

Abstract
Background
Diabetes is the most common metabolic disease with many chronic complications, and cognitive disorders are one of the common complications in patients with diabetes. Previous studies have showed that autophagy played important roles in the progression of metabolic syndrome, diabetes and other diseases. So we investigated whether aged diabetic mice are prone to be associated with the cognitive and affective disorders and whether Beclin-1-mediated autophagy might be involved in thepahological process.
Methods
High-fat diet/streptozotocin (STZ) injection-induced diabetic C57 mice were adopted in this study. Cognitive disorders were detected by Morris water maze and fear conditional test. Affective disorders were detected by tail suspension test and forced swimming test. Magnetic resonance imaging was applied to observe changes of morphology and metabolism in the brain. The 18 F-fluorodeoxyglucose positron emission tomography (FDG-PET) was used to assess metabolism changes in the brain of aged diabetic mice. Autophagy were evaluated by Beclin- 1, LC3II/I and P62, which were detected by western blot analysis and observed by electron microscopy.
Results
1. Compared with control group, diabetes mice showed significantly decreasing abilities in spatial memory and conditioned fear memory (all P < 0.05), and increasing tendency of depression (P < 0.05). 2. MRI showed that the majority of elderly diabetic mice were associated with multiple cerebral small vessel disease. Some even showed hippocampal atrophy, ventricular dilatation and leukoaraiosis. 3. FDG-PET-CT discovered that the glucose metabolism in the amygdala and hippocampus was significantly decreased compared with normal aged mice (P < 0.05). 4. Electron microscopy found that, although autophagy bodies was not widespread, and there was no significant difference between the two groups, yet compared with normal aged mice, apparent cell edema, myelinated tow reduction and intracellular lipofuscin augmentation existed in elderly diabetic mice brain. 5. The level of p62 was increased in the STZ-induced diabetic mice hippocampus and striatum, and beclin1 protein expression were significantly decreased in diabetic mice hippocampus compared with normal aged mice (P < 0.05). There was a upward trend of the ratio of LC3II/I in hippocampus, cortex and striatum, but no statistically difference between the two groups.
Conclusion
Compared with normal aged mice, diabetic aged mice were apt to cerebral small vessel disease and associated with cognitive and affective disorders, which may be related to the significantly reduced glucose metabolism in hippocampus and amygdala. Beclin1 mediated autophagy in hippocampus probably played an important role in cognitive and affective disorders of STZ-induced aged diabetic mice.
Background
Diabetes is the most common metabolic disease with many chronic complications. Studies on animal models and clinical diabetic patients have revealed that abnormalities appear in both chemical and ultra-structural levels in brain and in neurological behaviors including learning/memory disability and the increasing risk in depression [1]. Oxidative stress [2–7], chronic inflammation [8–10] and autophagic imbalance [11–14] are considered to be most likely causes to induce brain dysfunction [15–17]; however, whether autophagic imbalance is involved in diabetes-related cognitive and affective disorders remains unclear.
Autophagy, a self-eating mechanism, is a crucial cleaning system for aggregated proteins and dysfunctional organelles [18]. It is linked to the degradation of damaged materials by acid hydrolases within the lysosomal system. There is mounting evidence that the control of autophagocytosis is impaired in many neurodegenerative diseases, which display abnormal protein aggregation [19–21]. There are three different pathways of autophagic uptake and processing of cellular constituents, i.e., macro-, micro- and chaperone-mediated autophagy [22]. Beclin1, an adaptor protein via its interacting proteins called the Beclin1 interactome, can either stimulate or suppress the onset of autophagy. Beclin1 is one of the core elements to regulate autophagy. The expression level and post-translational modifications of Beclin1 interactome components can crucially control the degree of cellular autophagy.
Up to date, few researches have investigated the relationship between diabetes-related cognitive and affective disorders and Beclin1-mediated autophagy. The main purpose of this study was to verify whether long-term diabetes could induce cognitive and affective disorders in elderly mice, and to explore whether changes of Beclin1 protein and autophagy could affect this pathogenesis.
Methods
Animal grouping
Male C57BL/6 mice were purchased from ProMedican in Shanghai, China. Mice were raised under a 12-h light/dark cycle and humidity- and temperature-controlled environment with adlibitum access to water and standard laboratory chow. All the animal experiments were approved by Fudan University Animal Care and Use Committee. The 3-weeks aged mice were randomly divided into a control group (n = 12) and a diabetic group (n = 12).
Model making
Control mice were given with normal diet. The model mice were given high fat diet (HFD, Shanghai SLAC company) for 12 weeks. After that, 45 mg/kg Streptozocin (STZ, Enzo life sciences) were intraperitoneally injected for two weeks. Blood glucose in caudal venous of mice were detected after STZ injection. Mice with random blood glucose >11.1 mmol/L were considered as diabetic mice. Then the control and diabetic mice were respectively raised with normal diet and HFD (Fig. 1a).
Fig. 1.
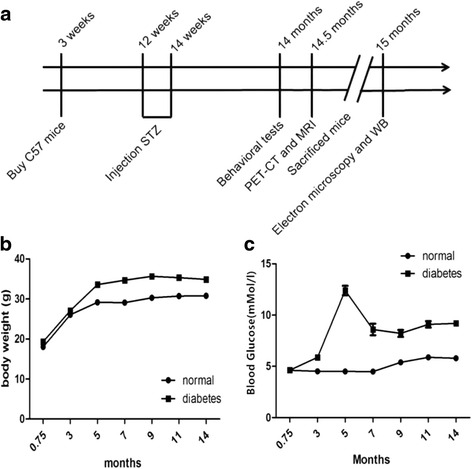
HFD/STZ treatment upregulated body weight and blood sugar in mice. a Experimental time-flow diagram. The body weight (b) and blood glucose (c) of mice were measured before death. Blood samples were collected from tails of mice under fasting condition (*P < 0.05, n = 12 for each group)
Morris water maze test
Learning and reference/working memory were evaluated by the Morris water maze test as previously described [23]. Groups were blinded to the examiners. In brief, swimming paths were video-tracked with a camera fixed on the ceiling of the room and analyzed by the software (Coulbourn, USA). A training session was carried out before the hidden platform test sessions. Mice were given 60 seconds free swimming and guided to climb onto the hidden platform and allowed to remain there for 30 seconds before returning to their cages. On the hidden platform test, the mice had 5 sessions at 20-min intervals per day on the following 4 consecutive days (day 1 to 4). During each session mice were released from randomly assigned 3 starting points and swam for 60 s. On the probe test at day 5, the hidden platform was removed and the mice swam freely for 60 s. The number of times the mice crossed the original platform location was recorded. On the visible platform test which was performed after the probe test on day 5, the platform was elevated 5 cm above the water surface level and placed in a different position. The mice were given four sessions of a visible trial with an inter-session.
Conditioned fear test
Conditioned fear (fear conditioning) formation is not only a process of learning and memory, but also the formation of an emotional process. The Study on the fear of the fear model used in the rodent fear memory, and the freeze is considered to be an objective indicator of fear [24].
Mice (14-months old) were subjected to fear conditioning test. Fear conditioning test was performed as described previously [25]. Briefly, a mouse was placed in a plexiglas training chamber whose floor had a stainless grid for shock delivery. After a 3-min baseline exploratory period in the chamber, mice received 3 tone (2000 Hz, 90 db)–shock (0.7 mA, 2 s) pairings separated by 1 min. Each training chamber was cleaned with 95% ethyl alcohol before the placement of a mouse and was illuminated only with a 10 W bulb in a dark experimental room. Twenty-four hours after the training session, mouse was placed again in the training chamber for 8 min without tone and foot shock. Each animal’s freezing behavior was scored every 8 s during the 8 min observation period. The percentage of time in freezing behavior was calculated using the formula of 100*f/n, where f was the number of freezing events in the 8 min and n was the total number of 8-s observation period in 8 min. One hour later, the mouse was placed in a new chamber without a stainless floor grid. This chamber was cleaned with the lemon fresh pine-sol each time after use and room light was turned on during the test. After a 3-min exploratory period in this new chamber, a 30-s tone (2000 Hertz, 90 db) was applied. The mouse was then left in the chamber for another 1 min. The percentage of time in freezing was calculated by the formula of 100*f/30, where f is the total freezing time during the 30-s observation period.
Tail suspension test
Tail suspension test was performed as described previously [26]. Briefly, animals were suspended by the tail from a ledge with adhesive tape (approximately 1 cm from the tip of the tail). The distance between the tip of the tail of the mouse and the floor was approximately 30 cm. Each animal was partitioned to avoid interference during the test. Immobility was defined as the absence of movement for 6 min. Each mouse in the test was recorded by a videocamera and scored by a blinded experimenter.
Forced swimming test
Forced swimming test was performed as described previously [27]. Briefly, animals were placed in an open cylindrical container (total volume, 2500 mL; height, 20 cm; diameter,14 cm) filled with 10 cm of water (25 °C). Immobility was defined as mouse ceasing struggling, remaining floating motionless in water, and making only movement necessary to keep its head above water. The duration of immobility in the last 4 min of the (total) 6 min of swimming time was recorded with a videocamera and scored by a blinded experimenter.
Magnetic resonance imaging scanning
We use Bruker 3.1 7 T superconducting magnetic resonance imaging with the diameter of the 47 mm microscope coil to do mouse MR imaging study. Before MR scan, the mice were anesthetized with isoflurane. After that, the mice were fixed on the fixed frame, and the T1, T2 and enhanced scan were performed. Scanning parameters: echo time 20s; repetition time 2000 ms; band width 333333.3hz; layer 16; layer thickness 0.8 mm; image size 64; scanning field of view 21600 ~ 15000 mm.
PET/CT scanning
Prior to the PET scans, the mice were fasted for 12 h. Each mouse was injected approximately 0.5 mCi (18.5 MBq) 18 F-FDG in less than 0.5 ml via tail vein. The injection was completed in less than 1 min and the injection site was pressed for 30 s to prevent leakage. The exact injection time and the radio activity of the syringe both before and after the injection, together with their measure time, were recorded for dosage calibration. After an uptake period of 40 min, the mouse was anaesthetized with agasmixture of 1% isoflurane and oxygen (1 L/min) and then prone positioned in the gantry with a mask covering its mouth to ensure continuous gas inhalation throughout the scan. The PET signals were acquired from approximately 45 min post-injection for 10 min inaInveon MM micro PET/CT scanner (Siemens Co., Ltd, Knoxville, TN, USA) designed for high resolution imaging of small laboratory animals. The following CT scan was performed for localization and attenuation correction. The raw PET data were binned into a single frame, and the multiple planar reconstruction (MPR) images were obtained by OSEM3D/MAP reconstruction methods with iterations of OSEM3D = 2 and iterations of MAP = 18. The final voxel size was 0.776 × 0.776 × 0.796 mm3, and the matrix was 128 × 128 × 159. PMOD 3.4 software (PMOD Technology, Switzerland) was used to analyze the images. Automatic rigid matching was performed after rough manual co-registration in the PFUS module. A Gaussian kernel with full width of half maximum (FWHM) of 0.6 mm × 0.6 mm × 0.6 mm was used to smooth the image during normalization. After that, Mirrione atlas was overlapped on the normalized PET images so as to obtain the SUV mean of all 19 mouse brain regions in this standardized template. Finally, each regional SUV mean was divided by that of the brain stem to derive an SUVR for statistical analysis purpose.
Transmission electron microscopy
Transmission electron microscopy (TEM, PHILIPS CM-120, Netherlands) Brain tissues were perfused with 2.5% glutaraldehyde perfusate (25% glutaraldehyde and 0.2 M phosphate buffer with 3mMMgCl2, pH 7.4), followed by fixation with 2.5% glutaraldehyde as previously described [28]. And then concentional of fixation solution, dehydrated, embedded in paraffin, sliced and 3% uranyl acetate and lead citrate double staining. The CM-120 PHILIPS was observed and photographed under transmission electron microscope.
Western Blot
Western Blot was performed as described previously [29]. The hippocampus tissues from mice were homogenized using RIPA (20mgorganizationwith 200 μl RIPA). The tissue lysates were centrifuged at 12,000 rpm for 5 min and the supernatant were collected to determine the protein concentrations by a bicinchoninic acid protein assay (Beyotime Institute of Biotechnology, China). Membranes were reprobed with an antibody specific against GAPDH as an internal control. The specific primary antibodies included: a rabbit polyclonal antibody for BECN1 (1:500; SantaCruz, USA), LC3 (1:200; cell signaling technology, USA), p62 (1:3000, abcam, England), and GAPDH (1: 2000, Boster, China). At last, X-ray films were used to Photosensitive, developing and fixing in dark room.
Statistical analysis
All statistical analysis was performed using PRISM software (GraphPad, La Jolla, CA). All data were represented as mean ± SEM and analyzed by student t-test using treatment (diabetes vs.control). P values less than 0.05 were considered significant.
Results
HFD/STZ treatment induces upregulation in both body weight and blood glucose level in mice
Before grouping, all the mice had similar body weight and random blood glucose levels. After STZ injection, the level of blood glucose in HFD/STZ-treated mice was significantly increased till 14 months later. As showed in Fig. 2, HFD/STZ-treated mice showed significant rise in both body weight (Fig. 1b, P < 0.05) and blood sugar level (Fig. 1c, P < 0.05) compared with those in normal diet-treated control mice. These results indicated that HFD/STZ-treatment successively induced diabetic mice model in 14-month-aged elderly mice.
Fig. 2.
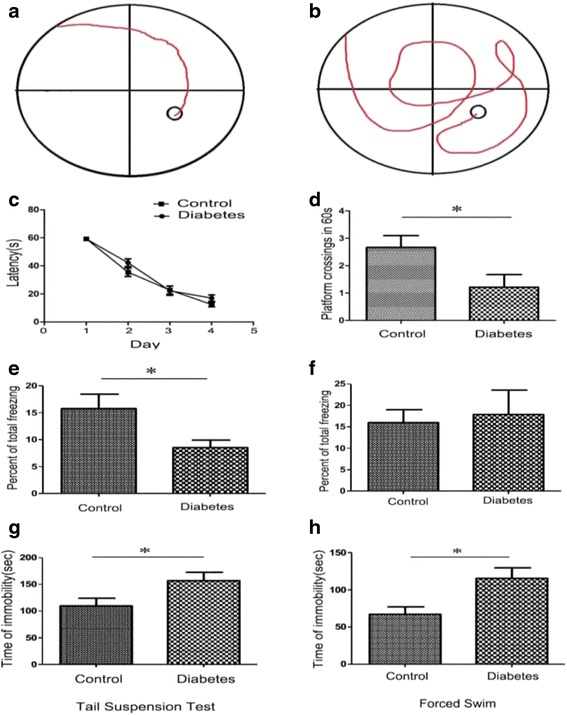
HFD/STZ treatment casused cognitive impairment and depressive symptoms in mice. a and b, typical swimming traces of control mice (a) and diabetic mice (b) in Morris watermaze test. c, No difference were found between the two groups in the latency time during training trials. d, During testing phase, the frequency of platform-crossing in diabetic mice decreased remarkably when compared with control mice. (*P < 0.05, n = 12 for each group). Gand F, cued and contextual fear memory was evaluated using the fear conditioning test. Percent of time in total freezing during the contextual test (e) and tone test (f) was represented. (*P < 0.05, n = 12 for each group). Gand H, effects of diabetes on immobility time in the tail suspension test (g) and the forced swimming test (h). The immobility time in diabetes group was significantly higher than that of control group. (*P < 0.05, n = 12 for each group)
Cognitive impairment and depressive symptoms in elderly diabetic mice
We performed Morris water maze test to evaluate the spatial acquisition ability. As showed in Fig. 2, the number of platform crossing significantly decreased in diabetic mice (Fig. 2d, P < 0.05), indicating that HFD/STZ-induced diabetes induced remarkable reduction in the spatial learning/memory ability.
In the fear conditioning test, we assessed changes in cued and contextual fear memory in diabetic mice. As showed in Fig. 2e, when compared with control mice, the percentage of time of total freezing behavior decreased significantly in diabetic mice (P < 0.05). For the tone test, no obvious difference was observed between two groups, indicating that HFD/STZ-induced diabetic mice had reduction in fear conditioning memory ability.
By using tail suspension and forced swimming, we also detected the depressive status in the mice. As showed in Fig. 2g and h, the time of ‘immorbility’ in diabetic mice remarkably increased during both tail suspension test (Fig. 2g) and forced swimming test (Fig. 2h).
These results indicated that abilities of HFD/STZ-induced diabetic mice decreased in both spatial memory acquisition and fear conditioning memory. Meanwhile, these diabetic mice represented more severe depressive behaviors than the control mice.
Glucose metabolism reduction and structural abnormalities in the amygdala and hippocampus of elderly diabetic mice
To investigate whether regional functional changes exist in the brain of diabetic mice, we further evaluated the FDG uptake ability by using PET-CT scaning. As showed in Fig. 3a, the standard uptake value of FDG in both hippocampus and amygdala were much less in diabetic mice than those in control mice (P < 0.05).
Fig. 3.
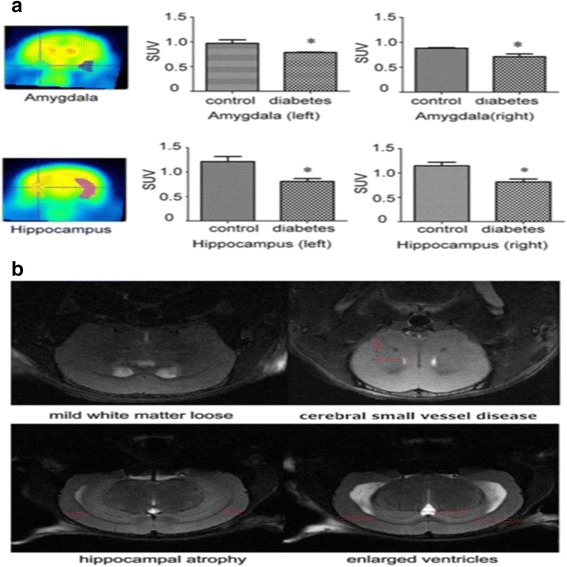
a Standard uptake value (SUV) of FDG in the Hippocampus and Amygdala in diabetes group and control group. The SUV of FDG in diabetes group were much less than those in the control (*P < 0.05, n = 3 for each group). b Magnetic resonance imaging showed mild white matter loose, cerebral small vessel disease, hippocampal atrophy and ventricle enlargement in the brain of diabetic mice
Magnetic resonance imaging showed that the majority of elderly diabetic mice were associated with multiple lacunar infarction which indicated cerebral small vessel disease. Some even appeared hippocampus atrophy, ventricle enlargement in parts of elderly diabetic mice (Fig. 3b).
We also performed electron microscopy to observe the ultrastructural changes in the hippocampus. As showed in Fig. 4, mice in diabetic group appeared a slight increase without significant differences in autophagosome, but there were more intracellular lipofuscin and less myelinated nerve fibers in diabetic hippocampus, in accompanied with apparent hippocampal edema.
Fig. 4.
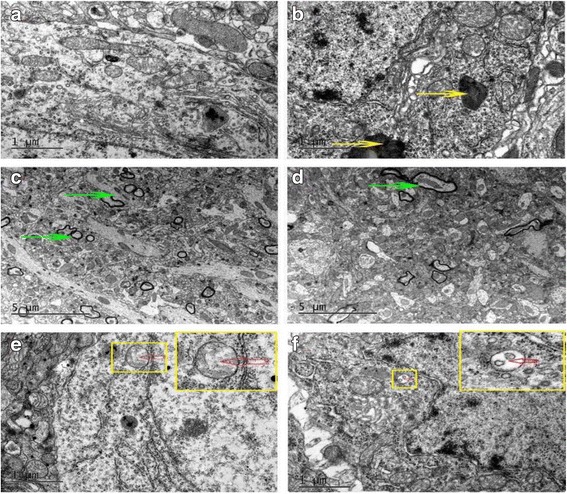
Alterations in hippocampus ultrastructure was examined by transmission electron microscope. Autophagosomes (red arrow) existed in both control and diabetic mice were no significantly decrease in diabetes mice. Diabetes mice have more lipofuscin (yellow arrow) and fewer myelinated nerve fibers (green arrow) in the hippocampus. a, c, e were control groups and (b, d, f) were diabetes groups
These results suggested that the brain function reduction and structural abnormalities existed in the amygdala and hippocampus of elderly diabetic mice.
Autophagy was upregulated in the hippocampus, cortex and striatum of elderly diabetic mice
By using western blot analysis, we detected the expression levels of p62, LC3 and Beclin1 in hippocampus, cortex and striatum. Our results showed that the level of p62 had a increased trend in the STZ-induced diabetic mice hippocampus and striatum without markedly difference, and the ratio of the Beclin1 protein level decreased significantly in the diabetic hippocampus (Fig. 5). There was a upward trend of the ratio of LC3II/LC3I in hippocampus, cortex and striatum, but no statistically difference between the two groups.
Fig. 5.
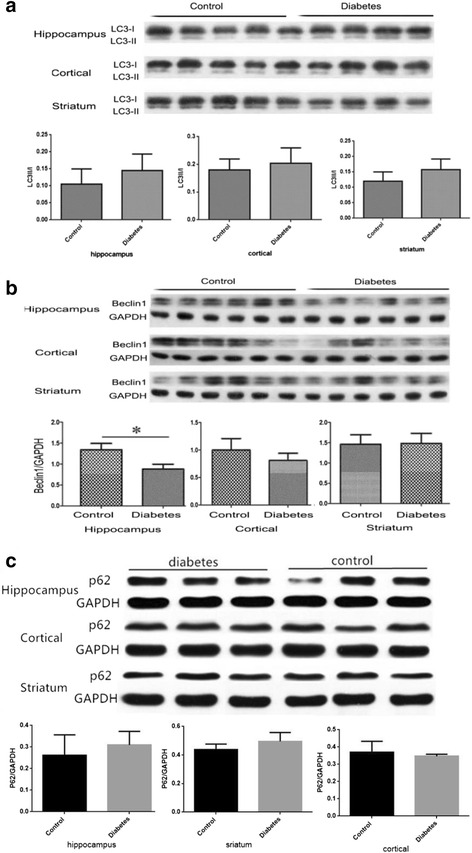
a The protein levels of LC3II/LC3I in Hippocampus, Cortex and Striatum were measured by Western Blotting, The graphs showed the relative density of LC3-II to LC3-I. (n = 4 for diabetic group, n = 5 for control group). b The protein levels of Beclin1 in Hippocampus, Cortex and Striatum were measured by Western Blotting, The graphs showed the relative density of Beclin1 to GAPDH. Note that the protein level of Beclin1 decreased significantly in the hippocampus of diabetic mice (*P < 0.05 n = 4 for diabetic group, n = 5 for control group). c The protein levels of p62 in Hippocampus, Cortex and Striatum were measured by Western Blotting, The graphs showed the relative density of p62/GAPDH
Discussion
Diabetes can produce long-term negative effects, such as high blood glucose, hypoglycemia, free radical-mediated oxidative stress, brain blood vessel reconstruction disorder and abnormal brain insulin [30, 31], thus, diabetes may represent a metabolic state in which neuroprotective and neuromodulatory effects of insulin in the Central Nervous System were disrupted [32], and causes brain damage. At present, the pathogenesis of diabetes related cognitive dysfunction is not clear. According to previous studies, diabetes related cognitive dysfunction consists of 2 types: One is vascular cognitive dysfunction which is caused by cerebrovascular disease; the other type shows apparent memory decline in association with atrophy in hippocampus and amygdala, but with no obvious cerebrovascular disease. In our study, we found that diabetic mice was prone to be accompanied by both cerebral small vessel disease and hippocampus atrophy, which was likely to be an important cause of diabetes related cognitive dysfunction, and maybe a predictor of cognitive impairment in patients with diabetes. This means that the pathogenesis of diabetic cognitive impairment include both of the two as showed before. Additionally, HFD/STZ-induced diabetic mice also showed apparent glucose uptake reduction in hippocampus and amygdala, indicating that vascular independent dementia might also be responsible for the cognitive decline.
In the present study, diabetes related cognitive impairment showed not only the decline of memory ability, but also apparent depressive symptoms. These emotional decline were corresponding to the glucose uptake reduction in hippocampus and amygdala, since the two brain regions were important for emotional integration. Current studies had confirmed that the hippocampal volume of depressed patients was significantly reduced [33–35], and the most serious damaged regions of depressed patients were hippocampus and amygdala [36]. Therefore, abnormalities in hippocampus and amygdala might induce emotional imbalance such as depression symptoms in diabetic mice. Few researches reported that diabetes induced reduction in metabolism of the amygdala, and our study suggested that the elderly diabetic mice had the dysfunction of glucose metabolism in the amygdala, which might be one of the pathological mechanisms of cognitive impairment. Therefore, these findings provided a new and interesting clue to find the intrinsic causes of memory impairment in patients with diabetes.
Autophagy played important roles of maintaining normal tissue and cellular homeostasis [37, 38]. Previous studies have showed that autophagy played important roles in the progression of metabolic syndrome, diabetes and other diseases. The pathogenesis of these diseases was, to some extent, related to the dysfunction of autophagic removement of damaged organelles [39]. It was found that the efficiency of autophagy function decreases with aging. Recent researches showed that type 2 diabetes could lead to autophagy dysfunction of vascular cells and neuronal cells, cause or aggravate cerebral vascular lesions and vascular dementia process, and eventually resulted dysfunction of memory [40–44].
The protein of p62, a protein associated with autophagosomes and degraded in the autolysosome, is commonly regarded as autophagy substrate protein [45, 46], as a mainly negative regulator of autophagy, activated mTORC1 inhibits autophagy induction through blocking of the initial inducer of autophagy [47, 48]. The p62 is the scaffold protein implicated in selective autophagy which is induced by the stress of cells. It plays an important role in the mechanism of diabetes. Beclin1 is not only a central positive regulator in the early stage of autophagy, but also one of the core proteins of autophagy, apoptosis and inflammatory reaction [49]. In recent years, scientists have discovered that Beclin1 protein levels decreased significantly in the brain regions of neurodegenerative diseases [49, 50], suggesting that Beclin1 may be involved in the cognitive dysfunction in Alzheimer’s disease. Our study found that the p62 protein expression in the brain tissue of elderly diabetic mice had a increased trend and the result meant that the autophagy in STZ-induced diabetic mice was downregulated. At the same time, the Beclin1 protein decreased markedly in hippocampus. Meanwhile, the result of LC3II/I showed that the diabetes group had a upward trend without statistical difference. These results indicating that the expression changes of p62 and Beclin1 might play different roles in the pathogenesis of autophagy. Based on our present study, we concluded that the autophagy was reduced in diabetic mice. Previous studies found that Akt plays a role in induced endothelial injury in diabetes [31], and that the NF-kB pathway is a mechanism of cerebral ischemia and hypoxia [51]. Next step, we will explore whether Akt and NF-kB pathway has an effect on diabetic cognitive dysfunction.
Conclusions
The results of this study suggested that the elderly diabetic mice were prone to complicated with cognitive and affective disorders, which was probably caused by hippocampal atrophy, glucose metabolism disorders in the hippocampus and amygdala, and cerebral small vessel disease. Compared with normal aged mice, diabetic aged mice were apt to complicate cerebral small vessel disease with cognitive and affective disorders. Beclin1 mediated autophagy might play an important role in the process of its occurrence and development. The intrinsic molecular mechanism still need further deep-going research in the future.
Acknowledgments
Not applicable.
Funding
This work was supported by grants from the National Natural Scientific Foundation, China (81170322, 81571361, U1632120), Shanghai Natural Scientific Foundation, China (11ZR1405300), and Shanghai Municipal Outstanding Talent Development Foundation (2012052).
Availability of data and materials
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
Authors’ contributions
Z-FG, X-LZ, and X-MZ contributed to collecte and analyzed the data and performed the experiments; YZ, Y-MW, Q-LG, GJ collected and analyzed the data; G-FW, N-NW, HY, and Z-YY provided important criticisms of the manuscript draft; H-GZ, J-CG and Y-CL conceived and designed the experiments. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Footnotes
Zhu-Fei Guan, Xiu-Ling Zhou and Xiao-Ming Zhang are co-first authors.
Contributor Information
Zhu-Fei Guan, Email: guanzhufei@163.com.
Xiu-Ling Zhou, Email: hsyy_xiulingzhou@163.com.
Xiao-Ming Zhang, Email: Zxming29@163.com.
Yu Zhang, Email: zy_huashan@163.com.
Yan-Mei Wang, Email: wyanmei90125@163.com.
Qi-Lin Guo, Email: 14211010007@fudan.edu.cn.
Guo-Feng Wu, Email: 961766707@qq.com.
Na-Na Wang, Email: 1083956314@qq.com.
Hao Yang, Email: 836525728@qq.com.
Zhong-Yu Yu, Email: 16211220017@fudan.edu.cn.
Hou-Guang Zhou, Phone: +86-21-52887292, Email: zhg7376@163.com.
Jing-Chun Guo, Phone: +86-21-54237231, Email: jingchunguo@shmu.edu.cn.
Ying-Chao Liu, Phone: +86-531-87938911, Email: fdlyc@aliyun.com.
References
- 1.Biessels GJ, Kappelle AC, Bravenboer B. Cerebral function in diabetes mellitus. Diabetologia. 1994;37:643–50. doi: 10.1007/BF00417687. [DOI] [PubMed] [Google Scholar]
- 2.Beckman KB, Ames BN. The free radical theory of agingmatures. Physiol Rev. 1998;78:547–81. doi: 10.1152/physrev.1998.78.2.547. [DOI] [PubMed] [Google Scholar]
- 3.Bonnefont-Rousselot D. Glucose and reactive oxygen species. Curr Opin Clin Nutr Metab Care. 2002;5:561–8. doi: 10.1097/00075197-200209000-00016. [DOI] [PubMed] [Google Scholar]
- 4.Jackson GR, Werrbach-Perez K, Pan Z. Neurotrophin regulation of energy homeostasis in the central nervous system. Dev Neurosci. 1994;16:285–90. doi: 10.1159/000112121. [DOI] [PubMed] [Google Scholar]
- 5.Dugan LL, Sensi SL, Canzoniero LMT. Mitochondrial production of reactive oxygen species in cortical neuronsfollowing exposure to N-methyl-D-aspartate. J Neurosci. 1995;15:6377–88. doi: 10.1523/JNEUROSCI.15-10-06377.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407:802–9. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- 7.Moreira PI, Cardoso SM, Pereira CM. Mitochondria as a therapeutic targetin Alzheimer’s disease and diabetes. CNS Neurol Disord: Drug Targets. 2009;8:492–511. doi: 10.2174/187152709789824651. [DOI] [PubMed] [Google Scholar]
- 8.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tabas RD. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–90. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meng Y, Yong Y, Yang Y. Autophagy alleviates neurodegeneration caused by mild impairment of oxidative metabolism. J Neurochem. 2013;126:805–18. doi: 10.1111/jnc.12268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh R, Kaushik S, Wang Y. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–5. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donohue TM., Jr Autophagy and ethanol-induced liver injury. World J Gastroenterol. 2009;15:1178–85. doi: 10.3748/wjg.15.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gonzalez CD, Lee M, Marchetti P. The emergingrole of autophagy in the pathophysiology of diabetes mellitus. Autophagy. 2011;7:2–11. doi: 10.4161/auto.7.1.13044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bergamini E, Cavallini G, Donati A. The role of macroautophagy in the ageing process, anti-ageingintervention and age-associated diseases. Int J Biochem Cell Biol. 2004;36:2392–404. doi: 10.1016/j.biocel.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 15.Whitmer RA, Sidney S, Selby J. Midlife cardiovascular risk factors and risk of dementia in late life. Neurology. 2005;64:277–81. doi: 10.1212/01.WNL.0000149519.47454.F2. [DOI] [PubMed] [Google Scholar]
- 16.Beeri MS, Silverman JM, Davis KL. Type 2 diabetes is negatively associated with Alzheimer’s disease neuropathology. J Gerontol. 2005;60:471–475.4. doi: 10.1093/gerona/60.4.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miles WR, Root HF. Psychologic tests appliedin diabetic patients. Arch Intern Med. 1992;30:767–70. doi: 10.1001/archinte.1922.00110120086003. [DOI] [Google Scholar]
- 18.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–41. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 19.Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120:4081–91. doi: 10.1242/jcs.019265. [DOI] [PubMed] [Google Scholar]
- 20.Garcı´a-Arencibia M, Hochfeld WE, Toh PP. Autophagy, a guardian against neurodegeneration. Semin Cell Dev Biol. 2010;21:691–8. doi: 10.1016/j.semcdb.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nixon RA, Yang DS. Autophagy failure in Alzheimer’s disease – locating the primary defect. Neurobiol Dis. 2011;43:38–45. doi: 10.1016/j.nbd.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Todde V, Veenhuis IJ. Autophagy: principles and significance in health and disease. Biochim Biophys Acta. 2009;1792:3–13. doi: 10.1016/j.bbadis.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 23.Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 24.Li X, Inoue T, Abekawa T, Weng S. 5-HT1A receptor agonist affects fear conditioning through stimulations of the postsynaptic 5-HT1A receptors in the hippocampus and amygdala. Eur J Pharmacol. 2006;532:74–80. doi: 10.1016/j.ejphar.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 25.Kim JJ, Fanselow MS. Modality-specific retrograde amnesia of fear. Science. 1992;256:675–82. doi: 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- 26.Porsolt RD, Bertin A, Jalfre M. Behavioural despair in mice: a primary screening test for antidepressants. Arch Int Pharmacodyn Ther. 1977;229:327–36. [PubMed] [Google Scholar]
- 27.Azizi-Malekabadi H, Hosseini M, Pourganji M. Deletion of ovarian hormones induces a sickness behavior in rats comparable to the effect of lipopolysaccharide. Neurol Res Int. 2015;2015:627–42. doi: 10.1155/2015/627642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gonzales KK, Pare JF, Wichmann T. GABAergic inputs from direct and indirect striatal projection neurons onto cholinergic interneurons in the primate putamen. J Comp Neurol. 2013;521:2502–22. doi: 10.1002/cne.23295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Młyniec K, Budziszewska B, Holst B. GPR39 (zinc receptor) knockout mice exhibit depression-like behavior and CREB/BDNF down-regulation in the hippocampus. Int J Neuropsychopharmaco. 2015;18:1–8. doi: 10.1093/ijnp/pyu002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brands MW, Bell TD, Gibson B. Nitric oxide mayprevent hypertension early in diabetes by counteracting renalactions of superoxide. Hypertension. 2004;43:57–63. doi: 10.1161/01.HYP.0000104524.25807.EE. [DOI] [PubMed] [Google Scholar]
- 31.ZhouHG LL, Zhang Y. Glutathione prevents free fatty acids-induced oxidative stres and apoptosis in human brain vascular endothelial cells throug Akt pathway. CNS Neurosci Ther. 2013;19:252–61. [DOI] [PMC free article] [PubMed]
- 32.Golimstok A, Cámpora N, Rojas JI. Cardiovascular risk factors and frontotemporal dementia: a case–control study. Transl Neurodegener. 2014;3:13. doi: 10.1186/2047-9158-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Harten B, Leeuw FE, Weinstein HC. Brain imaging in patients with diabetes: a systematic review. Diabetes Care. 2006;29:2539–48. doi: 10.2337/dc06-1637. [DOI] [PubMed] [Google Scholar]
- 34.Logroscino G, Kang JH, Grodstein F. Prospective study of type 2 diabetes and cognitive decline in women aged 70–81 years. BMJ. 2004;328:548. doi: 10.1136/bmj.37977.495729.EE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Puyd J, Vaslin A, Mottier V. Postischemic treatment of neonatalcerebral ischemia should target autophagy. Ann Neurol. 2009;66:378–89. doi: 10.1002/ana.21714. [DOI] [PubMed] [Google Scholar]
- 36.Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neumbiol Dis. 2008;32:329–39. doi: 10.1016/j.nbd.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 37.Bernales S, Schuck S, Walter P. ER-phagy: selective autophagy of the endoplasmic reticulum. Autophagy. 2007;3:285–7. doi: 10.4161/auto.3930. [DOI] [PubMed] [Google Scholar]
- 38.Rodriguez-Enriquez S, Kim I, Currin RT. Tracker dyes to probe mitochondrial autophagy (mitophagy) in rat hepatocytes. Autophagy. 2006;2:39–46. doi: 10.4161/auto.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cai D, Liu T. Inflammatory cause of metabolic syndromevia brain stress and NF-κB. Aging. 2012;4:98–115. doi: 10.18632/aging.100431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cavallini G, Donati A, Taddei M. Evidence for selective mitochondrial autophagy and failure in aging. Autophagy. 2007;3:26–7. doi: 10.4161/auto.3268. [DOI] [PubMed] [Google Scholar]
- 41.Pickford F. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid accumulation in mice. J Clin Invest. 2008;118:2190–9. doi: 10.1172/JCI33585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.He C, Levine B. The Beclin 1 interactome. Curr Opin Cell Biol. 2010;22:140–9. doi: 10.1016/j.ceb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Akisaki T. Cognitive dysfunction associates with white matter hyperintensities and subcortical atrophy on magnetic resonance imaging of the elderly diabetes mellitus Japanese elderly diabetes intervention trial (J-EDIT) Diabetes Metab Res Rev. 2006;22:376–84. doi: 10.1002/dmrr.632. [DOI] [PubMed] [Google Scholar]
- 44.Reeks-Nielsen E, Lundbaek K, Rafaelsen OJ. Pathological changes in the central and peripheral nervous system of young long-term diabetics. Diabetologia. 1966;1:233–41. doi: 10.1007/BF01257917. [DOI] [PubMed] [Google Scholar]
- 45.Sahani MH, Itakura E, Mizushima N. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy. 2014;10:431–41. doi: 10.4161/auto.27344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Viiri J, Amadio M, Marchesi N. Autophagy activation clears ELAVL1/HuR-mediated accumulation of SQSTM1/p62 during proteasomal inhibition in human retinal pigment epithelial cells. PLoS One. 2013;8:e69563. doi: 10.1371/journal.pone.0069563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Newton PT, Vuppalapati KK, Bouderlique T. Pharmacological inhibition of lysosomes activates the MTORC1 signaling pathway in chondrocytes in an autophagy-independent manner. Autophagy. 2015;11:1594–607. doi: 10.1080/15548627.2015.1068489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Castets P, Ruegg MA. MTORC1 determines autophagy through ULK1 regulation in skeletal muscle. Autophagy. 2013;9:1435–7. doi: 10.4161/auto.25722. [DOI] [PubMed] [Google Scholar]
- 49.Jaeger PA, Wyss-Coray T. Beclin 1 complex in autophagy and Alzheimer disease. Arch Neurol. 2010;67:1181–5. doi: 10.1001/archneurol.2010.258. [DOI] [PubMed] [Google Scholar]
- 50.Mijnhout GS, Scheltens P, Diamant M. Diabetic encephalopathy: a concept in need of adefinition. Diabetologia. 2006;49(6):1447–8. doi: 10.1007/s00125-006-0221-8. [DOI] [PubMed] [Google Scholar]
- 51.Ji G, Liu M, Zhao XF. NF-κB signaling is involved in the effects of intranasally engrafted human neural stem cells on neurofunctional improvements in neonatal Rat hypoxic–ischemic encephalopathy. CNS Neurosci Ther. 2015;21:926–35. doi: 10.1111/cns.12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.