr WG, Ohashi PS. miR-155 upregulation in dendritic cells is sufficient to break tolerance in vivo by negatively regulating SHIP1. J Immunol 2015; 195:4632-40; PMID:26447227; http://dx.doi.org/ 10.4049/jimmunol.1302941 [DOI] [PubMed] [Google Scholar]
- 47.Casey AE, Laster WR Jr., Ross GL. Sustained enhanced growth of carcinoma EO771 in C57 black mice. Proc Soc Exp Biol Med 1951; 77:358-62; PMID:14854049; http://dx.doi.org/ 10.3181/00379727-77-18779 [DOI] [PubMed] [Google Scholar]
- 48.Nachat-Kappes R, Pinel A, Combe K, Lamas B, Farges MC, Rossary A, Goncalves-Mendes N, Caldefie-Chezet F, Vasson MP, Basu S. Effects of enriched environment on COX-2, leptin and eicosanoids in a mouse model of breast cancer. PLoS One 2012; 7:e51525; PMID:23272114; http://dx.doi.org/25311112 10.1371/journal.pone.0051525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jia X, Yu F, Wang J, Iwanowycz S, Saaoud F, Wang Y, Hu J, Wang Q, Fan D. Emodin suppresses pulmonary metastasis of breast cancer accompanied with decreased macrophage recruitment and M2 polarization in the lungs. Breast Cancer Res Treat 2014; 148:291-302; PMID:25311112; http://dx.doi.org/ 10.1007/s10549-014-3164-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Incio J, Tam J, Rahbari NN, Suboj P, McManus DT, Chin SM, Vardam T, Batista A, Babykutty S, Jung K et al.. PlGF/VEGFR-1 signaling promotes macrophage polarization and accelerated tumor progression in obesity. Clin Cancer Res 2016; 22:2993-3004; PMID:26861455; http://dx.doi.org/ 10.1158/1078-0432.CCR-15-1839 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.