

In this study, Somasundaram et al. provide in vitro and in vivo evidence for a leukemic state that can be transferred between lineages even though the alteration in cell identity involves dramatic changes in gene expression and epigenetic state. They show that primary pro-B leukemia cells from mice carrying heterozygous mutations in either or both the Pax5 and Ebf1 genes, commonly mutated in human leukemia, can be converted into T lineage leukemia cells.
Keywords: B-ALL, lineage conversion, transcription factors
Abstract
Even though leukemia is considered to be confined to one specific hematopoietic cell type, cases of acute leukemia of ambiguous lineage and patients relapsing in phenotypically altered disease suggest that a malignant state may be transferred between lineages. Because B-cell leukemia is associated with mutations in transcription factors of importance for stable preservation of lineage identity, we here investigated the potential lineage plasticity of leukemic cells. We report that primary pro-B leukemia cells from mice carrying heterozygous mutations in either or both the Pax5 and Ebf1 genes, commonly mutated in human leukemia, can be converted into T lineage leukemia cells. Even though the conversion process involved global changes in gene expression and lineage-restricted epigenetic reconfiguration, the malignant phenotype of the cells was preserved, enabling them to expand as T lineage leukemia cells in vivo. Furthermore, while the transformed pro-B cells displayed plasticity toward myeloid lineages, the converted cells failed to cause myeloid leukemia after transplantation. These data provide evidence that a malignant phenotype can be transferred between hematopoietic lineages. This has important implications for modern cancer medicine because lineage targeted treatment of leukemia patients can be predicted to provoke the emergence of phenotypically altered subclones, causing clinical relapse.
Leukemia commonly involves a single blood cell lineage with expansion of lymphoid progenitor cells (B- or T-cell lymphoblastic leukemia) or myeloid progenitors (myeloid leukemia) (Jennings and Foon 1997; Craig and Foon 2008). The involvement of a single lineage of cells is well in line with the idea that leukemia is a monoclonal disease initiated in a lineage-restricted progenitor. However, even though this would appear to be generally applicable, a fraction of the hematopoietic malignancies appears to involve multiple lineages (Jennings and Foon 1997; Craig and Foon 2008; Manola 2013). These malignancies, denoted as acute leukemia of ambiguous lineage (ALAL), can be manifested as either bilineal leukemia, involving several lineages, or biphenotypic disease, with expansion of cells displaying combined expression of normally lineage-restricted surface markers (Jennings and Foon 1997; Craig and Foon 2008; Manola 2013). Additionally, rather dramatic phenotypic changes with preserved cytogenetic features have been reported from patients experiencing relapse of disease (Dorantes-Acosta and Pelayo 2012). This is especially notable after treatment of pediatric B lineage acute lymphoblastic leukemia (B-ALL) using genetically manipulated T lymphocytes (chimeric antigen receptor T [CAR-T] cells) targeting CD19-positive cells because 13 out of 20 children experiencing relapse did so with CD19− leukemia cells (Gardner et al. 2016; Park et al. 2016). Even though the phenotypic changes could be explained by malignant transformation of multipotent progenitors, this could also be a consequence of genetic alterations causing lineage instability in committed progenitors.
Stable B lymphocyte identity depends on a network of transcription factors (Somasundaram et al. 2015), including PAX5, EBF1, and IKZF1 (IKAROS), all of which are crucial for stable B lineage commitment (Mikkola et al. 2002; Cobaleda et al. 2007; Reynaud et al. 2008; Nechanitzky et al. 2013; Ungerback et al. 2015). Interestingly, mutations of transcription factors involved in the regulation of cell identity are prominent in B-ALL, where heterozygote inactivation of PAX5, EBF1, or IKZF1 is detected in a substantial fraction of the patients (Kuiper et al. 2007; Mullighan et al. 2007, 2008).
In order to explore whether disruption of a transcription factor network in leukemia cells causes lineage plasticity to generate biphenotypic or bilineal leukemia, we decided to explore the ability of mouse B-ALL cells carrying disruptions in fate-determining regulatory networks (Prasad et al. 2015) to adopt alternative cell fates. This revealed that either constitutive activation of Notch1 signaling or externally mediated Notch activation resulted in lineage switch of B-ALL cells to T lineage cells displaying a pronounced developmental block and expansion capacity in vitro and in vivo. The B lineage leukemia cells could be converted into myeloid cells; however, these displayed a mature phenotype with pronounced ability to respond to LPS and severely reduced ability to expand and generate malignant disease in vivo. This reveals that secondary genetic alterations as well as environmental signals may impact the phenotype of B-ALL cells and that the malignant state, including the block of differentiation, can be transferred between lymphoid lineages.
Results
Constitutive activation of the Notch signaling pathway in pro-B leukemia cells causes lineage conversion and expansion of CD19-negative leukemia cells in vivo
In order to explore whether a leukemic pro-B cell can be converted into a T-lineage cell, we decided to explore this ability in a mouse model carrying a combined heterozygous loss of the Pax5 and Ebf1 genes (Ungerback et al. 2015). The majority of these mice develops monoclonal or oligoclonal B-lineage leukemias before the age of 30 wk (Supplemental Fig. S1; Prasad et al. 2015) that, after transplantation, display a stable phenotype with expansion of leukemic pro-B cells in the bone marrow (BM) and spleens of the recipients (Prasad et al. 2015). In order to explore whether constitutive activation of Notch signaling, a feature commonly detected in human T-ALL (Weng et al. 2004; Lobry et al. 2011), would change the phenotype of the leukemia cells, we transduced four independently derived primary pro-B leukemia samples from lymph nodes (LNs) collected from leukemic Pax5+/− Ebf1+/− (TH) mice (#377, #436, #619, and #655) with a retrovirus encoding a constitutively active Notch1 protein (ICN1) (Pui et al. 1999). Cultivation of the transduced tumor cells on OP9 stroma cells induced Thy1.2 expression on three of the tumors transduced with the GFP control vector (Fig. 1A,B). In contrast, the ICN1-expressing cells down-regulated the expression of CD19 (Fig. 1A,B), yielding between 50% and 95% Thy1.2+CD19− cells. Exploring CD3 expression on the surface of Thy1.2+ cells revealed a low frequency of CD3+ cells. Quantitative PCR (qPCR) analysis of CD19−Thy1.2+ cells revealed down-regulation of the B-cell-restricted Igll1 gene, while the levels of the T-lineage-associated Cd3e and Lck genes were increased as compared with what we observed in the CD19+ cells (Fig. 1C). RNA sequencing (RNA-seq) experiments using two tumors (#377 and #619) and in vitro differentiated B and T cells explored with unsupervised cluster analysis revealed that, while the control virus transduced cells clustered with the normal CD19+ cells, the cells transduced with the ICN1-expressing virus clustered with the T-lineage cells (Fig. 1D). Identifying differentially expressed genes (Supplemental Table S1–S4) linked ICN1 expression to down-regulation of B-lineage-restricted genes, including Pax5, Cd79α, Cd79β, Igll1, and Cd19, and up-regulation of genes associated with T-lineage development such as Gata3, Tcf7, and Cd3e. Performing a gene ontology (GO) term analysis of the genes with increased expression in the ICN1 transduced cells revealed a significant enrichment of genes linked to T-cell differentiation and function (Fig. 1E; Supplemental Tables S3, S7), while the GO terms identified for the down-regulated genes included B-cell activation (GO: 0042113) and the B-cell receptor signaling pathway (GO: 005853) (Supplemental Table S7–S8). qPCR analysis of genomic DNA from ICN1 transduced tumor cells allowed for detection of TCRβ DJ rearrangement at a level comparable with that of in vitro differentiated normal T-lineage cells (Fig. 1F), while TCRβVDJ recombination was not observed in vitro. To verify that the generated T-lineage cells were derived from the transformed pro-B-cell tumor, we explored VDJ recombination events in the immunoglobulin heavy chain (IgH) locus in CD19− Thy1.2+ and CD19+ cells generated from tumor #377 (Fig. 1G). This revealed that the CD19− Thy1.2+ cells carried a VDJ rearranged IgH similar to what we observed in the CD19+ cells, supporting that the cells represent the same leukemic clone.
Figure 1.
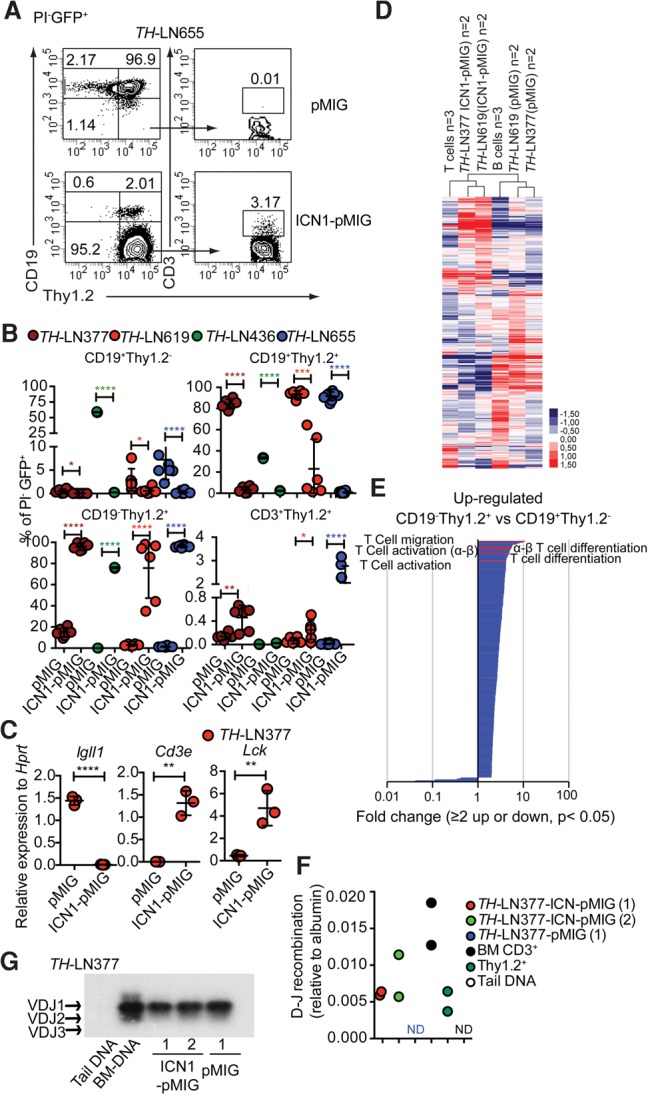
Constitutively active Notch signals in Pax5+/−Ebf1+/− pro-B leukemia cells cause lineage conversion from the pro-B-cell stage to the early T-cell stage in vitro. (A) Representative FACS plots from in vitro differentiation of TH-LN655 with or without a constitutively active Notch1 signal. (B) Graph displaying the percentage of PI−GFP+ events for the indicated populations generated from four TH pro-B tumors (#377, #436, #619, and #655) after 14 d of coculture on OP9 stroma with or without a constitutive Notch1 signal. Statistical analysis was based on unpaired Student's t-test. Asterisks in the same color as the tumor depicted in the figure indicate significance. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (****) P < 0.0001. (C) qPCR analysis after 14 d of OP9 coculturing of cells transduced with either ICN1-pMIG or pMIG. Each dot represents one well from cell cultures analyzed in triplicate qPCR reactions. Statistical analysis was performed using unpaired Student's t-test. (**) P < 0.01; (****) P < 0.0001. (D) Heat map displaying an unsupervised hierarchical clustering analysis of RNA-seq data from primary in vitro differentiated T cells (T cells) or B cells (B cells) as well as CD19+Thy1.2+ cells (pMIG transduced cells) and CD19−Thy1.2+ (ICN1-pMIG transduced) tumor cells 14 d after transduction. Data were normalized to 10 million mapped reads in log scale and were visualized by Java TreeView following hierarchical clustering in cluster 3 (log transform, centering genes on mean, average linkage cluster). (E) GO enrichment analysis of genes up-regulated twofold or more in ICN1 transduced cells with a statistic significance (P < 0.05) in D. Significantly (P < 0.05) enriched (twofold or more) or impoverished (≤0.5-fold) annotations from a biological process are shown. The X-axis indicates the value of fold enrichment. Some terms associated with T-cell development are highlighted. (F) qPCR data detecting the presence of TCRβ DJ recombination events in ICN1 or control transduced tumor cells. Primary sorted CD3+ BM cells (BM CD3+), Thy1.2+ in vitro differentiated T-lineage cells (Thy1.2+), and mouse tail DNA were included as positive and negative controls, respectively. The data were normalized to the amplification of a genomic fragment from the albumin gene and present two experiments with duplicate qPCR reactions. (G) IgH VDJ recombination was analyzed by PCR using genomic DNA from CD19+Thy1.2+ (pMIG) and CD19−Thy1.2+ (ICN1-pMIG) cells from TH-LN377. Generated PCR products were visualized by Southern blot. Wild-type mouse tail DNA and BM DNA were used as negative and positive controls, respectively.
Even though it is possible to identify childhood B-ALL leukemia that, similar to our mouse model, contains combined heterozygous deletion of PAX5 and EBF1 (Mullighan et al. 2007; Prasad et al. 2015), it is rare in comparison with single-heterozygous mutations in either the PAX5 or EBF1 gene (Mullighan et al. 2007; Prasad et al. 2015). While we rarely found leukemia development in single-heterozygote mice, a few cases were found among our animals, creating the possibility of exploring whether leukemia cells from single-heterozygote mice display T-lineage plasticity. To this end, we infected two pro-B-cell tumors from Pax5+/− mice and two from Ebf1+/− animals with ICN1 and explored the development of CD19−Thy1.2+ cells (Supplemental Table S9). This revealed that one of the Ebf1+/− and one of the Pax5+/− tumors gave a robust conversion of cells into a CD19−Thy1.2+ phenotype, while few CD3+ cells were generated. Hence, activation of Notch signaling in B-cell tumors carrying disruption in critical lineage-specifying transcription factors results in a switch of the tumor phenotype.
In order to investigate whether ICN1-expressing tumor cells could expand to generate a leukemic disease in vivo, we transplanted ICN1 transduced LN cells from three individual leukemic mice (#278, #327, and #377) and pMIG-GFP transduced LN cells from two individual mice (#327 and #377) into wild-type C57BL6 mice. After 3–4 wk, several of the transplanted mice displayed disease symptoms, including a partial paralysis also observed after transplantation of primary tumors (Prasad et al. 2015). Analysis of the spleens from the mice transplanted with cells transduced with a GFP control virus revealed a peripheral expansion of CD19+ cells (Fig. 2A,B), well in line with the previously observed stable phenotype of these tumors (Prasad et al. 2015). Analyzing the expansion of the ICN1 transduced tumor cells in the spleen revealed that, even though all of the animals displayed some expansion of CD19+ cells (Fig. 2A,B), all three tumors generated Thy1.2+CD19− cells, of which only a fraction were CD3+, suggesting that the cells are developmentally blocked. Analyzing the cellular composition of the thymus supported the notion that developmentally blocked CD19−Thy1.2+ cells expand after transplantation. However, in contrast to what we observed in the spleen, the thymus contained a fraction of CD19−Thy1.2+ cells generated from the GFP control transduced B-lineage tumor cells. qPCR analysis of sorted pMIG-GFP transduced CD19+ cells as well as ICN1-pMIG transduced CD19−Thy1.2+ cells from the thymi of the transplanted mice revealed that, while the CD19+ populations expressed Igll1, only the CD19−Thy1.2+ cells expressed the T-lineage-associated Cd3e and Gata3 and high levels of Lck (Fig. 2C). qPCR analysis using genomic DNA detected TCRβ DJ as well as low levels of TCRβ VDJ recombination in the ICN1 transduced cells (Fig. 2D). The B-cell origin of the cells was verified in one of the tumors by IgH VDJ recombination analysis, revealing a dominant expansion of a leukemic cell carrying a VDJ1 recombination event in the CD19+ as well as the CD19−Thy1.2+ populations (Fig. 2E). Hence, the lineage identity of tumor cells can be influenced by the local microenvironment to generate two phenotypically distinct but clonal tumors in the host.
Figure 2.
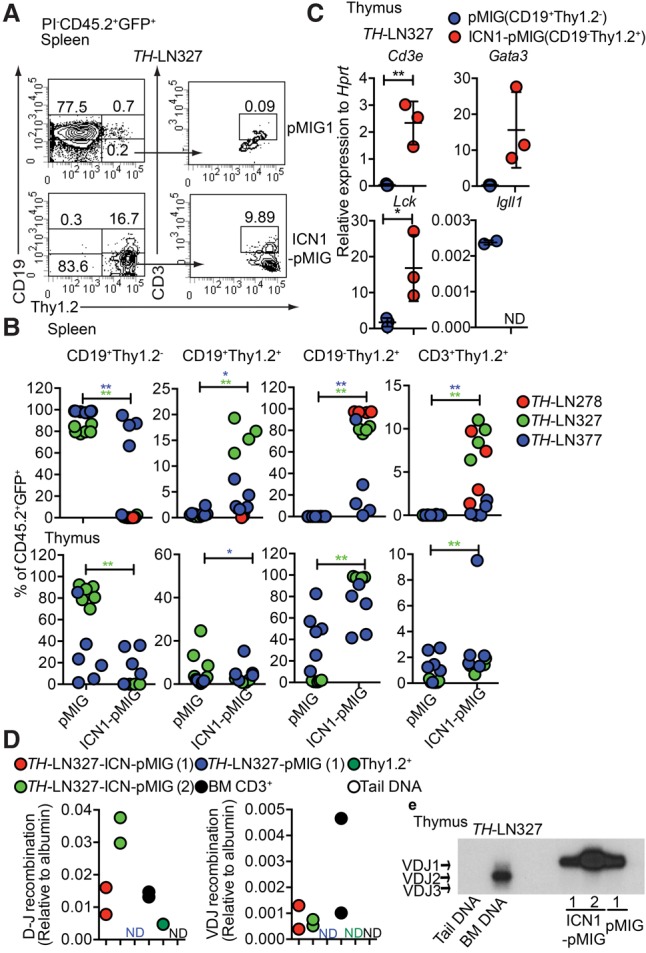
Constitutive activation of the Notch signaling pathway in Pax5+/−Ebf1+/− pro-B leukemia cells causes lineage conversion and expansion of CD19−Thy1.2+ leukemia in vivo. (A) Representative FACS plot of the spleen from a CD45.1 mouse transplanted with ICN1-pMIG or pMIG transduced TH-LN327 (CD45.2) cells. The numbers represent the percentage of total PI−CD45.2+GFP+ events. (B) Percentage of the indicated cell populations (% of PI−CD45.2+GFP+ events) in the spleens and thymi of transplanted mice. Each dot indicates one transplanted mouse, and the color code indicates one independently generated tumor. Statistical analysis was performed using Mann-Whitney U-test. (*) P < 0.05; (**) P < 0.01. Asterisks in the same color as the tumor depicted in the figure indicate significance. (C) qPCR analysis of CD19+Thy1.2+ (pMIG) and CD19−Thy1.2+ (ICN1-pMIG) cells sorted from the thymus of a mouse transplanted with TH-LN327 cells. Statistical analysis was performed using unpaired Student's t-test. (*) P < 0.05; (**) P < 0.01. (D) qPCR data displaying TCRβ DJ and VDJ recombination events in ICN1 or control tumor cells. Primary sorted CD3+ BM cells (BM CD3+), Thy1.2+ in vitro differentiated T-lineage cells (Thy1.2+), and mouse tail DNA were included as positive and negative controls, respectively. The data were normalized to the amplification of a genomic fragment from the albumin gene and present two experiments with duplicate qPCR reactions. (E) Southern blot of PCR products from VDJ recombination events in the IgH locus using genomic DNA from CD19+Thy1.2+ (pMIG) and CD19−Thy1.2+ (ICN1-pMIG) cells sorted from the thymus of a mouse transplanted with TH-LN377. Wild-type mouse tail DNA and BM DNA were used as negative and positive controls, respectively.
External Notch signaling is sufficient to cause lineage switch of leukemic pro-B cells
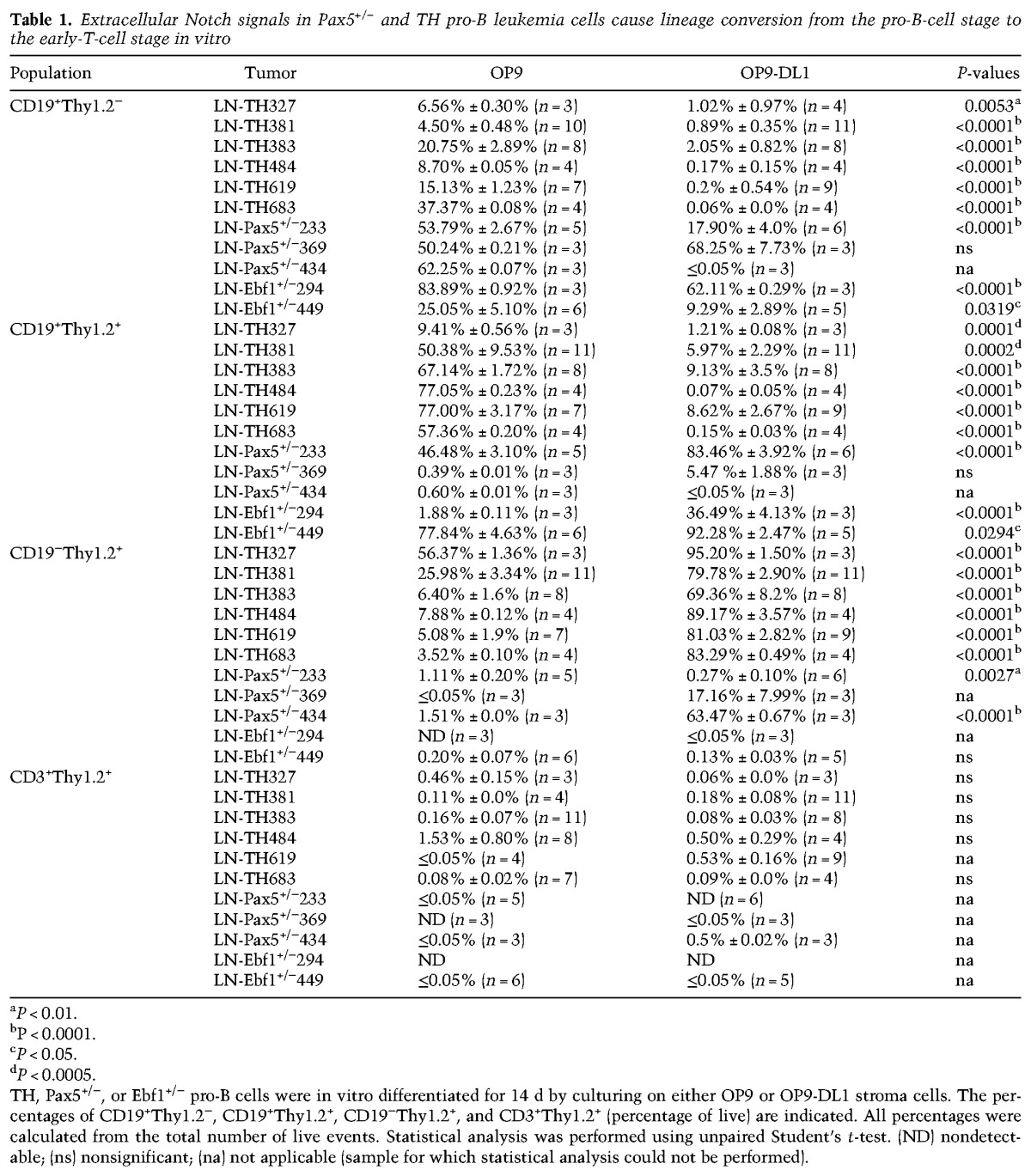
To explore whether a Notch signal is sufficient to convert the lineage identity of the tumor cells, we performed in vitro cultures on either OP9 or OP9-DL1 cells, the latter expressing the Notch ligand Delta1 (Schmitt and Zuniga-Pflucker 2002). While CD19 expression was retained when the cells were cultured on OP9, OP9-DL1 influenced the generation of CD19−Thy1.2+ cells, with few cells displaying surface expression of CD3 (Fig. 3A; Table 1). Cultivation of three Pax5+/− and two Ebf1+/− tumors under OP9-DL1 conditions for 14 d resulted in the development of Thy1.2+CD19− cells in two of the Pax5+/− tumors (Table 1), verifying the plasticity in single Pax5 heterozygote tumor cells. In order to explore the importance of the reduced PAX5 dose in the conversion process, TH tumor cells were transduced with either a GFP control virus or a virus encoding PAX5. The cells transduced with control- and PAX5-encoding virus expanded as CD19+ cells under B-cell conditions (Supplemental Fig. S3). However, while incubation of the transduced cells on OP9-DL1 cells allowed the control transduced cells to expand as CD19−Thy1.2+ cells, the tumor cells with ectopic expression of PAX5 were unable to grow and generate CD19− cells. Hence, a high level of PAX5 expression is incompatible with T-lineage switch in the tumor cells.
Figure 3.
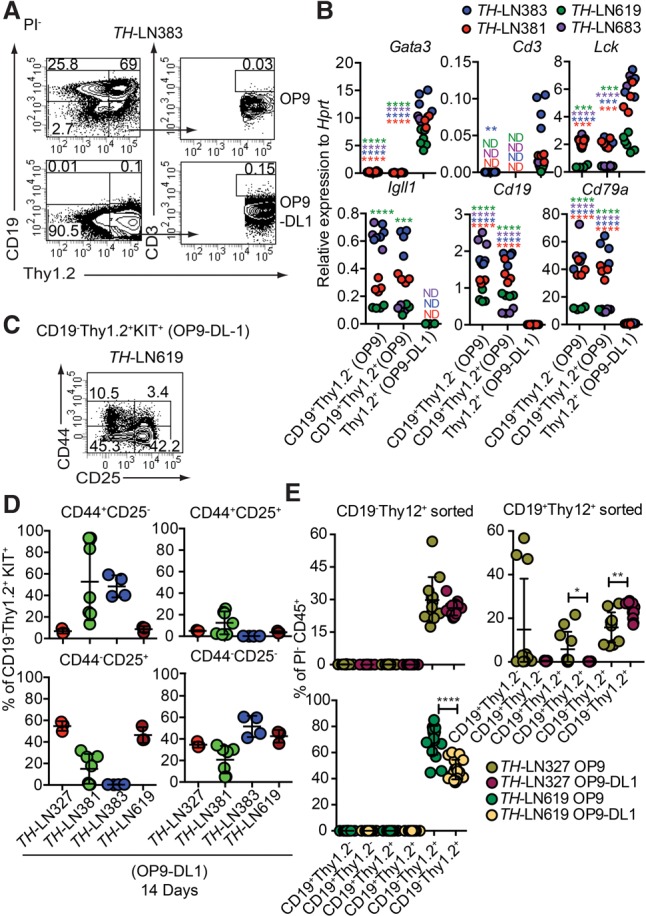
Extracellular Notch signals are sufficient to cause lineage switch of leukemic pro-B cells to T-lineage cells. (A) Representative FACS plots from TH-LN cultured with OP9 or OP9-DL1 for 14 d. The percentages were calculated from total PI− events. (B) qPCR analysis from TH tumor cells cocultured with OP9 (CD19+ cells) or OP9-DL1 (CD19−Thy1.2+ cells) for 14 d. Each dot indicates a biological replicate analyzed by triplicate qPCR reactions, and the color of the dot indicates one independently generated tumor. Statistical analysis was performed using unpaired Student's t-test. Asterisks in the same color as the tumor depicted in the figure indicates significance. (**) P < 0.01; (***) P < 0.001; (****) P < 0.0001. (C) Representative FACS plot of TH-LN619 cells after 14 d of in vitro differentiation on OP9-DL1 displaying the percentage of cells (% of live events) positive, negative, or double positive for CD44/CD25. (D) Diagrams displaying expression of CD44 and CD25 analyzed by flow cytometry in cells from four TH tumors. (Red) Tumor #327; (green) tumor #381; (blue) tumor #383; (dark red) tumor #619. The percentage of the indicated cell type was calculated from the total number of CD19−Thy1.2+KIT+ events. (E) Diagrams displaying the percentage of the indicated populations out of PI− events after reseeding of either CD19−Thy1.2+ or CD19+Thy1.2+ cells on OP9 or OP9-DL1 cells followed by coculture for 10 d. Each dot indicates one culture, and statistical analysis was performed using Mann-Whitney U-test. (*) P < 0.05; (**) P < 0.01; (****) P < 0.0001.
Table 1.
Extracellular Notch signals in Pax5+/− and TH pro-B leukemia cells cause lineage conversion from the pro-B-cell stage to the early-T-cell stage in vitro
qPCR analysis of populations from OP9- and OP9-DL1-cultured cells from both transheterozygous and single-heterozygous tumor cells revealed increased expression of T-cell genes and reduced expression of B-lineage genes in association with loss of CD19 expression (Fig. 3B; Supplemental Fig. S2). The formation of CD3+ cells was limited; however, the major fraction of the cells coexpressed KIT (Supplemental Fig. S4) and CD25 (Fig. 3C,D), while fewer cells expressed CD44. Hence, even though high KIT expression normally is limited to the DN1 and DN2 stages, the low expression of CD44 indicates that the major part of the cells resides in the DN2–DN4 stage (Rothenberg 2014; Yui and Rothenberg 2014). Even though it would appear as if the converted cells cannot be assigned to any one specific DN stage, the lack of CD3 expression and high expression of KIT support the idea that the cells display an early block of differentiation. Hence, environmental signals may impact the lineage identity of a leukemic cell.
The efficiency of the conversion process was explored by limiting dilution experiments, revealing that the cloning frequency of the B-cell tumors on OP9-DL1 cells was about one-fifth of that on OP9 cells, with the absolute majority of the clones generated on the OP9-DL1 cells containing CD19−Thy1.2+ cells (Supplemental Fig. S5). To explore the stability of the process, we incubated tumor cells on OP9-DL1 cells for 10 d, after which CD19+Thy1.2+ and CD19−Thy1.2+ were reseeded on either OP9 or OP9-DL1 cells (Fig. 3E). After an additional 10 d of culture, the cellular content was analyzed by FACS, revealing that, while the CD19+Thy1.2+ cells were able to generate CD19+ cells, the CD19−Thy1.2+ population generated only CD19− cells even under B-cell conditions. Hence, the conversion process is robust and induces a stable change of lineage identity.
Ectopic expression of C/EBP proteins results in conversion of leukemic pro-B cells into monocytic cells lacking in vivo expansion potential
It has been reported that the expression of C/EBP proteins causes a phenotypic conversion of B-lineage tumor cells to generate myeloid cells (Xie et al. 2004; Bussmann et al. 2009) with reduced malignant potential (Rapino et al. 2013; McClellan et al. 2015). To explore the ability of Pax5+/−Ebf1+/− to generate myeloid cells, we transduced normal wild-type cells as well as single Pax5+/−, Ebf1+/−, or Pax5+/−Ebf1+/− pro-B tumor cells with a retrovirus encoding the transcription factor C/EBPβ (Fig. 4A,B; Supplemental Fig. S6). The majority of the C/EBPβ transduced cells developed into a CD19− state in a matter of 5 d, and, even though we noted a transient formation of CD19+CD11b+ biphenotypic progenitors, these cells were virtually lost after 5 d of incubation, generating CD19−CD11b+ cells (Fig. 4A,B). May-Giemsa staining of the converted cells revealed that the absolute majority displayed a nuclear morphology of monocytes (M) rather than that of granulocytes (Fig. 4A). IgH VDJ analysis supported their B-lineage origin (Fig. 4C), and qPCR analysis revealed a down-regulation of Igll1 and up-regulation of the myeloid gene Myleo-peroxidase (Mpo) in the converted cells (Fig. 4D). The C/EBPβ transduced cells were able to mount a response to LPS stimulation, reflected in induced expression of Il6, Il1β, Il12p40, and TNFα mRNA (Fig. 4E; Supplemental Fig. S7), revealing functional features of monocytes.
Figure 4.
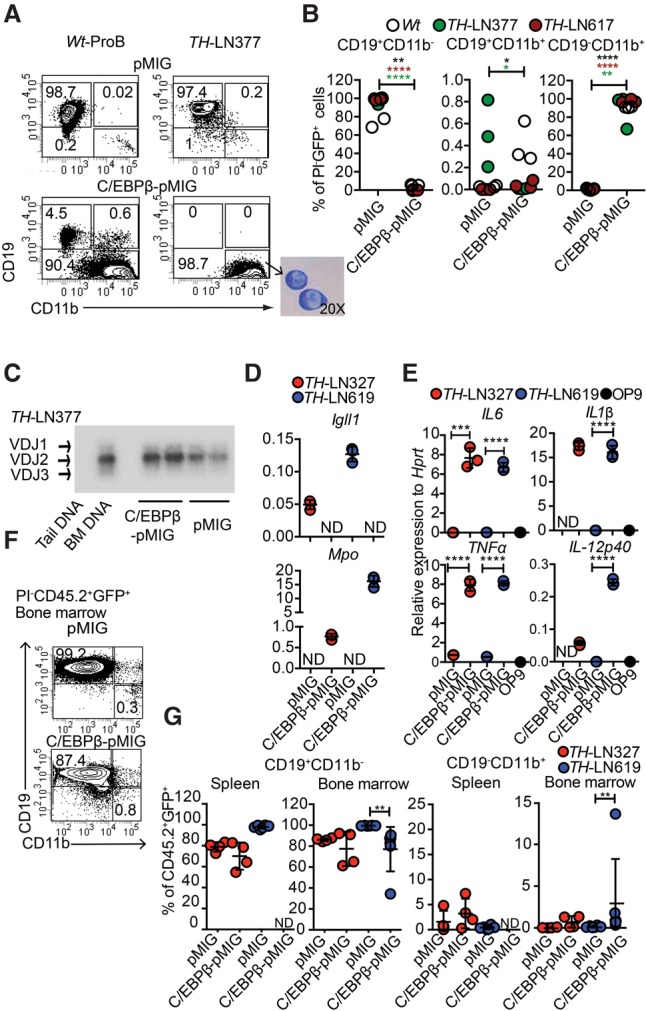
C/EBPβ expression in leukemic pro-B cells results in lineage conversion into LPS-responsive monocytic cells but not a myeloid leukemia. (A) Representative FACS plots from in vitro differentiated TH-LN and wild-type pro-B cells transduced with either C/EBPβ-pMIG or pMIG on OP9 coculture. The picture at the bottom of the panel displays a May-Grunwald-Giemsa staining of leukemic pro-B cells converted into CD19−CD11b+ cells. (B) Diagrams displaying the percentage of the indicated population out of total GFP+ cells 5 d after transduction with C/EBPβ-pMIG or pMIG. Each dot represents one well, and the cells used are indicated by individual color. Statistical analysis was performed using unpaired Student's t-test. The asterisk in the same color as the cell depicted in the figure indicates significance. (*) P < 0.05; (**) P < 0.01; (****) P < 0.0001. (C) IgH VDJ recombination in genomic DNA from CD19+CD11b− (pMIG) and CD19−CD11b+ (C/EBPβ-pMIG) of TH-LN377 analyzed by PCR followed by Southern blot. Wild-type mouse tail DNA and BM DNA were used as negative and positive controls, respectively. (D) C/EBPβ-pMIG and pMIG transduced TH-LN cells were analyzed by qPCR after 5 d of in vitro differentiation. (ND) Nondetectable. (E) qPCR analysis detecting the expression of the indicated genes in in vitro differentiated cells (as in D) after 18 h of treatment with 100 ng/mL LPS. Each dot represents a biological replicate analyzed by triplicate qPCR reactions, and the colors indicate independently derived tumors. (ND) Nondetectable. Statistical analysis was performed using unpaired Student's t-test. (***) P < 0.001; (****) P < 0.0001. (F) Representative FACS plot indicating the percentage of GFP+CD45.2+ cells of CD19+CD11b− and CD19−CD11b+ in the BM of CD45.1 recipient mice transplanted with pMIG or C/EBPβ-pMIG transduced TH-LN (CD45.2 donor) cells. (G) Percentages of CD19+CD11b− and CD19−CD11b+ cells (% of GFP+CD45.2+) from the spleens and BM of transplanted mice were plotted. Each dot indicates one mouse, and the color indicates tumor number as depicted in the figure. Statistical analysis was performed using Mann-Whitney U-test. (**) P < 0.01; (ND) nondetectable.
In order to explore whether the CD19−CD11b+ population could cause a malignant expansion generating a myeloid leukemia in vivo, we transduced leukemic pro-B cells with either control- or C/EBPβ-encoding retrovirus, cultivated the cells in vitro for 5 d, and transplanted sorted CD19+CD11b− cells from the control and CD19−CD11b+ cells from the C/EBPβ-expressing cells. Three weeks to 4 wk after transplantation, the mice receiving the control transduced cells developed signs of disease with expansion of GFP+CD19+CD11b− leukemic pro-B cells in the BM and spleens (Fig. 4F,G). GFP+ cells were rare or absent in the spleens of mice transplanted with the converted CD19−CD11b+ cells. Furthermore, the major part of the GFP+ cells detected was CD19+CD11b− (Fig. 4F,G), arguing against the converted cells being able to generate a myeloid-like leukemia in vivo.
This finding is in line with existing literature (Rapino et al. 2013; McClellan et al. 2015); however, because C/EBP proteins may act as tumor suppressors (Pabst et al. 2001; Nerlov 2004), continued ectopic expression of C/EBP proteins from the retroviral insertions might counteract the formation of myeloid leukemia. In order to explore this possibility, we took advantage of a C/EBPα retrovirus carrying the protein fused to a tamoxifen (4-OHT)-responsive estrogen receptor (ER) protein, allowing us to modulate the nuclear localization of the protein via the addition of 4-OHT. The conversion process was totally dependent on the addition of 4-OHT to the cultures (Fig. 5A,B, left panel), and the retraction of the drug resulted in reduced in vitro expansion of the converted CD19−CD11b+ cells (Fig. 5B, right panel). Exploring changes in RNA expression patterns associated with the generation of CD19− cells by unsupervised cluster analysis of RNA-seq data from 4-OHT-treated control transduced (pMIG) cells or cells transduced with the C/EBPα-ER-expressing virus revealed that the latter clustered together with in vitro differentiated normal CD11b+ cells rather than with the normal CD19+ cells (Fig. 5C; Supplemental Tables S1, S2). GO analysis identified genes associated with myeloid cell function among the genes with increased expression in the C/EBPα-ER-expressing cells (Fig. 5D; Supplemental Table S5, while genes associated with B-cell differentiation (GO: 0030183) and B-cell activation (GO: 0042113) were found among the down-regulated genes (Supplemental Table S6).
Figure 5.
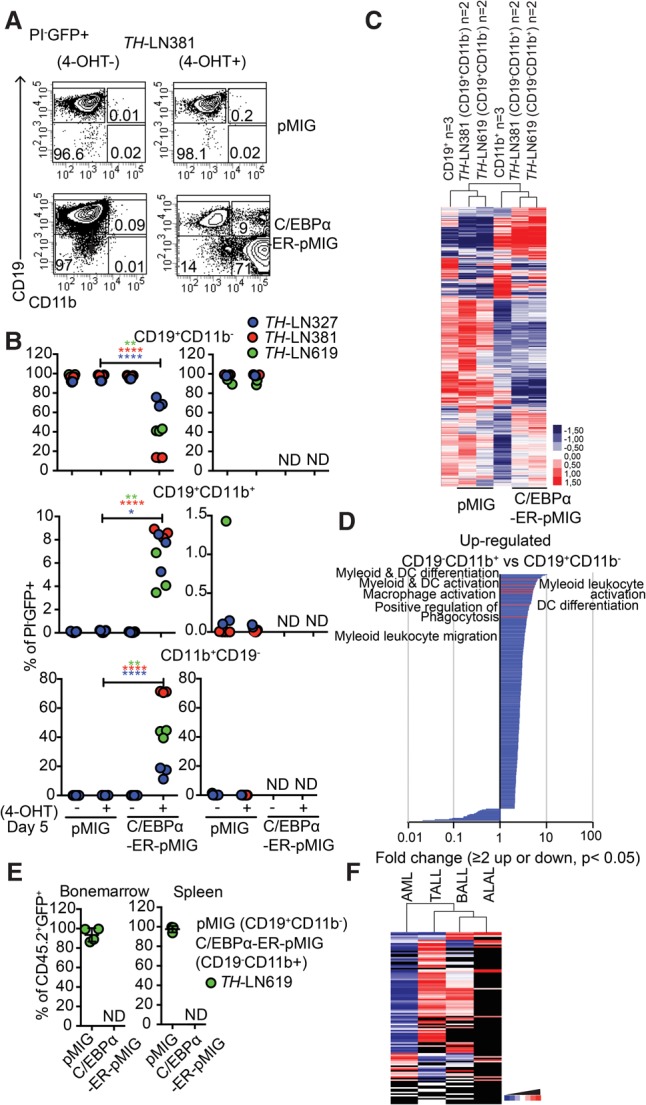
Reduced malignant myeloid potential in converted pro-B leukemia cells is not explained by sustained nuclear activity of C/EBPα proteins. (A) Representative FACS plots from in vitro differentiation of OP9 cultured TH -LN cells transduced with C/EBPα-ER-pMIG or pMIG 5 d after transduction. (B) Diagrams displaying the percentages of the indicated cell types out of PI−GFP+ cells after in vitro differentiation (as in A) of three different tumors in the presence or absence of 4-OHT. Each dot represents a well from cell culture. Statistical analysis was performed using unpaired Student's t-test. (*) P < 0.05; (**) P < 0.01; (****) P < 0.0001; (ND) nondetectable. The right panel displays the cellular content of cultures when CD19+CD11b− from pMIG or CD19−CD11b+ from C/EBPα-ER-MIG transduced cells was live-sorted after in vitro differentiation for 5 d on OP9 stroma and further cultured either with or without 4-OHT for another 5 d. Diagrams displaying the percentages of the indicated cell types out of PI−GFP+ cells. Each dot represents one well, and the colors indicate different tumors. (C) Heat map displaying an unsupervised hierarchical clustering analysis of RNA-seq data from primary in vitro differentiated CD11b+ cells or B cells as well as CD19+CD11b− cells (pMIG transduced cells) and CD19−CD11b+ (CEBPα-ER-pMIG transduced) tumor cells 5 d after transduction. Data were normalized to 10 million mapped reads in log scale and were visualized by Java TreeView following hierarchical clustering in cluster 3 (log transform, centering genes on mean, average linkage cluster). (D) GO enrichment analysis of genes up-regulated or down-regulated twofold or more with a statistic significance. P < 0.05. Significantly (P < 0.05) enriched (twofold or more) or impoverished (≤0.5-fold) annotations from biological process are shown. The X-axis indicates the value of fold enrichment. Some terms associated with myeloid cell development are highlighted. (E) FACS analysis of the spleens and BM from mice transplanted with CD19+CD11b− pMIG or CD19−CD11b+ C/EBPα-ER-pMIG transduced TH-LN619 cells. Percentages of CD19+CD11b− (pMIG) and CD19−CD11b+ (C/EBPα-ER-pMIG) of PI−CD45.2−GFP+ cells were plotted. Each dot indicates an individual transplanted mouse. (ND) Nondetectable. (F) Heat map displaying the relationship in mutational landscapes as estimated using data from the COSMIC (Catalogue Of Somatic Mutations In Cancer) cancer gene census database-reported mutations in B-ALL (n = 1303), T-ALL (n = 1846), acute myeloid leukemia (AML) (n = 26022), and ALAL (n = 52). Genes with mutations less common than a frequency of 0.1% were excluded from the list. The remaining list of mutations in each tumor type was subjected to hierarchical clustering using cluster 3 (log transform, center genes on mean, average linkage clustering of genes, and arrays) and was visualized in a heat map using Java TreeView. Black indicates absent value.
Transplantation of pMIG transduced control cells and C/EBPα-ER converted CD19−CD11b+ cells revealed that while the control cells expanded as CD19+CD11b− cells, we were unable to detect GFP+ cells in mice transplanted with the C/EBPα-ER converted cells (Fig. 5E). Hence, the lack of establishment of myeloid leukemia from converted leukemic pro-B cells is unlikely to be a consequence of tumor suppressor activity from the ectopically expressed C/EBP proteins but rather is a consequence of the leukemic state not being easily transferable between a lymphoid lineage and a myeloid lineage in this model system.
An easier transition of a leukemic state between lymphoid lineages could be a result of higher similarity between the mutational landscapes in lymphoid malignancies as compared with that of acute myeloid leukemia (AML). To explore this possibility, we extracted the frequency and abundance of mutated genes in human T-ALL, B-ALL, AML, and ALAL from the COSMIC (Catalogue Of Somatic Mutations In Cancer) cancer gene census database. Analyzing these data by unsupervised cluster analysis (Fig. 5F) revealed that the AML samples constituted a separate branch of the hierarchical tree and that the lymphoid malignancies displayed a higher degree of similarity in the mutational landscape.
The conversion of leukemic pro-B cells into T or myeloid lineages involves epigenetic changes associated with the binding of lineage-specific transcription factors
To explore whether the phenotypic alterations observed in the lineage conversion processes were reflected in changes of the epigenetic landscape, we performed ATAC-seq (assay for transposase-accessible chromatin [ATAC] combined with sequencing) analysis of CD19+ pro-B tumor cells (tumors #619 and #381) converted into either T-lineage cells or macrophages as well as of in vitro differentiated nontransformed B, T, or CD11b+ myeloid cells. Comparing the epigenetic landscape in the CD19+ control cells and the converted CD19− cells suggested that the conversion process has a major impact on chromatin accessibility. The transition into a myeloid state was reflected in increased accessibility at ∼3000 sites and reduced accessibility at 6000 sites, and the conversion into T-lineage cells increased accessibility at 2500 sites and reduced accessibility at ∼16,000 sites (Fig. 6A). The major parts of the sites were located in either intergenic or intronic regions; however, T-lineage conversion involved reduced access at almost 4000 promoters. Annotation of the identified peaks to genes allowed us to match changes in accessibility to changes in gene expression patterns, revealing that a substantial part of the epigenetic changes could be linked to significant changes in mRNA levels. Over 50% of the genes up-regulated upon conversion into myeloid cells and 80% of those displaying significantly increased as well as decreased expression upon conversion into T-lineage could be annotated to changes in accessibility (Fig. 6B). Even though we could see a higher fraction of genes associated with regions of increased accessibility among the up-regulated genes and vice versa, this correlation was not absolute.
Figure 6.
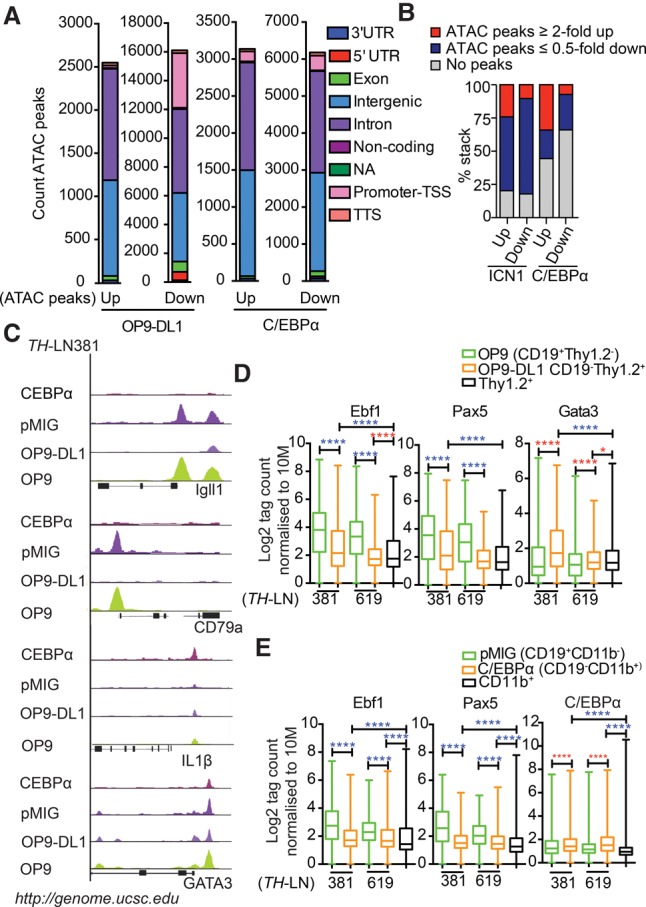
The conversion of leukemic pro-B cells into T or myeloid lineages involves epigenetic changes associated with the binding of lineage-specific transcription factors. (A) Diagrams displaying the genomic distribution of differential ATAC-seq peaks in in vitro differentiated TH-LN cells. Sorted GFP+CD19+CD11b− and GFP+CD19−CD11b+ from pMIG or C/EBPα-ER-pMIG transduced cells treated with 4-OHT and CD19+Thy1.2+ and CD19−Thy1.2+ populations after coculturing on OP9 or OP9-DL1 stromal cells. (B) Diagrams displaying the link between changes in mRNA levels (as estimated by RNA-seq analysis) and changes in chromatin accessibility (as investigated by ATAC-seq analysis). (C) Peak tracks generated from BedGraph files of ATAC tag coverage in merged duplicate samples created using HOMER and visualized on Igll1, CD79α, IL1β, and Gata3 genes using the University of California at Santa Cruz (UCSC) genome browser (Kent et al. 2002). (D,E) ATAC tags were annotated on peaks from ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) data retrieved from Gene Expression Omnibus (GEO) mapped to mm10 and reanalyzed for peak finding using HOMER (EBF1 [GSE69227], PAX5 [GSE38046], GATA3 [GSE31235], and C/EBPα [GSM537983]). Box plots of log2 tags (means from duplicates) on these peaks were quantified. Statistics was tested using one-way ANOVA with Sidak's multiple comparison test. Significant up-regulation and down-regulation are shown with red and blue asterisks, respectively; (*) P < 0.05; (****) P < 0.0001.
Among the promoters with reduced accessibility upon either T-lineage or myeloid conversion were several known to regulate the expression of B-cell genes, including Igll1 and Cd79a (Fig. 6C). Exploring the ATAC-seq peaks at these promoters revealed a drastic reduction in accessibility upon lineage conversion (Fig. 6C). Because EBF1 and PAX5 are involved in the regulation of a large set of genes defining the early B-lineage-restricted genetic program (Zandi et al. 2008; Treiber et al. 2010; Gyory et al. 2012; Vilagos et al. 2012), we explored the overall changes in chromatin accessibility at EBF1-binding sites (GSE69227) or PAX5-binding sites (GSE38046) as determined by CHIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) analysis of wild-type mouse pro-B cells (Fig. 6D,E; Supplemental Table S10–S13), revealing a significant decrease in accessibility after lineage conversion.
The accessibility to the Il-1β promoter (Fig. 6C) rendered responsive to LPS activation in the cells converted into M-lineage cells (Fig. 4E; Supplemental Fig. S7) was selectively increased in the M-converted cells, and the accessibility to C/EBPα-binding sites (GSM537983) (Heinz et al. 2010) revealed increased accessibility in the CD11b+ as compared with the original B cells (Fig. 6E). Accessibility of the Gata3 promoter was decreased in the M-converted cells but was high in the T-converted cells (Fig. 6C). However, accessibility in the original tumor cells indicated that this gene is epigenetically available in B-lineage progenitors. Matching ATAC-seq data to ChIP-seq data of GATA3 binding in FLDN2b T-cell progenitors (GSE31235) (Zhang et al. 2012b) revealed a significantly increased epigenetic accessibility in the T-cell as compared with the original B-cell tumors (Fig. 6D). Hence, the conversion of pro-B tumor cells into other lineages involves changes in the epigenetic landscape.
In order to explore the progression of the conversion process, we followed the formation of CD19− cells from Ebf1+/−Pax5+/− B-lineage tumor cells. Both the T-lineage and myeloid conversion processes generated intermediate states with combined expression of CD19 and Thy1.2 or CD11b (Fig. 7A,D). The presence of these cells was most prominent after 6 and 2 d, respectively, indicating that they represent a transient cellular state. Analyzing RNA-seq data from these intermediate stages by unsupervised hierarchical cluster analysis suggested that their gene expression pattern displayed a higher overall similarity to that of CD19+ B-cell progenitors than the fully converted CD19− cells (Fig. 7B,E). However, exploring accessibility at EBF1- and PAX5-binding sites suggested a reduced accessibility already in the biphenotypic cells (Fig. 7C,F). This was accompanied by increased accessibility at C/EBPα sites in the CD19+CD11b+ cells and GATA3-binding sites in CD19+Thy1.2+ cells. The accessibility at TCF7 (TCF1) sites was not increased before the cells reached a CD19−Thy1.2+ stage. Hence, even though we could observe minor changes in chromatin accessibility when comparing normal nontransformed wild-type B-lineage cells with their normal or transformed Ebf1+/−Pax5+/− counterparts (Supplemental Fig. S8), the lineage conversion process is reflected in additional major changes of the epigenetic landscape.
Figure 7.
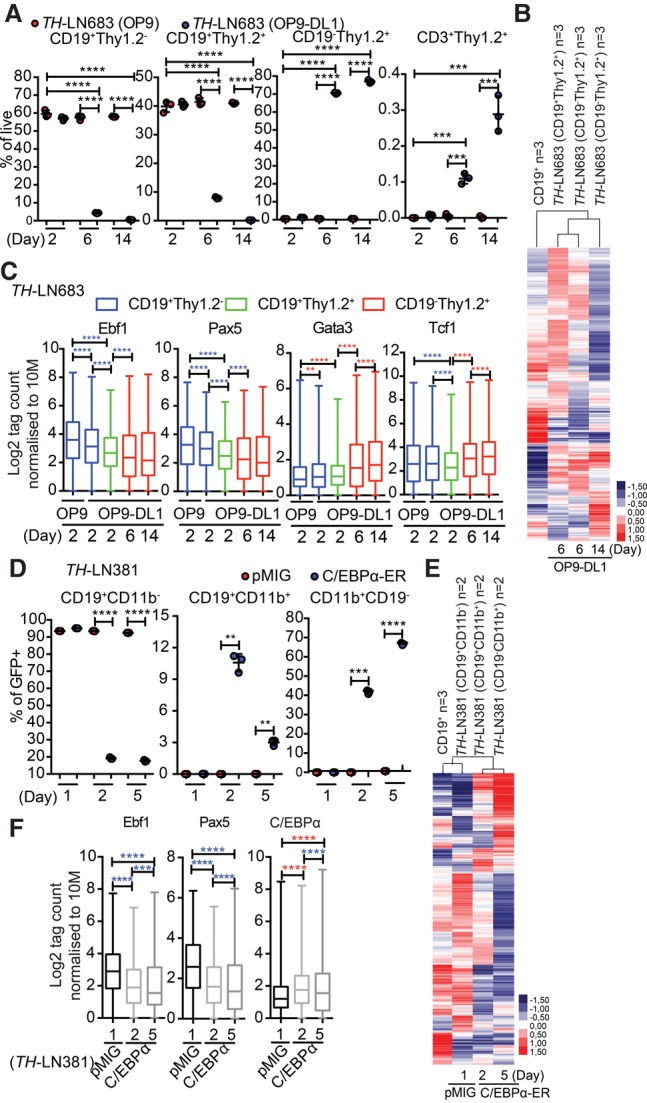
The conversion of B-ALL cells into T or myeloid lineages proceeds through a transient biphenotypic state. (A) Graph displaying the percentage (% of PI−GFP+) of the indicated cells generated from a TH pro-B tumor (#683) after 2, 6, or 14 d of coculture on OP9 or OP9-DL1 stroma. Statistical analysis was performed using unpaired Student's t-test. (***) P < 0.001; (****) P < 0.0001. (B) Heat map displaying an unsupervised hierarchical clustering analysis of RNA-seq data from primary in vitro differentiated B-cells, CD19+Thy1.2+ cells generated by cultivation of a TH pro-B tumor (#683) on OP9-DL1 cells for 6 d, and CD19−Thy1.2+ cells generated after 6 or 14 d of incubation. Data were normalized to 10 million mapped reads in log scale and were visualized by Java TreeView following hierarchical clustering in cluster 3 (log transform, centering genes on mean, average linkage cluster). (C) Box plots displaying the relative chromatin accessibility at EBF1-, PAX5-, GATA3-, or TCF7-binding sites as assayed by ATAC-seq analysis of the indicated cell types after 2, 6, and 14 d of incubation. ATAC tags were annotated on peaks from ChIP-seq data retrieved from GEO mapped to mm10 and reanalyzed for peak finding using HOMER (EBF1 [GSE69227], PAX5 [GSE38046], GATA3 [GSE31235], and TCF7 [TCF1] [GSE46662]). Box plots of log2 tags (means from duplicates) on these peaks were quantified. Statistics were tested using one-way ANOVA with Sidak's multiple comparison test. Significant up-regulation and down-regulation are shown with red and blue asterisks, respectively. (**) P < 0.01; (***) P < 0.001; (****) P < 0.0001. (D) Graph displaying the percentage (% of PI−GFP+) of CD19+CD11b−, CD19+CD11b+, and CD19−CD11b+ cells generated from a TH pro-B tumor (#683) transduced with either control GFP virus or a virus encoding CEBPα-ER 1, 2, or 5 d after the addition of 4-OHT. Statistical analysis was performed using unpaired Student's t-test. (***) P < 0.001; (****) P < 0.0001. (E) Heat map displaying an unsupervised hierarchical clustering analysis of RNA-seq data from primary in vitro differentiated B cells and CD19+CD11b−, CD19+CD11b+, and CD19−CD11b+ cells generated as in D and analyzed as in C. (F) Box plots displaying the relative chromatin accessibility at EBF1-binding, PAX5-binding, or CEBPα-binding (GSM537983) sites as assayed by ATAC-seq analysis of CD19+CD11b−, CD19+CD11b+, and CD19−CD11b+ generated as in D. ATAC tags were annotated on peaks from ChIP-seq data retrieved from GEO EBF1 (GSE69227), PAX5 (GSE38046), and C/EBPα (GSM537983) and were analyzed as in C.
Discussion
We report here that leukemic pro-B cells carrying a disruption in regulatory networks controlling lineage identity display plasticity in response to either secondary genetic events or signals in the microenvironment. The change in lineage features involves genetic reprogramming and alterations in the epigenetic landscape, arguing that the observed processes are a result of genuine lineage conversion. It has been reported that fluctuations in transcription factor dose in MYC-induced B-cell lymphoma results in phenotypic changes involving down-regulation of B-cell markers and induced expression of myeloid markers such as CD11b and F4/80 (Yu et al. 2003). Upon transplantation of the converted cells, they generated mainly B-lineage lymphoma, suggesting that the observed phenotype was a transient cellular state (Yu et al. 2003). Even though we observed a similar phenomena transplanting B-cell leukemia cells converted to M-lineage cells, we observed an apparent delay in the expansion of leukemia cells (Figs. 4G, 5E) more in line with data suggesting that conversion of the B-lineage cells to myeloid lineages reduces the malignancy of the cells (Rapino et al. 2013; McClellan et al. 2015) in these experimental settings.
In leukemia patients, it is more common to find ALAL with shared B and myeloid characteristics (Jennings and Foon 1997; Craig and Foon 2008; Manola 2013), and the dose of PAX5 has also been suggested to be involved in the formation of biphenotypic leukemia, which is characterized by a combined expression of B-lineage and myeloid lineage markers (Simmons et al. 2012). Even though this would be well in line with the role for PAX5 in silencing the myeloid program in B-cell progenitors (Nutt et al. 1999; Tagoh et al. 2006), we observed only a transient formation of biphenotypic cells from the Pax5+/−Ebf1+/− pro-B tumor cells (Figs. 4A,B, 5A,B, 7D), indicating that reduced dose as a consequence of loss of one Pax5 allele is not sufficient to generate myeloid plasticity of biphenotypic cells.
It is interesting to note that B leukemia cells converted into macrophages not only silenced the B-lineage program but also developed an ability to mount a robust response to LPS. This would indicate that the cells had gained at least some degree of functional maturation. This was in contrast to what we observed for the B-lineage leukemia cells converted into T-lineage cells. Even though these cells down-regulated the B-lineage program, the cells appeared unable to proceed in the differentiation pathway to generate CD3+ cells. Despite the fact that the surface phenotype of the converted cells was not completely compatible with any single defined normal differentiation stage and was rather heterogeneous, the lack of CD3 on the surface in combination with expression of KIT and CD25 on the majority of cells suggests that they were arrested in the DN2 stage (Rothenberg 2014; Yui and Rothenberg 2014). However, the cells did express low or undetectable levels of CD44, a marker normally expressed on DN2 cells. Even though this would be compatible with the phenotype of DN2b cells (Rothenberg 2014; Yui and Rothenberg 2014), the generated cells could not easily be assigned to any single normal stage of development. In any case, the relatively small amounts of CD3+ cells generated from the converted leukemia cells in vivo were in sharp contrast to the massive generation of CD3+ cells after transplantation of nontransformed Pax5+/−Ebf1+/− pro-B cells into Rag-1-deficient recipients (Ungerback et al. 2015). The difference in differentiation capacity between the converted T and M cells could also be reflected in their ability to generate a malignant expansion in the transplanted hosts. Even though care always should be taken in the interpretation of negative results, the inability of the M-converted cells to expand in vivo was in sharp contrast to the massive expansion of CD19−Thy1.2+ cells that we observed in the T-lineage-converted cells. The peripheral expansion of these immature T-lineage cells largely resembled what would be observed in a progenitor T-cell leukemia, suggesting that the leukemic state can be inherited from the B lineage to the T lineage. This would be in line with the finding that several of the genes and pathways, including IL-7 signaling, found modulated in human B-ALL are also targeted in T-ALL (Mullighan et al. 2009; Collins-Underwood and Mullighan 2010; Zhang et al. 2011, 2012a; Roberts et al. 2012).
While our data provide evidence that a leukemic state can be transferred between lymphoid lineages, it is difficult to estimate whether and, if so, how frequently lineage conversion processes are present in human leukemia. Monosomy 5 or deletion of 5q [−5/del(5q)], harboring the EBF1 gene (Milatovich et al. 1994), is found in both children and adults with the B+T or M+T immunophenotype (Manola 2013). Exploring the copy number status of PAX5 and EBF1 in a set of human T-ALL cell lines or primary leukemia samples (Supplemental Fig. S9) revealed that nine out of 17 cell lines and eight out of 67 primary T-ALL samples displayed loss of one copy of either or both of the genes. However, the deletions observed in the T-ALL involves large regions of the chromosome, while those detected in B-ALL often are smaller and more focused to the EBF1 or PAX5 genes (Mullighan et al. 2007). Furthermore, out of 118 human T-cell leukemias, 22% displayed recombination events in the IgH loci (Szczepanski et al. 1999). The majority of these events (61%) was monoallelic recombination events, but biallelic DJ as well as some rare VDJ recombination events could also be detected. Even though IgH recombination, especially the earliest event involving DJ recombination, can be detected also in a fraction of the normal T cells, and the genetic loss of PAX5 and EBF1 is part of larger deletions, this supports a close relationship between lymphoid leukemias as well as the possibility of transfer between states in human disease.
The finding that B-cell tumor-derived T-lineage cells can be detected in the thymi of transplanted mice (Fig. 2B), while such cells are not detected in the BM or spleens, should be considered in light of the fact that leukemia treatment is progressing from general cytotoxic treatment that targets all hematopoietic cells into more restricted drugs targeting, for instance, a lineage-specific surface marker (Jabbour et al. 2015; Maude et al. 2015). This is a most welcome development, since it drastically reduces the side effects of the treatment; however, in a scenario in which a B-ALL has generated a small fraction of leukemic cells of another lineage, these may escape treatment to cause recurrent disease with a different phenotype. This may be an underlying explanation for the observation that a large fraction of patients experiences relapse with CD19− leukemia after treatment with CAR cells targeting CD19 on the B-ALL cell surface (Park et al. 2016). This problem is just beginning to be explored, and, in the few cases described, the relapses appear to involve the expansion of myeloid cells in one of the cases with clear relation to the original B-ALL (Gardner et al. 2016). Even though conversion into myeloid malignancies is not fully in line with our observations, the finding that the AML-like disease develops rapidly (Gardner et al. 2016) indicates that this is the consequence of expansion of existing lineage-converted subclones, well in line with our idea that a tumor clone may not be fully restricted to one lineage.
In all, we report here on the plastic nature of primary B-lineage leukemia cells and show that it is possible to transfer a leukemic state between lymphoid lineages. Even though it is difficult to estimate how much this plasticity may impact human leukemia, it is becoming increasingly clear that the same regulatory pathways that regulate lineage identity are also involved in malignant transformation. Hence, we may need to reconsider our thinking about leukemia as a disease with a defined and fixed lineage.
Materials and methods
Animal models
Pax5+/− (Urbánek et al. 1994) or Ebf1+/− (Lin and Grosschedl 1995) cells were all on a C57BL/6 (CD45.2) background, whereas the control animals used in transplantation were on C57BL/6 (CD45.1). Pax5+/− and Ebf1+/− mice were crossed to generate transheterozygotes for Pax5+/−Ebf1+/−. Animal procedures were performed with consent from the local ethics committee at Linköping University (Linköping, Sweden).
Cell culture
Normal lineage-negative Sca1+Kit+ cells or LN pro-B cells (Lin− CD11b, Gr1, Ter119, and CD11c) NK1.1−IgM−CD3−CD19+CD43+ were sorted and expanded in vitro by coculture on OP9 or OP9-DL1 stroma cells (Schmitt and Zuniga-Pflucker 2002) using OptiMEM supplemented with 10% heat-inactivated fetal calf serum, 25 mM HEPES, 50 µg/mL gentamicin, 50 µM β-mercaptoethanol, 10 ng/mL KIT ligand, 10 ng/mL Fms-like tyrosine kinase 3 ligand (FLT3L), and 10 ng/mL interleukin-7. Differentiation and transduction experiments were performed as detailed in the Supplemental Material.
FACS staining
FACS analysis was performed as in Ungerback et al. (2015). For details about antibodies and methods, see the Supplemental Material.
RNA-seq and data analysis
Total RNA was isolated from control CD19+ LN cells (pMIG), converted CD11b+ cells (C/EBPα-ER-pMIG), Thy1.2+ cells (ICN1-pMIG), and cells cultured on OP9 or OP9-DL1, and libraries were constructed using NuGEN's Ovation Ultralow library systems (NuGEN Technologies) and subsequently subjected to 76 cycles of NextSeq500 sequencing (Illumina). Each biological sample was processed and sequenced in duplicate or triplicate. For analysis of RNA-seq experiments, the reads were aligned to mouse reference genome (mm10/GRCm38) and analyzed using the HOMER platform. For details about sequencing and analysis, see the Supplemental Material. Gene lists (twofold up-regulated or down-regulated in converted cells, with a statistical significance P < 0.05) from RNA-seq experiments were uploaded to Panther Overrepresentation Test (release 2016/7/15; http://geneontology.org), and enrichment analyses were run with the GO database (released on September 24, 2016). Mus musculus genes from the database (22,322 genes) were used as references.
PCR and RT–PCR
qPCR analysis of sorted cells was performed as described previously (Mansson et al. 2008) using Assays-on-Demand probes. The TCRβ VDJ and DJ assays were adopted from Hamrouni et al. (2003), and the recombination events were quantified by real-time qPCR using FastStart Universal SYBR Green Master (ROX, Roche). Ig VDJ analyses were performed as in Schlissel et al. (1991). The obtained PCR products were further analyzed by Southern blot, and the correct products were detected by hybridization to a 32P-labeled internal J3 oligonucleotide. For details, see the Supplemental Material.
Transplantation procedures
Adoptive transfers were performed by tail vein injection. Nonirradiated CD45.1 animals were transplanted with ∼250,000 LN cells transduced with retrovirus for either ICN1-pMIG or pMIG for pro-B-to-T-cell experiments. For pro-B-to-myeloid experiments, 100,000 CD19−CD11b+ from C/EBPβ-pMIG/C/EBPα-ER-pMIG cells or CD19+CD11b− from pMIG transduced cells were transplanted into nonirradiated CD45.1 hosts.
ATAC-seq
Eighty-thousand cells from the cultures of CD19+ LN cells on OP9 or OP9-DL1 and control pMIG, C/EBPα-ER-pMIG, or ICN1-pMIG transduced CD19+ LN cells were washed in ice-cold PBS prior to ATAC-seq library preparation as described in Buenrostro et al. (2013). Libraries were single-end-sequenced on a NexSeq500. Each biological sample was processed for ATAC-seq and sequenced in triplicate. The data were mapped to mm10 using tag directories with reads mapped to the mitochondrial chromosome filtered out, and University of California at Santa Cruz (UCSC) BedGraph files normalized to 10 million total mapped reads were created using the HOMER platform (Heinz et al. 2010). ATAC-seq peaks were identified using findPeaks.pl in HOMER (Heinz et al. 2010). Published ChIP-seq data of EBF1-ChIP in pro-B cells (GSE69227), PAX5 (GSE38046) and GATA3-ChIP in T-cell progenitors (FLND2b; GSE31235), and TCF7 (TCF1; GSE46662) and C/EBPα-ChIP in thioglycolate-elicited macrophages (GSM537983) were retrieved from Gene Expression Omnibus (GEO) and remapped to mm10 using Bowtie2, and peaks were identified using HOMER with the -factor setting. Abundance of ATAC tags on the ChIP-seq peaks was analyzed in HOMER using the annotatePeaks command. For details, see the Supplemental Material.
Mutation analysis of human tumors
To explore the relationship in mutational landscapes, reported mutations in B-ALL, T-ALL, AML, and ALAL were extracted from the COSMIC cancer gene census database. Mutation frequency per reported tumor (AML, n = 26022; T-ALL, n = 1846; B-ALL, n = 1303; and ALAL, n = 52) was calculated. Genes with mutations less common than a frequency of 0.1% were excluded from the list. The mutation frequencies of the remaining genes in B-ALL, T-ALL, AML, and ALAL were subjected to hierarchical clustering using cluster 3 and were visualized in a heat map using Java TreeView. For details, see the Supplemental Material.
Supplementary Material
Acknowledgments
This work was supported by grants from the Swedish Cancer Society, the Swedish Childhood Cancer Foundation, the Swedish Research Council, including a Center grant to Hematolinne, the Knut and Alice Wallenbergs Foundation, and Linköping University. R.S., J.Å., J.U., K.O., and T.S. designed, conducted, and analyzed the experiments. H.L. and T.F. analyzed T-ALL data. M.S. designed the experiments and analyzed the data. All authors contributed to the writing of the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.285536.116.
References
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussmann LH, Schubert A, Vu Manh TP, De Andres L, Desbordes SC, Parra M, Zimmermann T, Rapino F, Rodriguez-Ubreva J, Ballestar E, et al. 2009. A robust and highly efficient immune cell reprogramming system. Cell Stem Cell 5: 554–566. [DOI] [PubMed] [Google Scholar]
- Cobaleda C, Jochum W, Busslinger M. 2007. Conversion of mature B cells into T cells by dedifferentiation to uncommitted progenitors. Nature 449: 473–477. [DOI] [PubMed] [Google Scholar]
- Collins-Underwood JR, Mullighan CG. 2010. Genomic profiling of high-risk acute lymphoblastic leukemia. Leukemia 24: 1676–1685. [DOI] [PubMed] [Google Scholar]
- Craig FE, Foon KA. 2008. Flow cytometric immunophenotyping for hematologic neoplasms. Blood 111: 3941–3967. [DOI] [PubMed] [Google Scholar]
- Dorantes-Acosta E, Pelayo R. 2012. Lineage switching in acute leukemias: a consequence of stem cell plasticity? Bone Marrow Res 2012: 406796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner R, Wu D, Cherian S, Fang M, Hanafi LA, Finney O, Smithers H, Jensen MC, Riddell SR, Maloney DG, et al. 2016. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 127: 2406–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyory I, Boller S, Nechanitzky R, Mandel E, Pott S, Liu E, Grosschedl R. 2012. Transcription factor Ebf1 regulates differentiation stage-specific signaling, proliferation, and survival of B cells. Genes Dev 26: 668–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamrouni A, Aublin A, Guillaume P, Maryanski JL. 2003. T cell receptor gene rearrangement lineage analysis reveals clues for the origin of highly restricted antigen-specific repertoires. J Exp Med 197: 601–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabbour E, O'Brien S, Ravandi F, Kantarjian H. 2015. Monoclonal antibodies in acute lymphoblastic leukemia. Blood 125: 4010–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings CD, Foon KA. 1997. Recent advances in flow cytometry: application to the diagnosis of hematologic malignancy. Blood 90: 2863–2892. [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. 2002. The human genome browser at UCSC. Genome Res 12: 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiper RP, Schoenmakers EF, van Reijmersdal SV, Hehir-Kwa JY, van Kessel AG, van Leeuwen FN, Hoogerbrugge PM. 2007. High-resolution genomic profiling of childhood ALL reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia 21: 1258–1266. [DOI] [PubMed] [Google Scholar]
- Lin H, Grosschedl R. 1995. Failure of B-cell differentiation in mice lacking the transcription factor EBF. Nature 376: 263–267. [DOI] [PubMed] [Google Scholar]
- Lobry C, Oh P, Aifantis I. 2011. Oncogenic and tumor suppressor functions of Notch in cancer: it's NOTCH what you think. J Exp Med 208: 1931–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manola KN. 2013. Cytogenetic abnormalities in acute leukaemia of ambiguous lineage: an overview. Br J Haematol 163: 24–39. [DOI] [PubMed] [Google Scholar]
- Mansson R, Zandi S, Anderson K, Martensson IL, Jacobsen SE, Bryder D, Sigvardsson M. 2008. B-lineage commitment prior to surface expression of B220 and CD19 on hematopoietic progenitor cells. Blood 112: 1048–1055. [DOI] [PubMed] [Google Scholar]
- Maude SL, Teachey DT, Porter DL, Grupp SA. 2015. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 125: 4017–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan JS, Dove C, Gentles AJ, Ryan CE, Majeti R. 2015. Reprogramming of primary human Philadelphia chromosome-positive B cell acute lymphoblastic leukemia cells into nonleukemic macrophages. Proc Natl Acad Sci 112: 4074–4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkola I, Heavey B, Horcher M, Busslinger M. 2002. Reversion of B cell commitment upon loss of Pax5 expression. Science 297: 110–113. [DOI] [PubMed] [Google Scholar]
- Milatovich A, Qiu RG, Grosschedl R, Francke U. 1994. Gene for a tissue-specific transcriptional activator (EBF or Olf-1), expressed in early B lymphocytes, adipocytes, and olfactory neurons, is located on human chromosome 5, band q34, and proximal mouse chromosome 11. Mamm Genome 5: 211–215. [DOI] [PubMed] [Google Scholar]
- Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, Girtman K, Mathew S, Ma J, Pounds SB, et al. 2007. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 446: 758–764. [DOI] [PubMed] [Google Scholar]
- Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J, Ma J, White D, Hughes TP, Le Beau MM, Pui CH, et al. 2008. BCR–ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 453: 110–114. [DOI] [PubMed] [Google Scholar]
- Mullighan CG, Zhang J, Harvey RC, Collins-Underwood JR, Schulman BA, Phillips LA, Tasian SK, Loh ML, Su X, Liu W, et al. 2009. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci 106: 9414–9418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechanitzky R, Akbas D, Scherer S, Gyory I, Hoyler T, Ramamoorthy S, Diefenbach A, Grosschedl R. 2013. Transcription factor EBF1 is essential for the maintenance of B cell identity and prevention of alternative fates in committed cells. Nat Immunol 14: 867–875. [DOI] [PubMed] [Google Scholar]
- Nerlov C. 2004. C/EBPα mutations in acute myeloid leukaemias. Nat Rev Cancer 4: 394–400. [DOI] [PubMed] [Google Scholar]
- Nutt SL, Heavey B, Rolink AG, Busslinger M. 1999. Commitment to the B-lymphoid lineage depends on the transcription factor Pax5 [see comments]. Nature 401: 556–562. [DOI] [PubMed] [Google Scholar]
- Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S, Behre G, Hiddemann W, Tenen DG. 2001. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-α (C/EBPα), in acute myeloid leukemia. Nat Genet 27: 263–270. [DOI] [PubMed] [Google Scholar]
- Park JH, Geyer MB, Brentjens RJ. 2016. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date. Blood 127: 3312–3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad MA, Ungerback J, Ahsberg J, Somasundaram R, Strid T, Larsson M, Mansson R, De Paepe A, Lilljebjorn H, Fioretos T, et al. 2015. Ebf1 heterozygosity results in increased DNA damage in pro-B cells and their synergistic transformation by Pax5 haploinsufficiency. Blood 125: 4052–4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pui JC, Allman D, Xu L, DeRocco S, Karnell FG, Bakkour S, Lee JY, Kadesch T, Hardy RR, Aster JC, et al. 1999. Notch1 expression in early lymphopoiesis influences B versus T lineage determination. Immunity 11: 299–308. [DOI] [PubMed] [Google Scholar]
- Rapino F, Robles EF, Richter-Larrea JA, Kallin EM, Martinez-Climent JA, Graf T. 2013. C/EBPα induces highly efficient macrophage transdifferentiation of B lymphoma and leukemia cell lines and impairs their tumorigenicity. Cell Rep 3: 1153–1163. [DOI] [PubMed] [Google Scholar]
- Reynaud D, Demarco IA, Reddy KL, Schjerven H, Bertolino E, Chen Z, Smale ST, Winandy S, Singh H. 2008. Regulation of B cell fate commitment and immunoglobulin heavy-chain gene rearrangements by Ikaros. Nat Immunol 9: 927–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts KG, Morin RD, Zhang J, Hirst M, Zhao Y, Su X, Chen SC, Payne-Turner D, Churchman ML, Harvey RC, et al. 2012. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell 22: 153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberg EV. 2014. Transcriptional control of early T and B cell developmental choices. Annu Rev Immunol 32: 283–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlissel MS, Corcoran LM, Baltimore D. 1991. Virus-transformed pre-B cells show ordered activation but not inactivation of immunoglobulin gene rearrangement and transcription. J Exp Med 1763: 711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt TM, Zuniga-Pflucker JC. 2002. Induction of T cell development from hematopoietic progenitor cells by delta-like-1 in vitro. Immunity 17: 749–756. [DOI] [PubMed] [Google Scholar]
- Simmons S, Knoll M, Drewell C, Wolf I, Mollenkopf HJ, Bouquet C, Melchers F. 2012. Biphenotypic B-lymphoid/myeloid cells expressing low levels of Pax5: potential targets of BAL development. Blood 120: 3688–3698. [DOI] [PubMed] [Google Scholar]
- Somasundaram R, Prasad MA, Ungerback J, Sigvardsson M. 2015. Transcription factor networks in B-cell differentiation link development to acute lymphoid leukemia. Blood 126: 144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepanski T, Pongers-Willemse MJ, Langerak AW, Harts WA, Wijkhuijs AJ, van Wering ER, van Dongen JJ. 1999. Ig heavy chain gene rearrangements in T-cell acute lymphoblastic leukemia exhibit predominant DH6-19 and DH7-27 gene usage, can result in complete V-D-J rearrangements, and are rare in T-cell receptor αβ lineage. Blood 93: 4079–4085. [PubMed] [Google Scholar]
- Tagoh H, Ingram R, Wilson N, Salvagiotto G, Warren AJ, Clarke D, Busslinger M, Bonifer C. 2006. The mechanism of repression of the myeloid-specific c-fms gene by Pax5 during B lineage restriction. EMBO J 25: 1070–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treiber T, Mandel EM, Pott S, Gyory I, Firner S, Liu ET, Grosschedl R. 2010. Early B cell factor 1 regulates B cell gene networks by activation, repression, and transcription-independent poising of chromatin. Immunity 32: 714–725. [DOI] [PubMed] [Google Scholar]
- Ungerback J, Ahsberg J, Strid T, Somasundaram R, Sigvardsson M. 2015. Combined heterozygous loss of Ebf1 and Pax5 allows for T-lineage conversion of B cell progenitors. J Exp Med 212: 1109–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbánek P, Wang Z-Q, Fetka I, Wagner EF, Busslinger M. 1994. Complete block of early B cell differentiation and altered patterning of the posterior midbrain in mice lacking Pax5/BSAP. Cell 79: 901–912. [DOI] [PubMed] [Google Scholar]
- Vilagos B, Hoffmann M, Souabni A, Sun Q, Werner B, Medvedovic J, Bilic I, Minnich M, Axelsson E, Jaritz M, et al. 2012. Essential role of EBF1 in the generation and function of distinct mature B cell types. J Exp Med 209: 775–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC. 2004. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306: 269–271. [DOI] [PubMed] [Google Scholar]
- Xie H, Ye M, Feng R, Graf T. 2004. Stepwise reprogramming of B cells into macrophages. Cell 117: 663–676. [DOI] [PubMed] [Google Scholar]
- Yu D, Allman D, Goldschmidt MH, Atchison ML, Monroe JG, Thomas-Tikhonenko A. 2003. Oscillation between B-lymphoid and myeloid lineages in Myc-induced hematopoietic tumors following spontaneous silencing/reactivation of the EBF/Pax5 pathway. Blood 101: 1950–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yui MA, Rothenberg EV. 2014. Developmental gene networks: a triathlon on the course to T cell identity. Nat Rev Immunol 14: 529–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandi S, Mansson R, Tsapogas P, Zetterblad J, Bryder D, Sigvardsson M. 2008. EBF1 is essential for B-lineage priming and establishment of a transcription factor network in common lymphoid progenitors. J Immunol 181: 3364–3372. [DOI] [PubMed] [Google Scholar]
- Zhang J, Mullighan CG, Harvey RC, Wu G, Chen X, Edmonson M, Buetow KH, Carroll WL, Chen IM, Devidas M, et al. 2011. Key pathways are frequently mutated in high risk childhood acute lymphoblastic leukemia: a report from the Children's Oncology Group. Blood 118: 3080–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, et al. 2012a. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481: 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JA, Mortazavi A, Williams BA, Wold BJ, Rothenberg EV. 2012b. Dynamic transformations of genome-wide epigenetic marking and transcriptional control establish T cell identity. Cell 149: 467–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.