
Fig. 1.
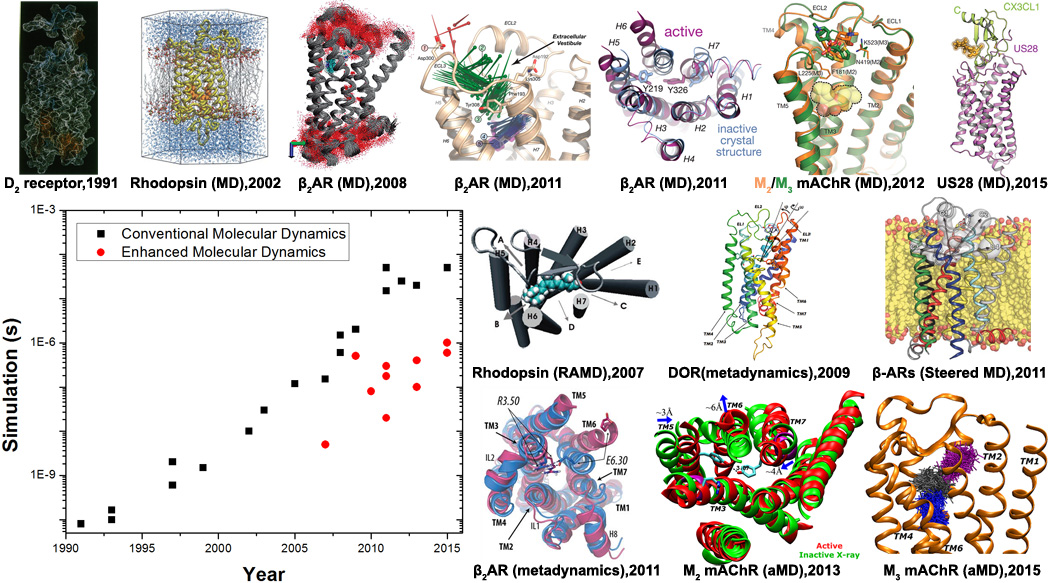
Advances in conventional and enhanced molecular dynamics (MD) simulations of GPCR signaling to date. GPCR structural images are adapted from conventional MD simulations on a homology model of the D2 dopamine receptor in 1991[8], rhodopsin using its X-ray structure in 2002[14,15], flexibility and internal hydration of β2AR following determination of the X-ray structure in 2008[17], drug binding and deactivation processes of β2AR in 2011[32,40], ligand binding to the M2 and M3 mAChRs in 2012[33] and constitutive activity of US28 bound by CX3CL1 chemokine in 2015[49]. Enhanced MD simulations have also been applied to explore structural dynamics of GPCRs. For example, ligand unbinding has been investigated through random acceleration MD (RAMD) simulations on rhodopsin in 2007[27] and β2AR in 2009[28], metadynamics simulations of δ-opioid receptor (DOR) in 2009[31] and steered MD simulations of the b-ARs in 2011[30]. Moreover, GPCR activation processes have been characterized through combined metadynamics and adiabatic biasing MD (ABMD) simulations of rhodopsin in 2010[50] and β2AR in 2011[42], as well as aMD simulations of M2 mAChR in 2013[59] and M3 mAChR in 2015[51]. In addition, ligand binding to the M3 mAChR captured through aMD simulations was reported in 2015[36].