

Abstract
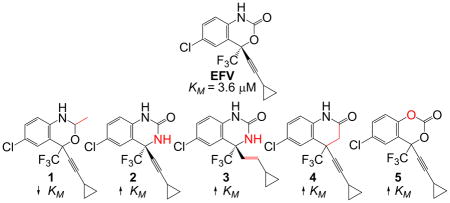
Previously, we observed that the oxazinone ring is important for CYP2B6 activity toward efavirenz ((4S)-6-Chloro-4-(2-cyclopropylethynyl)-1,4-dihydro-4-(trifluoromethyl)-2H-3,1-benzoxazin-2-one), a CYP2B6 substrate used to treat HIV. Here, to further understand the structural characteristics of efavirenz that render it a CYP2B6 substrate, we test the importance of each heteroatom of the oxazinone ring. We assembled a panel of five analogues: 6-Chloro-4-(2-cyclopropylethynyl)-1,4-dihydro-2-methyl-4-(trifluoromethyl)-2H-3,1-benzoxazine (1), (4S)-6-Chloro-4-[(1E)-2-cyclopropylethenyl]-3,4-dihydro-4-(trifluoromethyl)-,2(1H)-quinazolinone (2), (4S)-6-Chloro-4-(2-cyclopropylethynyl)-3,4-dihydro-4-(trifluoromethyl)-2(1H)-quinazolinone (3), 6-Chloro-4-(cyclopropylethynyl)-3,4-dihydro-4-(trifluoromethyl)-2(1H)-quinolinone (4), and 6-Chloro-4-(cyclopropylethynyl)-4-(trifluoromethyl)-4H-benzo[d][1,3]dioxin-2-one (5). Metabolism of 1–5 was investigated using human liver microsomes, individual P450s, and mass spectrometry or UV absorbance detection. Steady-state analysis of CYP2B6 metabolism of 1–5 showed KM values ranging from 0.3 to 3.9 fold different than observed for efavirenz (KM of 3.6 ± 1.7 μM). The lowest KM values approximating 1 μM, were observed for metabolism of 1, while the largest KM, 14 ± 6.4 μM, was found for 4. Our work reveals that analogues with heteroatom changes in the oxazinone ring are still CYP2B6 substrates, though the changes KM suggest altered substrate binding.
Keywords: cytochromes P450, drug metabolism, efavirenz, structure-activity relationships
Graphical Abstract
A panel of efavirenz analogues with single heteroatom changes in the oxazinone ring were assessed for their ability to be metabolized by human cytochrome P450 2B6 (CYP2B6). Though each analogue was still found to be a substrate for CYP2B6, KM values associated with product formation by CYP2B6 were determined to be quite different from efavirenz.
Introduction
The cytochromes P450 (P450s) are a superfamily of heme-containing monooxygenases that participate in the metabolism of xenobiotic compounds, such as drugs and environmental toxins. P450 substrates are generally nonpolar molecules that are rendered more polar by insertion of an oxygen atom, resulting in a product that can be more readily excreted from the body. Cytochrome P450 2B6 (CYP2B6) represents the only bona fide member of family 2B in humans. Other well-studied 2B subfamily members, each sharing at least 75% amino acid sequence identity with CYP2B6, include rat CYP2B1, rabbit CYP2B4, and mouse Cyp2b10. CYP2B6 plays a prominent role in the metabolism of many clinically relevant substrates including the chemotherapeutic pro-drug cyclophosphamide,[1] the antidepressant bupropion,[2] the opioid maintenance drug methadone,[3] and the HIV non-nucleoside reverse transcriptase inhibitor efavirenz (EFV).[4] Over 100 different single nucleotide polymorphisms have been identified in CYP2B6, representing 38 distinct variant alleles.[5] These variants are associated with a broad spectrum of phenotypes from low to high activity towards CYP2B6 substrates.[6] Some variants of CYP2B6 have been associated with decreased response to cyclophosphamide-based therapies,[6a, 7] elevated plasma levels of methadone,[8] and of EFV.[9]
EFV is a commonly prescribed drug used to treat HIV type 1. As a non-nucleoside reverse transcriptase inhibitor, EFV inhibits the activity of HIV reverse transcriptase via binding at an allosteric site.[10] CYP2B6 is the primary enzyme responsible for the formation of 8-hydroxyefavirenz (8-OH EFV), the most abundant product of phase 1 EFV metabolism in humans.[4a, 11] EFV has been shown to modulate the expression of CYP2B6 via nuclear receptor activation[12] as well as to inactivate CYP2B6 protein via mechanism-based inactivation.[4b] Thus, in addition to being a CYP2B6 substrate, EFV impacts both CYP2B6 expression and activity. In the absence of crystal structures of CYP2B6 in complex with EFV, little is known about the regions of the EFV chemical structure that make important contacts with the CYP2B6 active site and may contribute to the high activity of CYP2B6 against EFV. Previously, we demonstrated an intact oxazinone ring to be important for metabolism of EFV by CYP2B6.[13] Additionally, in those studies, we found that an analogue lacking only the carbonyl oxygen of EFV was a better CYP2B6 substrate than EFV itself. Here we continue this work by examining the impact of each heteroatom position of the EFV oxazinone ring on metabolism by CYP2B6. In doing so, we found that CYP2B6 is tolerant of single atom changes in the oxazinone ring; however, these changes impact the KM associated with the metabolism of these substrates. Having observed the consequences of these structural changes for CYP2B6, we tested whether another CYP2B enzyme, rat CYP2B1, displayed an activity profile similar to that of CYP2B6 against EFV analogues. We found a striking similarity between the activity of these CYP2B enzymes towards EFV analogues that was not observed with other P450s of the 1A, 2A, 2C, 2D, or 3A families, suggesting the ability to metabolize certain EFV analogues may be conserved in CYP2B family members.
Results
Our interest in the oxazinone ring of EFV led us to acquire a targeted panel of EFV analogues with discreet changes in the oxazinone ring (Figure 1). We first performed incubations with pooled human liver microsomes in order to identify P450-dependent metabolites of these analogues. Using ultra high-performance liquid chromatography tandem mass spectrometry (uHPLC-MS/MS), we detected monooxygenated metabolites formed from each analogue. Two monooxygenated metabolites (m/z 332) were formed from 1, which has a methyl group in place of the carbonyl oxygen of efavirenz (Figure 1). Metabolite 1-M1 displayed a retention time (RT) of 5.85 min while 1-M2 displayed a RT of 7.10 min (Figure 2A.i and 2A.ii). Fragmentation of 1-M1 (theoretical m/z: 332.0659, experimental m/z: m/z 332.0658) using a high resolution accurate mass instrument revealed fragment ions of m/z 290.0550, 272.0444, 232.0128, and 178.0416 resulting from a loss of C2H2O, C2H4O2, C5H8O2, and C5H5F3O2, respectively. Metabolite 1-M2 (theoretical m/z: 332.0659, experimental m/z: 332.0660) possessed fragment ions of m/z 288.0394, 268.0332, 248.0074, and 184.0755. These fragments suggested a loss of C2H4O, C5H4, C5H8O, and C3H4ClF3O, respectively. Based on the fragmentation, we propose that 1-M1 resulted from a single oxygen insertion at the methyl group of the oxazine ring (Figure 3A). We further propose that 1-M2 resulted from oxygen insertion on the benzene ring at the 5, 7, or 8 position (Figure 3B).
Figure 1.
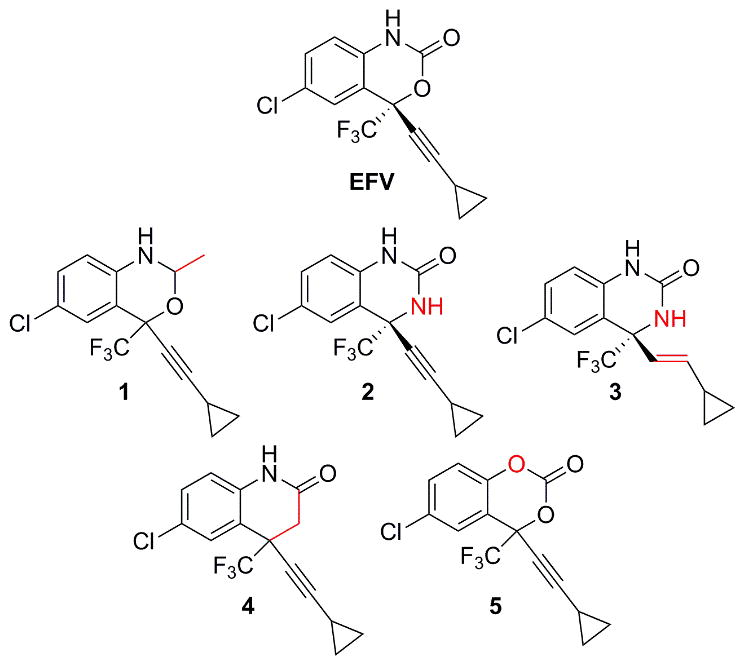
Efavirenz (EFV) and analogues 1–5 used in this study. Structural departures from EFV are denoted in red.
Figure 2.

Extracted ion chromatograms of metabolites formed from incubations with human liver microsomes and analogues 1–4 and UV chromatogram from incubations with analogue 5. Metabolites were named in order of elution beginning with M1: 1-M1 at 5.85 min using 332>234 (A.i), metabolite 1-M2 at 7.10 min using 332>220 (A.ii), metabolite 2-M1 and -M2 at 5.42 and 5.65 min, respectively, using 331>265 (A.iii), metabolite 3-M1 at 5.65 min using 333>248 (A.iv), Metabolites 4-M1 and -M2 at 6.01 and 6.42 min, respectively, using 330>288 (A.v), and metabolite 5-M1 at 11.41 min (B). Analogue 5 appears in B at 12.13 min. Peaks shown in the chromatograms that are not labeled as metabolites appeared in the absence of NADPH and/or the absence of parent compound.
Figure 3.

High resolution accurate mass spectra and proposed fragment assignments for 1-M1 (A), 1-M2 (B), 2-M1 (C), 2-M2 (D), 3-M1 (E), 4-M1 (F), and 4-M2 (G). Samples were obtained from incubations with 0.5 mg/mL human liver microsomes and 10 μM of the indicated substrate for 30 minutes at 37 °C in the presence of 100 mM potassium phosphate, pH 7.4, and an NADPH regenerating system. Spectra were collected using a Q-Exactive mass spectrometer. Data are representative results from three independent incubations. Corresponding triple quadrupole MS2 spectra for these metabolites can be found in Figure S1.
Analogue 2, which has a nitrogen atom rather than the oxygen atom within the oxazinone ring of efavirenz, also displayed two monooxygenated metabolites (m/z 331, Figure 2A.iii) from incubations with human liver microsomes. Metabolite 2-M1 (theoretical m/z: 331.0456, observed m/z: 331.0454), with a RT of 5.42 min, showed fragments of m/z 288.0392, 264.9983, 261.0418, and 184.0755. We propose these fragments resulted from a loss of CHNO, C5H6, CHF3, and C2HClF3O, respectively. With a RT of 5.65 min, 2-M2 (theoretical m/z: 331.0456, observed m/z: 331.0454) had the same characteristic fragments as 2-M1, suggesting that perhaps the oxygen insertions were in close proximity. These data lead us to propose that 2-M1 and 2-M2 both resulted from oxygen insertions at separate positions on the benzene ring (Figures 3C and 3D).
Similar to 2, 3 has the same modification to the oxazinone ring of efavirenz, but 3 also has a trans-alkene in place of the efavirenz alkyne. One monooxygenated metabolite with m/z of 333 was detected from human liver microsome incubations with 3 (Figure 2A.iv). With a retention time of 5.65 min, 3-M1 (theoretical m/z: 333.0612, experimental m/z: 333.0609) displayed characteristic fragments of m/z 290.0550, 264.9982, and 248.0081. Our proposed fragments suggest these ions resulted from a loss of CHNO, C5H6, and C4H5NO, respectively. Based on these proposed fragment structures, we predict 3-M1 was formed from oxygen insertion at the 5, 7, or 8 position of the benzene ring (Figure 3E).
Whereas 2 and 3 have a nitrogen in place of the oxygen atom within the oxazinone ring of EFV, analogue 4 has a carbon atom at this same position. Two monooxygenated metabolites (m/z 330) were observed to be formed from incubations with this analogue and human liver microsomes (Figure 2A.v). With a RT of 6.01 min, metabolite 4-M1 (theoretical m/z: 330.0503, experimental m/z: 330.0500) displayed characteristic fragment ions of m/z 288.0394, 264.0029, 226.0859, and 184.0755. Our interpretation of these fragments suggests that they resulted from a loss of C2H2O, C5H6, CHF3, and C3H2ClF3O. Metabolite 4-M2 (theoretical m/z: 330.0503, experimental m/z: 330.0500) had a RT of 6.42 min and displayed characteristic ions of m/z 288.0394, 262.0427, 252.0632, and 226.0859. We propose these fragment ions resulted from a loss of C2H2O, C5H6, C2H3ClO, and CF3, respectively. Based on our structural predictions and the observation that the elution times differ between 4-M1 and 4-M2 to the same degree as observed for 2-M1 and 2-M2, we predict that 4-M1 and 4-M2 also resulted from separate oxygen insertions on the benzene ring (Figures 3F and 3G). In the absence of NMR data, the structures proposed for metabolites formed from 1–4 are tentative. Detection of 5 using uHPLC-MS/MS was not successful; therefore, we employed uHPLC-UV to visualize 5 and any potential metabolites. Using this method, one prominent metabolite was detected from incubations with 5 and pooled human liver microsomes (Figure 2B). In the absence of a mass spectrum for this metabolite, no structural predictions can be made regarding the site(s) of oxygen insertion.
Having determined which metabolites can be formed from these analogues, we wanted to test whether analogues 1–5 were also CYP2B6 substrates. To answer this question, we incubated each analogue (10 μM) with cDNA-expressed CYP2B6 (10 nM) and measured formation of each metabolite using selected reaction monitoring or UV detection (for 5). We found that CYP2B6 readily metabolized analogues 1–5 from our panel (Figure 4), indicating that the changes to the oxazinone ring did not prevent these compounds from being metabolized by CYP2B6. Compared to the other P450s in our panel, activity of CYP2B6 was found to result in the most 1-M1, 2-M2, and 3-M1 formation under the given reaction conditions (Figure 4). Conversely, formation of 1-M2 was most abundant with CYP3A4 and CYP3A5 while CYP1A2 activity resulted in high amounts of 4-M2 (Figure 4). The finding that analogues in our panel are also substrates for P450s in the families 1A and 3A is consistent with the fact that CYP1A2, CYP3A4, and CYP3A5 have previously been shown to metabolize efavirenz.[4a] The formation of 5-M1 by CYP2B6 and CYP1A2 seemed relatively equal, while CYP1A1 was also observed to catalyze the formation of 5-M1. Only two metabolites identified in our experiments in human liver microsomes were not found to be formed by CYP2B6 (2-M1 and 4-M1). However, these metabolites were produced by other P450s (Figure 4). Of note, no other P450 tested displayed a pattern of activity against our panel of efavirenz analogues that was similar to that of CYP2B6.
Figure 4.

Intensities of metabolites formed from incubations with 1 (A.i), 2 (A.ii), 3 (A.iii), 4 (A.iv), and 5 (B). Each analogue (10 μM) was incubated for 30 min with 10 nM of the indicated cDNA-expressed P450 in 100 mM potassium phosphate pH 7.4 and an NADPH regenerating system. Metabolite formation was measured by triple quadrupole uHPLC-MS/MS (A.i–A.iv) or uHPLC-UV (B). Data represent the mean ± SEM for three independent experiments.
Since we found 1–5 to be CYP2B6 substrates, we next sought to probe whether the kinetic constants for metabolism of these substrates by CYP2B6 might reveal more subtle impacts of these single atom substitutions on CYP2B6 activity. Results from our previous work with other EFV analogues suggested differences in binding affinity,[13] thus we sought to explore whether these analogues also differed from EFV in their affinity for CYP2B6. In order to explore this question, we performed product formation kinetic analysis with 1–4 using uHPLC-MS/MS and with 5 using uHPLC-UV detection, and compared the results to the KM associated with 8-OH EFV formation (Table 1).
Table 1.
Estimated Michaelis constants for product formation by CYP2B6
| Product measured | KM (μM) [a] | Fold Change |
|---|---|---|
| 8-OH EFV | 3.6 ± 1.7 | 1 |
| 1-M1 | 1.4 ± 0.76 | 0.4 |
| 1-M2 | 1.0 ± 0.53 | 0.3 |
| 2-M2 | 11 ± 4.9 | 3.0 |
| 3-M1 | 13 ± 3.4 | 3.6 |
| 4-M2 | 14 ± 6.4 | 3.9 |
| 5-M1 | 5.5 ± 2.1 | 1.5 |
KM values are reported as the mean ± SEM for three independent experiments collected in duplicate.
We found the formation of 8-OH EFV to be characterized by a KM of 3.6 ± 1.7 μM, which is similar to KM values obtained in the reconstituted system[4b, 15] and in another study also using cDNA-expressed CYP2B6.[4a] By comparison, the KM values for metabolism of analogues 1–5 by CYP2B6 were different (Table 1). Production of metabolite 1-M2 displayed the lowest KM of 1.0 ± 0.53 μM, which is an approximate 3.3-fold decrease compared to 8-OH EFV and suggests that CYP2B6 has a higher affinity for this analogue than for EFV. Conversely, CYP2B6 seems to have a lower affinity for 2, 3, and 4, as the formation of 2-M2, 3-M1, and 4-M2 were characterized by KM values 3.0 to 3.9 times higher than for 8-OH EFV (11 ± 4.9, 13 ± 3.4, and 14 ± 6.4 μM, respectively). Interestingly, the KM for formation of 5-M1 differed by only a factor of 1.5 from 8-OH EFV (5.5 ± 2.1 μM) indicating a similar affinity of CYP2B6 for 5 and EFV.
Since we observed that CYP2B6 was the only P450 in our panel that metabolized 1–5, we wanted to test whether this activity profile might be conserved in a closely related enzyme in the CYP2B family. To this end, we explored the activity of the well-studied rat enzyme CYP2B1, which shares 76% amino acid sequence identity with CYP2B6, against our panel of EFV analogues (1–5). CYP2B1 produced the same metabolites from 1–5 as we observed for CYP2B6 (Figure S2). We then incubated CYP2B1 with each of the eight EFV analogues reported previously.[13] We again found that CYP2B1 mirrored CYP2B6 activity towards our entire panel of EFV analogues, including M2 formation from E-dihydroefavirenz, M4 and M5 formation from the pentynyl analogue, and M2 formation from the oxazine analogue. Moreover, with the exception of 2-M2, 3-M1, 4-M2, and 5-M1; metabolite formation from our panel of EFV analogues differed by less than 3-fold between CYP2B6 and CYP2B1 (Figure 5). Like CYP2B6, CYP2B1 did not form metabolites from the two methoxyphenyl analogues or the methoxybenzamide analogue. Interestingly, CYP2B1 displayed activity towards the both (R) and (S) -5-Chloro-α-(cyclopropylethynyl)-2-amino-α-(trifluoromethyl) benzenemethanol analogues, which we did not observe previously with CYP2B6.[13] We then developed a more sensitive selected reaction monitoring method to detect metabolites from these analogues and found CYP2B6 to also form a monooxygenated metabolite from these enantiomers (Figure S3). Taken together, these data suggest that CYP2B1 and CYP2B6 share a parallel profile of activity towards EFV analogues.
Figure 5.

Comparison of metabolite formation by CYP2B1 and CYP2B6. CYP2B1 or CYP2B6 (10 nM) were incubated with EFV or EFV analogues (10 μM) in the presence of an NADPH regenerating system for 30 minutes and the formation of the indicated metabolite measured by uHPLC-MS or uHPLC-UV (5-M1 only). Mean metabolite peak height from three independent incubations with CYP2B6 was set at 100% (represented by a dashed line). Values in the graph represent the metabolite formed by CYP2B1 in three independent experiments and are expressed as a mean percent of metabolite formed by CYP2B6 ± SEM.
Discussion
In this study, we have explored the impact of single heteroatom changes in the oxazinone ring of EFV on CYP2B6 activity towards these compounds. Though analogues 1–5 were all found to be substrates for CYP2B6, the KM values corresponding to the metabolism of 1–5 indicate perturbations of the affinity of CYP2B6 for the substrate. Our data also show that the substrate specificity observed for CYP2B6 and our panel of EFV analogues also extends to another CYP2B enzyme, rat CYP2B1. Together these findings further support a role for the heteroatom composition of the oxazinone ring in CYP2B6 metabolism of EFV.
Multiple reports characterizing CYP2B6 activity towards many different substrates have been published to date (reviewed in [6]). In addition, several computational approaches have sought to predict potential CYP2B6 substrates based on the structural characteristics of known substrates.[16a–e] Each of these computational approaches determined that lipophilicity, molecular size, and molecular shape were important considerations for predicting CYP2B6 substrates. Compared to EFV, whose theoretical log P is estimated to be 3.68, 1–5 have similar log P values ranging from 3–4. Since our analogue panel was developed around minor changes to the oxazinone ring, the molecular size and shape were nearly identical for each analogue compared to EFV. Thus, our findings that these analogues are substrates for CYP2B6 is in agreement with previous computational work, however, our observations here demonstrate that minor structural changes that are not captured by changes in log P or molecular size and shape still contribute to substrate binding.
Since no crystal structures of EFV bound to CYP2B6 have been reported to date, additional computational work in the form of molecular modeling provides the best available visualization of EFV in the CYP2B6 active site.[17] This molecular model shows that hydrogen bonding between CYP2B6 residue E301, various water molecules, and EFV largely account for the calculated interaction energy. Moreover, this network of hydrogen bonds is reported to involve interactions with heteroatoms within the oxazinone ring. Our observation that KM values for the metabolism of 1–4 differed from those for 8-OH EFV formation suggest that changes in the oxazinone ring resulted in altered affinities of these analogues for CYP2B6. The lowest KM values were obtained from incubations with 1, which has a methyl group in place of the carbonyl oxygen of EFV. Interestingly, 1-M1 was the only metabolite we found in this panel that resulted from oxygen insertion at a site other than the benzene ring, indicating that this analogue must adopt a different conformation in the CYP2B6 active site. We previously showed that elimination of this position altogether, resulted in a lower KM and higher Vmax compared to EFV.[13] Our data obtained here demonstrate that addition of a hydrophobic methyl group also results in tighter binding, as is suggested by the lower KM value. Thus, our work here further supports that the presence of a carbonyl oxygen atom seems to decrease the affinity of CYP2B6 for EFV. In the case of the methyl analogue, the added stereocenter on the heterocyclic ring would result in an additional non-polar group projecting out of the plane of the heterocyclic ring. This methyl group might then be able to participate in intermolecular interactions perpendicular to the heterocyclic ring. The carbonyl oxygen of EFV would be unable to adopt this conformation due to the restriction of the double bond. Instead the carbonyl oxygen would lie only slightly out of the plane delineated by the heterocyclic ring, as has been previously observed in crystals of EFV.[18] Based on this, it is possible that the carbonyl oxygen atom largely in the plane of the heterocyclic ring clashes with nearby CYP2B6 residues. In the previously reported model of CYP2B6 and EFV, L363 and V367 are proposed to lie parallel to the carbonyl oxygen.[17] It is possible that the added methyl group forms hydrophobic interactions with these residues and thus further stabilizes 1 in the CPY2B6 active site. This specific possibility seems less likely for the formation of 1-M1 than for 1-M2 due to the proposed position of oxygen insertion.
An approximate three to four fold increase in KM was observed for both quinazolinone analogues (2 and 3) as well as the lone quinolinone analogue (4). Compared to EFV these analogues reflect changes to the oxygen atom within the oxazinone ring. In EFV, this position may play a role as a hydrogen bond acceptor, but in 2 and 3 this position may be a hydrogen bond donor. Moreover, in 4 the presence of the carbon atom completely abrogates the capacity for hydrogen bonding at this position. Since we observed a 3 to 4-fold effect on affinity of CYP2B6 for 2–4 compared to EFV, hydrogen bonding at this position may have a slight impact on the interaction of CYP2B6 and EFV.
Analogue 5 is the only analogue tested that reflects changes to the EFV nitrogen atom. Like 1–4, this dioxin-2-one analogue was still a CYP2B6 substrate, and 5-M1 exhibited a KM only 1.5 fold higher than 8-OH EFV. The alteration of the EFV nitrogen atom seems to have little effect when changed to an oxygen, indicating that a hydrogen bond donor is not crucial at this position.
Kinetic analysis of 1–5 revealed that each analogue was best characterized by the substrate inhibition equation rather than the Michaelis-Menten equation. Formation of 8,14 dihydroxyefavirenz by CYP2B6 is best characterized by the substrate inhibition equation,[4a] but 8-OH EFV formation has been repeatedly shown to fit well to Michaelis-Menten.[4a, 4b, 11, 15] This observed difference with our analogues is of interest and worthy of further study.
To our surprise, we did not observe any dihydroxylated metabolites from our panel. CYP2B6 has been shown to catalyze the formation of 8, 14 dihydroxyefavirenz,[4a] which we have also observed (data not shown), yet no dihydroxylated metabolites from 1–4 were seen in this study. The use of uHPLC-UV for detection of 5-M1 does not allow us to determine the m/z for this metabolite, thus we cannot rule out the possibility that this is a dihydroxylated metabolite. However, we do not observe any other metabolites formed from 5 with HLM or with individual P450s. We would expect that if 5-M1 resulted from two oxygen insertions, we would have also observed another peak corresponding to a monooxygenated metabolite. The fact that no dihydroxylated metabolites were found from 1–4 suggests that the heteroatom changes explored in this study seem to have an impact on sequential metabolism by CYP2B6.
Our finding that CYP2B1 and CYP2B6 display a similar activity profile towards our panel of EFV analogues suggests the toleration of changes to the EFV structure might be conserved in CYP2B enzymes. Indeed, we observed that none of the other nine P450s tested showed the same profile of selectivity towards EFV, analogues 1–5, and the eight analogues we characterized previously.[13] Moreover, the magnitude of metabolite formation from 2B6 and 2B1 differed by no more than six-fold across all 13 EFV analogues studied. In the case of 8-OH EFV production, CYP2B1 formed an approximate 48% of this metabolite compared to 2B6. The largest differences between 2B1 and 2B6 activity against our panel were observed for formation of 2-M2, 3-M1, 4-M2, and 5-M1 (16.4, 17.7, 20.4, and 23.1% of metabolite formation by 2B6, respectively). Three of these analogues, 2–4, represent changes to the oxygen atom within the oxazinone ring of EFV. It is possible that CYP2B1 is more sensitive to alterations at this position than we observed for CYP2B6. The smallest differences (<2-fold) between CYP2B1 and 2B6 metabolite formation were observed for 1-M1, 1-M2, M5 of the pentynyl analogue, and M2 of the oxazine analogue. These analogues share changes to the carbonyl oxygen or the cyclopropyl group of EFV, indicating that these positions may reside in similarly tolerant active site environments within CYP2B1 and CYP2B6. Additionally, CYP2B1 was found to form 2–3 fold more metabolite from (S) and (R) -5-Chloro-α-(cyclopropylethynyl)-2-amino-α-(trifluoromethyl) benzenemethanol analogues compared to 2B6. Further kinetics experiments with these two analogues may aid in determining the source of these differences and may provide a more detailed rationale for the increased activity of 2B1 compared to 2B6.
Since the results of this study include only two CYP2B family members, more CYP2B P450s need to be tested to see if they also display the same activity profile towards EFV analogues as CYP2B6 and CYP2B1. If a CYP2B family-wide pattern of activity against EFV analogues were to be established, this might imply the existence of an important series of conserved residues within CYP2B enzymes, but not other P450s.
Conclusion
We have determined that CYP2B6 maintains activity towards analogues of EFV that have discreet substitutions in the heterocyclic ring. These changes, however, result in differences in KM values. The substitution of a methyl group for the carbonyl oxygen increases the affinity of CYP2B6 towards this molecule compared to EFV, supporting the dispensability of the carbonyl oxygen for metabolism of EFV by CYP2B6. Additionally, atom substitutions to the internal oxygen and nitrogen atoms do not have strong KM impacts. This indicates that these heteroatoms are not critical for binding of EFV to CYP2B6. Finally, we found that changing the EFV nitrogen atom to oxygen did not prevent this molecule from being a CYP2B6 substrate and had virtually no impact on binding affinity. This work provides further insight into the functional interaction of CYP2B6 and EFV and lays the groundwork for a better understanding of 2B family enzyme activity towards EFV.
Experimental Section
Analogues
EFV, racemic 8-OH EFV, and EFV analogues were synthesized by Toronto Research Chemicals (Toronto, Canada) and were ≥ 97% pure, according to the manufacturer. The analogues used in this study were 6-Chloro-4-(2-cyclopropylethynyl)-1,4-dihydro-2-methyl-4-(trifluoromethyl)-2H-3,1-benzoxazine (analogue 1), (4S)-6-Chloro-4-(2-cyclopropylethynyl)-3,4-dihydro-4-(trifluoromethyl)-2(1H)-quinazolinone (2), (4S)-6-Chloro-4-[(1E)-2-cyclopropylethenyl]-3,4-dihydro-4-(trifluoromethyl)-,2(1H)-quinazolinone (3), 6-Chloro-4-(cyclopropylethynyl)-3,4-dihydro-4-(trifluoromethyl)-2(1H)-quinolinone (4), and 6-Chloro-4-(cyclopropylethynyl)-4-(trifluoromethyl)-4H-benzo[d][1,3]dioxin-2-one (5) (Figure 1). The synthesis for analogues 2 and 3 was originally reported by DuPont Pharmaceuticals.[14] We also made use of eight additional EFV analogues, which were reported in our previous publication.[13] These analogues were (S)-5-Chloro-α-(cyclopropylethynyl)-2-amino-α-(trifluoromethyl)benzenemethanol, (R)-5-Chloro-α-(cyclopropylethynyl)-2-amino-α-(trifluoromethyl)benzenemethanol, (E)-Dihydroefavirenz, rac 6-Chloro-1,4-dihydro-4-(1-pentynyl)-4-(trifluoromethyl)-2H-3,1-benzoxazin-2-one, (R)-5-Chloro-α-(cyclopropylethynyl)-2-[[(4-methoxyphenyl)methyl]amino]-α-(trifluoromethyl)benzenemethanol, (S)-5-Chloro-α-(cyclopropylethynyl)-2-[[(4methoxyphenyl)methyl]amino]-α-(trifluoromethyl)benzenemethanol, rac N-[4-Chloro-2-[3-cyclopropyl-1-hydroxy-1-(trifluoromethyl)-2-propynyl]phenyl]-4-methoxybenzamide, and (4S)-6-Chloro-4-(2-cyclopropylethynyl)-1,4-dihydro-4-(trifluoromethyl)-2H-3,1-benzoxazine.
Enzyme assays
Enzyme incubations were carried out essentially as previously described.[13] Reactions were pre-incubated in 100 mM potassium phosphate pH 7.4 containing 0.5 mg/mL pooled human liver microsomes (Xenotech LLC, Lenexa, KS) or 10 nM cDNA-expressed P450 (Supersomes ®, Corning, Corning, NY) and 10 μM of each analogue for 5 minutes at 37°C. Reactions were then initiated by the addition of an NADPH regenerating system (Corning, Corning, NY) for a total volume of 100 μL and allowed to proceed for 30 minutes at 37°C. After this incubation, the protein was precipitated by the addition of 100 μL ice-cold acetonitrile. Precipitated reactions were incubated on ice for 10 minutes then centrifuged for 10 minutes at 10,000 × g at 4 °C and the supernatant dried in a vacuum centrifuge. Samples were reconstituted in 100 μL methanol in preparation for mass spectrometry or UV detection.
For assays measuring product formation for determination of KM, pilot incubations were performed to determine the linearity with respect to enzyme concentration and time. CYP2B6 (5–20 nM) was incubated as above with eight concentrations (0.5–100 μM) of EFV or the indicated EFV analogue and metabolism allowed to occur in the presence of an NADPH regenerating system for 5–20 minutes. Substrate concentrations of 0.5–30 μM were used for 3 due to limited solubility in aqueous reaction buffer. An equal volume (100 μL) of ice-cold acetonitrile was added to terminate the reaction and precipitate protein. Reactions were then processed for mass spectrometry as above. In the absence of authentic metabolite standards, we were unable to quantitate product formation for metabolism of 1–5. With our goal of comparing relative affinities of these substrates for CYP2B6 in mind, we sought to obtain only KM values from analysing product formation.
In order to draw comparisons between enzymatic activity of CYP2B1 and CYP2B6, we sought to use a similar system as our experiments using cDNA-expressed CYP2B6. To that end, cDNA-expressed CYP2B1 (Supersomes ®, Corning, Corning, NY) were obtained. In contrast to our other experiments presented here, this system also contained cytochrome b5, thus, for comparison purposes, cDNA-expressed CYP2B6 containing b5 (Supersomes ®, Corning, Corning, NY) was tested side-by-side to CYP2B1. The molar ratio of CYP2B6:reductase:b5 was 1:10:2, as determined by the manufacturer. The molar ration of CYP2B1:reductase:b5 was 1:1:2, according to the manufacturer. These assays were performed and samples processed in the same manner described above.
Analyte Detection
Detection of EFV, previously reported EFV analogues, and EFV analogues 1–4 was achieved essentially as previously described.[13] Reconstituted samples were resolved with an Xterra C18 column (2.1 × 50 mm, 2.5 μm; Waters, Milford, MA) using a Dionex Ultimate 3000 uHPLC system coupled to a TSQ Vantage Triple Stage Quadrupole mass spectrometer (Thermo Scientific, Pittsburgh, PA) or a Q-Exactive benchtop Orbitrap mass spectrometer (Thermo Scientific, Pittsburgh, PA). The mobile phases used for all analogues except 5 were water with 0.1% formic acid (mobile phase A) and acetonitrile with 0.1% formic acid (mobile phase B). The gradient for efavirenz and all analogues except 5 was identical: 10% B from 0–0.5 min, 10–95% B from 0.5–10 min, 95% B from 10–10.5 min, 95–10% B from 10.5–10.6 minutes, and 10% B from 10.6–11.5 minutes. Positive ion mode was used for detection of 1–4 and corresponding metabolites. Detection of analogues and metabolites using our triple quadrupole instrument was first achieved with product ion mode. Upon selecting unique fragments from each observed analyte, we developed transitions for selected reaction monitoring in order to increase sensitivity (Table 2). The transition used to detect monooxygenated metabolites from (S) and (R) -5-Chloro-α-(cyclopropylethynyl)-2-amino-α-(trifluoromethyl)benzenemethanol was m/z 306>248. For Q-Exactive experiments, PRM scans were constructed for each metabolite using the proposed molecular formula (1 – C15H13ClF3NO2, 2 – C14H10ClF3N2O2, 3 – C14H12ClF3N2O2, and 4 – C15H11ClF3NO2. Fragmentation spectra were obtained with stepped NCE of 20, 30 and 40.
Table 2.
Selected reaction monitoring transitions used in detection of EFV, 1–4, and metabolites via triple quadrupole mass spectrometry.
| Analyte | Parent m/z | Selected fragment m/z |
|---|---|---|
| EFV | 314 | - |
| 8-OH EFV | 330 | 162 |
| 1 | 316 | 246 |
| 1-M1 | 332 | 234 |
| 1-M2 | 332 | 220 |
| 2 | 315 | 249 |
| 2-M1 and -M2 | 331 | 265 |
| 3 | 317 | 232 |
| 3-M1 | 333 | 248 |
| 4 | 314 | 272 |
| 4-M1 and -M2 | 330 | 288 |
Analogue 5 was not readily detectable via mass spectrometry. Therefore, we instead used UV to detect parent and to measure metabolite formation. Separation of 5 and its metabolite was accomplished with the same Xterra C18 column as above using an Acquity uHPLC system coupled to a photodiode array detector set at 220 nm (Waters, Milford, MA). The solvents for uHPLC-UV were water with 0.1% trifluoroacetic acid (mobile phase A) and acetonitrile with 0.1% trifluoroacetic acid (mobile phase B). The chromatographic program for 5 was 10% B from 0–0.5 minutes, 10–95% B from 0.5–15 minutes, 95% B from 15–16 minutes, 95–10%B from 16–16.1 minutes, and 10% B from 16.1–20 minutes. All solvents used for uHPLC-MS/MS and uHPLC-UV were of the highest grade commercially available.
Data Analysis
Reported KM values were estimated from fitting product formation data to the substrate inhibition equation using GraphPad Prism version 7 (GraphPad Software Inc., San Diego, CA). Initial data fits were made to the Michaelis-Menten equation; however, a better R2 value was found for substrate inhibition. For comparison of CYP2B1 and CYP2B6 activity towards EFV analogues, observed metabolite peak height from CYP2B6 incubations was set at 100% and metabolite peak height from CYP2B1 incubations normalized to this value.
Supplementary Material
Acknowledgments
This research was supported by NIH R01GM103853 to N.N.B. and NSF DGE-1232825 to P.M.C. The authors would like to thank Drs. Mark Marzinke and Teresa Parsons for assistance with uHPLC-UV experiments.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.Chang TK, Weber GF, Crespi CL, Waxman DJ. Cancer Res. 1993;53(23):5629–5637. [PubMed] [Google Scholar]
- 2.Hesse LM, Venkatakrishnan K, Court MH, von Moltke LL, Duan SX, Shader RI, Greenblatt DJ. Drug Metab Dispos. 2000;28(10):1176–1183. [PubMed] [Google Scholar]
- 3.Kharasch ED, Hoffer C, Whittington D, Sheffels P. Clin Pharmacol Ther. 2004;76(3):250–269. doi: 10.1016/j.clpt.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 4.a Ward BA, Gorski JC, Jones DR, Hall SD, Flockhart DA, Desta Z. J Pharmacol Exp Ther. 2003;306(1):287–300. doi: 10.1124/jpet.103.049601. [DOI] [PubMed] [Google Scholar]; b Bumpus NN, Kent UM, Hollenberg PF. J Pharmacol Exp Ther. 2006;318(1):345–351. doi: 10.1124/jpet.106.102525. [DOI] [PubMed] [Google Scholar]
- 5.Sim SC, Ingelman-Sundberg M. Hum Genomics. 2010;4(4):278–281. doi: 10.1186/1479-7364-4-4-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a Zanger UM, Klein K. Front Genet. 2013;4:24. doi: 10.3389/fgene.2013.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wang H, Tompkins LM. Curr Drug Metab. 2008;9(7):598–610. doi: 10.2174/138920008785821710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson GG, Lin K, Cox TF, Oates M, Sibson DR, Eccles R, Lloyd B, Gardiner LJ, Carr DF, Pirmohamed M, Strefford JC, Oscier DG, Gonzalez de Castro D, Else M, Catovsky D, Pettitt AR. Blood. 2013;122(26):4253–4258. doi: 10.1182/blood-2013-07-516666. [DOI] [PubMed] [Google Scholar]
- 8.Kharasch ED, Regina KJ, Blood J, Friedel C. Anesthesiology. 2015;123(5):1142–1153. doi: 10.1097/ALN.0000000000000867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a Rotger M, Tegude H, Colombo S, Cavassini M, Furrer H, Decosterd L, Blievernicht J, Saussele T, Gunthard HF, Schwab M, Eichelbaum M, Telenti A, Zanger UM. Clin Pharmacol Ther. 2007;81(4):557–566. doi: 10.1038/sj.clpt.6100072. [DOI] [PubMed] [Google Scholar]; b Rakhmanina NY, van den Anker JN. Expert Opin Drug Metab Toxicol. 2010;6(1):95–103. doi: 10.1517/17425250903483207. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Telenti A, Zanger UM. Annu Rev Pharmacol Toxicol. 2008;48:227–256. doi: 10.1146/annurev.pharmtox.48.113006.094753. [DOI] [PubMed] [Google Scholar]
- 10.Ren J, Milton J, Weaver KL, Short SA, Stuart DI, Stammers DK. Structure. 2000;8(10):1089–1094. doi: 10.1016/s0969-2126(00)00513-x. [DOI] [PubMed] [Google Scholar]
- 11.a Ogburn ET, Jones DR, Masters AR, Xu C, Guo Y, Desta Z. Drug Metab Dispos. 2010;38(7):1218–1229. doi: 10.1124/dmd.109.031393. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mutlib AE, Chen H, Nemeth GA, Markwalder JA, Seitz SP, Gan LS, Christ DD. Drug Metab Dispos. 1999;27(11):1319–1333. [PubMed] [Google Scholar]
- 12.Faucette SR, Zhang TC, Moore R, Sueyoshi T, Omiecinski CJ, LeCluyse EL, Negishi M, Wang H. J Pharmacol Exp Ther. 2007;320(1):72–80. doi: 10.1124/jpet.106.112136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cox PM, Bumpus NN. ACS medicinal chemistry letters. 2014;5(10):1156–1161. doi: 10.1021/ml500297n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corbett JW, Ko SS, Rodgers JD, Jeffrey S, Bacheler LT, Klabe RM, Diamond S, Lai CM, Rabel SR, Saye JA, Adams SP, Trainor GL, Anderson PS, Erickson-Viitanen SK. Antimicrob Agents Chemother. 1999;43(12):2893–2897. doi: 10.1128/aac.43.12.2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang H, Sridar C, Kenaan C, Amunugama H, Ballou DP, Hollenberg PF. J Pharmacol Exp Ther. 2011;338(3):803–809. doi: 10.1124/jpet.111.183111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a Ekins S, Vandenbranden M, Ring BJ, Gillespie JS, Yang TJ, Gelboin HV, Wrighton SA. J Pharmacol Exp Ther. 1998;286(3):1253–1259. [PubMed] [Google Scholar]; b Wang Q, Halpert JR. Drug Metab Dispos. 2002;30(1):86–95. doi: 10.1124/dmd.30.1.86. [DOI] [PubMed] [Google Scholar]; c Lewis DF, Lake BG, Dickins M. Xenobiotica. 2004;34(6):501–513. doi: 10.1080/00498250410001691316. [DOI] [PubMed] [Google Scholar]; d Leong MK, Chen YM, Chen TH. Journal of computational chemistry. 2009;30(12):1899–1909. doi: 10.1002/jcc.21190. [DOI] [PubMed] [Google Scholar]; e Lewis DF, Ito Y, Lake BG. Journal of enzyme inhibition and medicinal chemistry. 2010;25(5):679–684. doi: 10.3109/14756360903514149. [DOI] [PubMed] [Google Scholar]
- 17.Niu RJ, Zheng QC, Zhang JL, Zhang HX. J Mol Model. 2011;17(11):2839–2846. doi: 10.1007/s00894-011-0970-2. [DOI] [PubMed] [Google Scholar]
- 18.Ravikumar K, Sridhar B. Molecular Crystals and Liquid Crystals. 2009;515:190–198. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.