


Abstract
The connexin43 (cx43) gene was originally described as consisting of two exons, one coding for most of the 5′-untranslated region (5′-UTR), and the other for the protein sequence and 3′-UTR. We now report that in mouse four additional exons are expressed, all coding for novel 5′-UTRs. Altogether, we found nine different cx43 mRNA species (GenBank accession numbers NM010288, and AY427554 through AY427561) generated by differential promoter usage and alternative splicing mechanisms. The relative abundance of these different mRNAs varied with the tissue source. In addition, the different transcripts showed varying translational efficiencies in several cell lines, indicating the presence of cis-RNA elements that regulate cx43 translation. We propose that it is the promoter driving the expression of the cx43 gene that determines exon choice in the downstream splicing events in a cell-type-dependent fashion. This in turn will affect the translation efficiency of the transcript orchestrating the events that lead to the final expression profile of cx43. Since a similar organization of the cx43 gene was also observed in rat it is likely that the complex regulation of cx43 expression involving transcription, splicing and translation mechanisms is a common trait conserved during evolution.
INTRODUCTION
Connexin43 belongs to a family of transmembrane proteins that oligomerize to form hexameric structures called connexons. Head-to-head docked connexons provide low-resistance cell-to-cell channels between adjoining cells, clusters of which are called gap junctions. It is through gap junction channels that cells share ions, second messengers and small metabolites of up to 1 kDa in molecular mass (1). If gap-junctional communication is compromised, normal tissue function cannot be maintained and disease may ensue. Mutations in genes that encode gap-junctional proteins leading to abnormal patterns of connexin expression often correlate with diseases, such as Charcot-Marie-Tooth disease (CMTX) (2,3), deafness (4), heart failure (5–8) and cancer (9–12). Cx43 is expressed in multiple tissues including epithelium, myocardium, myometrium and other smooth muscles. Its relative abundance varies from tissue to tissue. The regulation of its expression in response to external stimuli or during development appears to be cell-type-specific (13–17). This suggests that complex mechanisms may be involved in the regulation of the cx43 gene.
Fishman et al. (18) and Fromaget et al. (19) showed that the levels of cx43 mRNA and protein concentrations increased 8–15-fold in the rat and mouse developing heart between the early fetal period and 6 weeks after birth. Maximum cx43 mRNA levels are reached 1 week after birth while the peak in cx43 protein does not occur until the third week of life. This temporal difference in the accumulation of mRNA and protein suggests that at early stages of heart development the levels of expression of cx43 protein are regulated at the post-stranscriptional level. In addition to the temporal differences, there are also regional differences. Immunohistochemistry and in situ hybridization in rodent fetal heart showed high expression of cx43 mRNA but no detectable protein in the subepicardial layer of the ventricular free wall. In contrast, a relatively faint signal corresponding to cx43 mRNA and a relative abundant expression of cx43 protein are observed in the myocardium of the atria and the ventricular trabeculations (18–21). Adult heart is also an example of regional regulation of cx43 expression. In the ventricle, cx43 protein is abundant in the trabeculae and weak in the epicardial myocardium, despite similar mRNA levels (22,23).
Another example of the complex regulation of the cx43 gene is found in the ovary (24). In the primordial follicle cx43 expression is restricted to the cumulus/granulosa cells, and no cx43 is expressed in the prophase-arrested oocyte. Maturation of the follicle involves a series of events that require varying levels of cx43 (13,14,16,24). During estrous cycle, the regulation of cx43 expression is dependent on the levels of gonadotropins. Exposure of granulosa cells to follicle stimulating hormone results in the upregulation of cx43 mRNA and protein. In contrast, luteinizing hormone (LH) elicits an inhibitory effect on cx43 synthesis in large preovulatory follicles. A decrease but not complete disappearance in cx43 mRNA is seen after long exposure to LH (13). Altogether, these studies suggest that the cx43 gene in the ovary is also regulated by cell-type-dependent transcriptional and post-transcriptional mechanisms.
The structure of the connexin genes was initially thought to be conserved for all members of this family. It was described as consisting of two exons interrupted by a single intron of variable size. The intron was found to be 10.5 kb for the murine (25) and 8.6 kb for the rat cx43 gene (15) as characterized by restriction digestion mapping and sequencing of clones isolated from genomic libraries. In this structure, exon1 encodes most of the 5′-untranslated region (5′-UTR) while exon2 contains only 13 bases of the 5′-UTR followed by the complete coding sequence and its 3′-UTR. Recently, a few exceptions to this common structure have been found in some connexin genes. The exceptions have been classified into two groups, one involving alternative splicing of 5′-UTRs, and the other splicing of the connexin coding region (26). Examples of the first group include the mammalian cx32 gene which consists of four exons, three of them representing alternative 5′-UTRs (27–29); the murine cx45 gene consisting of three exons, two of them consecutively spliced to form its 5′-UTR (30); the cx30 gene with three exons either alternatively or consecutively spliced (26); and the human cx40 gene with three exons two of them alternatively spliced to form two alternative 5′-UTRs (31). The second group includes the connexins hCx31.3, mCx36, hCx36 and mCx57 (26,32,33), but most of their gene structures have not been fully described.
Even though enough evidence exists to suggest the existence of regulation of translation of the connexin genes, not much is known about the mechanisms involved. Alternate 5′-UTRs are highly suggestive of translational control. In fact, a single point mutation in the nerve-specific 5′-UTR of the cx32 mRNA prevents its translation in patients with CMTX disease (34). In addition, internal ribosomal entry sites (IRESs) have been found in the 5′-UTRs of the cx32 and the cx43 genes (34,35), which have been proposed to function in maintaining connexin expression at times when cap-mediated translation is shut off (36). This together with our interest to understand the mechanisms of regulation of the cx43 gene prompted us to further investigate whether additional 5′-UTR sequences of the cx43 gene may exist. If such alternative 5′-UTRs exist, any investigation on connexin43 gene regulation would have to take them into account. As a first step, we searched the expressed sequence tags (ESTs) database for sequences coding for cx43 but varying at their 5′ ends. This initial search led us to discover eight previously unknown 5′ UTRs in mouse cx43 mRNA, which appear to exhibit different translational efficiencies. Furthermore, two additional cell-type-specific promoters were identified which are likely to be used to differentially regulate transcription of the cx43 gene. From this work, a complex gene structure emerges for cx43 which suggests a rather intricate regulation of cx43 expression.
MATERIALS AND METHODS
Database search and secondary structure prediction
GenBank vertebrate EST databases and rodent genomes were searched for cx43 sequences with the basic local alignment search tool (BLAST) developed by the National Center for Biotechnology Information (NCBI) (37). The secondary structure of each cx43 5′-UTR was predicted by the mFOLD program based on the Zuker algorithm at http://www.bioinfo.rpi.edu (38,39).
RT–PCR
An aliquot of 1–5 μg of total RNA from mouse and rat tissues isolated with Trizol® reagent (Invitrogen), and subsequently treated with RQ1 DNase (Promega), was used for first-strand cDNA synthesis with M-MuLV Reverse Transcriptase (NEB) and an antisense primer annealing to the cx43 coding region (5′-ACT GTT CAT CAC CCC AAG CTG-3′). The RNA template was subsequently cleaved with RNAse A, and one-tenth of the cDNA products were amplified using Taq polymerase in a standard PCR. A nested antisense primer within the cx43 coding region (mouse: 5′-AGC ACC GAC AGC CAC ACC TTC-3′ and rat: 5′-AGC ACT GAC AGC CAC ACC TTC-3′) and several exon-specific forward primers were employed in the subsequent PCRs (mouse: exon1As, 5′-AAA GCT CTG TGC TCC AAG TTA A-3′; exon1A, 5′-AAA GGC GTG AGG GAA GTA CCC-3′; exon1Bs, 5′-TGC GTT GCG TCT TTG ACT TAG G-3′; exon1C, 5′-TCT CTG GCA GAG TCT TGG TCG-3′; exon1A-1E, CAT TAA GTG AAA GAA CAG GGT CTC TC-3′; and rat: exon1As, 5′-AAG CTC TGC GCT CCA AGT TAG-3′; exon1A, 5′-GGA AGG CGT GAG GAA AGT ACC-3′; exon1B, GCG TTG CGT CTT TGG CTT AGG-3′; exon1C, 5′-ATG CTC ACT GGG CTT CTC TGG-3′). The PCR products were separated on a 2% agarose gel and visualized by ethidium bromide staining. Gel-purified products (GenElute minus EtBr spin columns; Sigma) were cloned into the pPCR-Script Amp vector (Stratagene) and sequenced. Sequencing was performed in an automated 337 DNA sequencer (PerkinElmer) in the DNA Core Facility of the University of Miami (Department of Biochemistry and Molecular Biology). For semiquantitative RT–PCR, an antisense primer annealing to the β-actin coding region (5′-ATA GCA CAG CTT CTC TTT GAT GTC-3′) was included in the first-strand synthesis reaction and a nested reverse (5′-TCA GGA TCT TCA TGA GGT AGT CTG-3′) and forward (5′-GAA TGG GTC AGA AGG ACT CCT ATG-3′) primers annealing to the β-actin coding region were included in the PCR amplification step. For semiquantitative RT–PCR of the chimeric cx43 5′-UTR-F-luc mRNA transfections, an antisense primer annealing to the F-luc coding region: 5′-TCA CTG CAT ACG ACG ATT CTG TG-3′ and the other two nested antisense primers, both of which anneal to the F-luc coding region: 5′-GCT GGA GAG CAA CTG CAT AAG GC-3′ and 5′-GAA GTA CTC AGC GTA AGT GAT GTC-3′, were used.
5′-RLM-rapid amplification of cDNA ends (RACE)
To map the transcription start sites for the cx43 transcripts, we used the 10–12-day-old mouse embryonic cDNA First Choice RACE-Ready cDNA system from Ambion. In this system only full-length, capped mRNA is reverse transcribed. To assure this, triple oligo(dT) selected, DNA-free, RNA template is treated with calf intestinal phosphatase (CIP) to remove 5′-phosphates from rRNA, tRNA and fragmented mRNA. The cap structure is then removed by treatment with the Tobacco Acid Pyrophosphatase (TAP) to ligate a synthetic RNA adapter oligonucleotide to the mRNA 5′ end with T4 RNA ligase. A random-primed reverse transcription reaction is then carried out to make a cDNA copy of the treated RNA. Finally, the cDNA is RNase treated and size selected by column chromatography. An aliquot of 0.5 ng of the cDNA was used as template for PCR amplification with an antisense primer annealing to the cx43 coding region (5′-ACT GTT CAT CAC CCC AAG CTG ACT-3′) and a primer containing the synthetic RNA adapter sequence. The products were further amplified by a subsequent nested PCR using exon-specific antisense primers (exon1As, 5′-TCT GGG CAC CTC CTC GTA AAA GC-3′; exon1Bs, 5′-TCT GGG CAC CTC AGT ATC CTA AGT-3′; exon1C, 5′-GGA CTG GTA AGA TTC ACT ACC TTC C-3′; and exon1A-1E, 5′-CAT GAT GGT GCA CGC CTA TAA TCC-3′). The final PCR products were analyzed on a 2% agarose gel and visualized by ethidium bromide staining. Gel-purified products (GenElute minus EtBr spin columns; Sigma) were cloned into the pPCR-Script Amp vector (Stratagene) and sequenced.
Reporter constructs
Generation of pGLp1(P1), pGLp2/2rev (P2/P2rev), pGLp3/3rev (P3/P3rev) and pGLc(C) reporter promoter constructs
A series of Firefly-luciferase expression constructs were created by cloning fragments of the promoter and intron region of the cx43 gene into the KpnI and either the HindIII or the BglII sites of the pGL2Basic vector (Promega). pGLp1 contained 244 nt corresponding to positions −263 and −19 with respect to the cx43 transcription start site. Similarly, pGLp2 contained 201 nt between positions +7 and +208; pGLp3(0.2) contained 251 nt between positions +1561 and +1812; pGLp3(1 kb) contained 1091 nt between positions +1561 and +2652; and pGLc contained 296 nt between positions +425 and +721. The rat genomic clone c433 (15) was used as the template for PCR amplification of all these fragments with the following primer sets: for pGLc-5′-GGT ACC AAT TTC CTA GAC TTG-3′ (sense) and 5′-AAG CTT AAT CCA AAA AAA AGG C-3′ (antisense); for pGLp1- 5′-GGT ACC TTT TAT CTG TGA GGA GTC-3′ (sense) and 5′-GGA TCC TTT TAA AAG TTT CAA GCC-3′ (antisense); for pGLp2-5′-GGT ACC TTT TAC GAG GTA TCA G-3′ (sense) and 5′-AAG CTT ATC AAA ACT TAC TCT TTC-3′ (antisense); for pGLp3 (0.2 kb) 5′-GGT ACC AAT CTG AAG TCT CTT CC-3′ (sense) and 5′-AAG CTT GCC TCA GCA CAC ATC AG-3′ (antisense); and for pGLp3 (1 kb)-5′-GGT ACC AGA AAA TAC GTA GAT CGG-3′ (sense) and same antisense as for pGLp3 (0.2 kb). For the reverse constructs, similar sets of primers with opposite restriction site tailings were used.
Generation of pSP-luc+[A]60-derived cx43 5′-UTR constructs
Each of the mouse cx43 5′-UTRs were subcloned into pSP-luc+[A]60 vector, a modified version of the pSP-luc+ vector (Promega) containing a poly(A) tail downstream of the Firefly-luciferase coding region which was kindly provided by Dr P. Marsden. A KpnI site and an NsiI site were used to insert each of the 5′-UTRs immediately upstream of the luciferase coding region. The NsiI site was created by mutating the luciferase start codon with the QuikChange® site-directed mutagenesis kit (Stratagene) and a set of mutagenic primers (5′-TCC TAG GAA GCT TTC CAT GCA TGA CGC CAA AAA CAT AAA G-3′ and its complementary primer). The strategy used to obtain the different 5′-UTRs comprises PCR amplification steps using the RLM-RACE products and/or the RT–PCR clones as templates with KpnI and NsiI tailed specific primers.
The control vector pSP-Rluc+[A]60 was constructed by replacing the Firefly-luciferase cDNA in pSP-luc+[A]60 with the Renilla-luciferase cDNA obtained by digesting the pRL-TK vector (Promega) with the NheI and the XbaI restriction enzymes. For pSP-luc+[A]60-UTR1A-1EmATG and pcDNA-UTR1A-1EmATG constructs, the upstream out of frame ATG in UTR1A-1E was mutated to AAG in the constructs pSP-luc+[A]60-UTR1A-1E and pcDNA-UTR1A-1E by site-directed mutagenesis. We used the QuikChange site-directed mutagenesis kit (Stratagene) and the following mutagenic primers: 5′-CGT GCA CCA TCA AGC CCG GCA CAA GAG-3′ and 5′-CTC TTG TGC CGG GCT TGA TGG TGC ACG-3′.
Generation of pcDNA-derived 5′-UTR constructs
Each of the pSP-luc+[A]60-UTR construct was digested with KpnI and EcoRI and the 5′-UTRs were subcloned into these same sites present in the multi-cloning site (MCS) of the pcDNA3.1(+) vector (Invitrogen).
Generation of SL2-UTR1A, -UTR1AL, -UTR1A-1E and SL2-EMCV bicistronic constructs
All the mouse cx43 5′-UTRs containing Exon1A, and the encephalomyocarditis virus (EMCV) internal ribosome entry site (IRES) element (positive control) were cloned into the EcoRI and XhoI sites of the bicistronic SL2 vector. As a negative control vector, the SL2M2 construct containing a mutated cx32 5′-UTR was used (35).
Cell lines, transient transfections and dual luciferase assays
Chinese hamster ovary (CHO) cells were maintained in Ham's F12K medium supplemented with 2 mM l-glutamine/10% FBS and adjusted to contain 1.5 g/l sodium bicarbonate. Mouse mammary epithelial HC-11 cells were maintained in RPMI 1640 medium supplemented with 2 mM l-glutamine, 10% FBS, 0.1 mg/ml gentamicin, 0.01 μg/ml EGF and 5 μg/ml of bovine insulin. Human cervix ephitheloid carcinoma HeLa cells were maintained in DMEM-F12 medium supplemented with 2 mM l-glutamine and 10% FBS. NIH3T3 mouse embryo fibroblasts were maintained in DMEM supplemented with 10% newborn calf serum and 2 mM l-glutamine. N2A mouse neuroblastoma cells were maintained in MEM supplemented with 10% FBS, 2 mM l-glutamine, 0.1 mM non-essential amino acids and 1 mM sodium pyruvate. The cells were transiently co-transfected with either 500 ng of each vector (pGL constructs), 1200 ng (pcDNA-derived constructs), 300 ng (SL2-derived constructs) and 15–50 ng of pRL-TK vector, as a control for transfection efficiency, per well of an either 6- or 24-well-dish using LipofectAMINE™ 2000 (Invitrogen) according to the manufacturer's recommendations. For mRNA transfections, 1 μg of F-luc mRNA and 0.05 μg of R-luc mRNA per well were used. Cells were lysed 48 h after transfection (or 10 h for mRNA transfections) and Firefly- and Renilla-luciferase activities were assayed with the Dual-Luciferase Reporter Assay System according to the manufacturer's instructions (Promega), and a TD-20/20 luminometer (Turner Designs) set to dual mode with a 2 s delay and a 10 s integration time. F-luc and R-luc mRNAs were in vitro transcribed from 1 μg of XhoI linearized pSP-luc+[A]60-UTR and pSP-Rluc+[A]60 constructs with the SP6 mMessage mMachine™ kit from Ambion. The quality of the capped mRNA was assessed by electrophoretic separation on a RNA Nano LabChip in a 2100 Bioanalyzer (Agilent Technologies, UM Microarray facility).
Ribonuclease protection assays (RPAs)
An aliquot of 10 μg of total mouse ovary RNA was analyzed with 250 pg of specific antisense RNA probe using the HybSpeed RPA kit according to the manufacturer's protocol (Ambion). The biotinylated antisense RNA probes contained 160 nt of the cx43 exon2 sequence directly downstream of 110 nt of either exon1C or exon1E sequence. The sequences were PCR amplified from the tissue RT–PCR products and ligated to an SP6 promoter adapter and subsequently amplified with the Lig'nScribe™ kit from Ambion. An antisense primer annealing to the mouse cx43 coding region: 5′-ACT GTT CAT CAC CCC AAG CTG ACT-3′ and an exon-specific sense primer annealing to the 3′ end of either exon1C: 5′-TAC AGG GCA GCC TTT GGA GG-3′ or exon1E: 5′-GGC TGT CCT GGA ACT CAC TTT G-3′ were used. For in vitro transcription, 100 ng of each SP6 template was used with the MAXIscript Transcription kit (Ambion) according to the manufacturer's protocol. A total of 4 nmol of Biotin-16-UTP and 6 nmol of unlabeled UTP were included in the reaction. After DNaseI digestion of the DNA template, the RNA probes were purified by gel electrophoresis (5% acrylamide/8 M urea), and elution into 0.5 M ammonium acetate/1 mM EDTA/0.2% SDS. For each probe, two control reactions where the probe was hybridized to 50 μg of yeast total RNA were included. In one reaction, RNase treatment followed hybridization (background) and in the other the RNase treatment was omitted (full-length probe).
RESULTS
Identification of alternate 5′-UTRs of the cx43 gene
In the mouse, the two known exons, one coding for 5′-UTR and the other for the remaining 13 nt of the 5′-UTR followed by the complete coding sequence and 3′-UTR, are located on chromosome 10 between 2 223 826 and 2 257 208 nt; they are separated by a 30 kb intron, according to the data available at GenBank (http://www.ncbi.nlm.nih.gov). However, restriction digestion and sequencing data on genomic clones had assigned a size of 10.5 kb to the mouse intron (21). This discrepancy of 20 kb could be due to an error in the assembly of chromosome 10 sequence or to DNA rearrangements that occurred during the generation or propagation of the genomic library.
A BLAST alignment of the cx43 cDNA sequence against the murine EST database revealed a long list of sequences that perfectly matched exon2 but differed upstream from it. All these sequences mapped to a region of chromosome 10 upstream of exon2 and they all contained consensus splice junctions at their 3′ ends. Based on this information, it appeared possible that multiple mRNA species with different 5′-UTRs are generated from the cx43 gene. To confirm the existence of these predicted transcripts, total RNA from a variety of tissues expressing cx43 was analyzed using RT–PCR. As antisense primer a sequence complementary to the coding region was used, and various forward primers were designed according to the putative 5′-UTRs derived from the EST analysis (for details see Materials and Methods). All the predicted exons were found in mRNA obtained from six different tissues (Figure 1A). It appeared that the relative abundance of some of the mRNA species varied according to the source of RNA and, therefore, must constitute a tissue-specific feature. It should be noted that the RT–PCR products representing UTR 1BL and UTR 1BS-1D run at the same position in the gel shown and thus needed to be identified by subcloning. Sequence analysis of the PCR products confirmed that each of the new exons, previously described as part of the cx43 intron, was directly linked to exon2 (coding sequence). The complete redefined exon–intron structure of the cx43 gene is shown in Figure 1B. It contains six exons, five of which code for different 5′-UTRs, and one for the coding region. Some of the transcripts contain combinations of two exons, others three exons (Figure 1C). In addition, there seems to be variation in the size of some of the exons.
Figure 1.
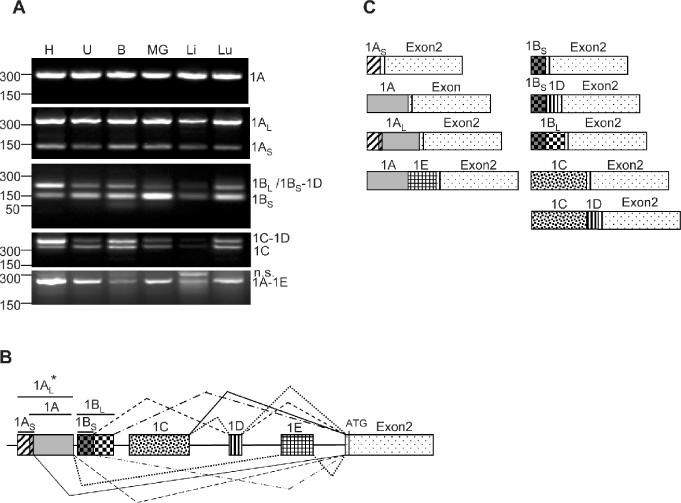
Identification of new connexin43 5′-UTRs in mouse. (A) RT–PCR of total RNA prepared from various mouse tissues including heart (H), uterus (U), brain (B), mammary gland (MG), liver (Li) and lung (Lu). A common antisense primer annealing to the cx43 coding region and variable forward primers specific to each of the new exons (see Materials and Methods) were used. The PCR products were analyzed in a 2% agarose gel and visualized by ethidium bromide staining. Subcloning and sequencing of the bands confirmed that the products contain the cx43 UTR1A, 1AL, 1AS, 1BL, 1BS-1D, 1BS, 1C-1D, 1C and 1A-1E sequences, as indicated on the right-hand side of the panels. n.s. indicates a non-specific PCR product. On the left-hand side, the positions of the molecular weight markers (PCR markers; Promega) are indicated. (B) Diagram showing the genomic organization of the new cx43 exons, and the splicing combinations found in mouse cx43 mRNAs. Exon1AS and exon1A partially overlap (11 nt). Asterisk indicates exon1AL splices directly to exon2. (C) Diagram showing the structure of the transcripts originated from the mouse cx43 gene.
To investigate whether the complex structure of the cx43 gene applied only to the murine gene or whether this was a general feature of the cx43 gene that is conserved across species, a BLAST search of the human and other eukaryotes EST databases was carried out using either the rat or the human cDNA sequence as the query. The search did not yield any hit in the human database other than the known 5′-UTR (GenBank accession no. NM000165). The rat EST database contained a single alternative 5′-UTR related to the mouse exon1C sequence (GenBank accession no. CB613999). The human and rat EST databases are much smaller and therefore less complete than the mouse EST database. Thus, the lack of hits does not rule out the existence of alternative 5′-UTRs.
RT–PCR on different rat tissues (Figure 2A) indicates that four of the six exons found in mouse are also present in rat. The primers for RT–PCR analysis were chosen by their sequence identity to the recently identified murine 5′-UTRs and by their relative position to exon2. No equivalents of the mouse exons 1D or 1E were found. However, novel mRNA species containing both a shorter and a longer version of the exon1C, not found in mouse, were present in rat. This shows that the cx43 gene in other species is transcribed into multiple mRNA species as well (Figure 2B). A new structure of the rat cx43 gene is depicted in Figure 2C. No RT–PCR was performed on human mRNA.
Figure 2.
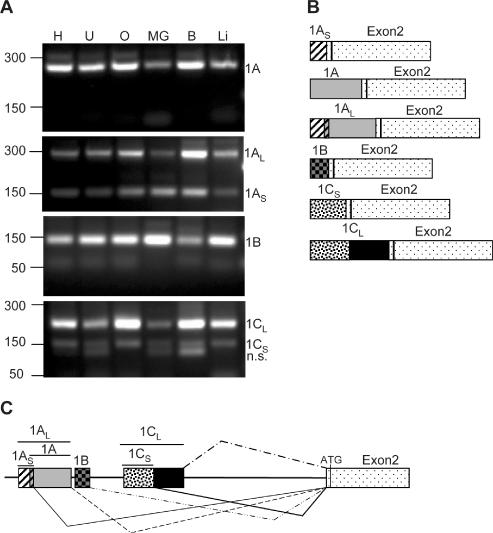
Identification of new connexin43 5′-UTRs in rat. (A) RT–PCR of total RNA prepared from various rat tissues including heart (H), uterus (U), ovary (O), mammary gland (MG), brain (B) and liver (Li). A common antisense primer annealing to the cx43 coding region and variable forward primers specific to each of the new exons (see Materials and Methods for sequence and details) were used. The PCR products were analyzed in a 2% agarose gel and visualized by ethidium bromide staining. Subcloning and sequencing of the bands confirmed that the products contain the cx43 UTR1A, 1AL, 1AS, 1B, 1CL and 1CS sequences, as indicated on the right-hand side of the panels. n.s. indicates a non-specific PCR product. On the left-hand side, the position of the molecular weight markers (PCR markers; Promega) is indicated. (B) Diagram showing the structure of the transcripts originated from the rat cx43 gene. (C) Diagram showing the genomic organization of the new exons and the splicing combinations found in rat cx43 mRNAs. Exons1AS and exon1A partially overlap (11 nt).
Cell-type-dependent promoter usage and alternative splicing generates a series of cx43 transcripts
Some of the newly identified exons, especially those that are located within the previously described large intron, are likely to be generated from different promoters. To determine the transcription start sites of these mRNA species, cDNA constructed from capped full-length oligo(dT)-purified mRNA from 10–12–day-old mouse embryos (Ambion) was subjected to 5′-RLM-RACE analysis. The positions of the reverse primers used in this experiment are indicated in Figure 3A. Sequencing of the obtained PCR products revealed that there are several transcription start sites for exon 1A corresponding to cytosines at positions −58, −13 and +1. The start site for exon1B is an adenine at position +215, and for exon1C a thymine at position +1793. The nucleotide positions were numbered with respect to the previously described start site (+1) (25). No primer was used for exon1D since this exon always appeared linked to either exon1B or exon1C. The two start sites found for transcription of exon1AS probably are controlled by the previously known cx43 promoter, which we now call P1. However, there must be two additional promoters downstream of P1: promoter P2 located within exon1A and promoter P3 located in the intron just upstream of exon1C. The P2 sequence had been previously proposed by Carystinos et al. (40) to function as an additional promoter of the cx43 gene.
Figure 3.
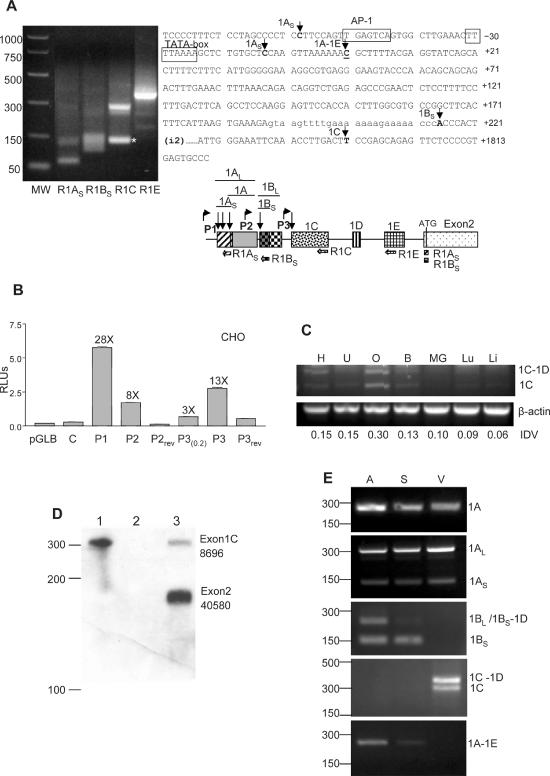
Cell-type-specific expression of the new cx43 exons from three different promoters. (A) RLM-RACE analysis of mouse embryo full-length capped mRNA. Sequencing of the PCR-purified products determined the transcription start sites (tss) for exons 1A, 1B and 1C. With the R1C primer an unspecific product of smaller size was obtained (*). PCR molecular markers (MW) (Promega) are shown. The positions of the tss are indicated by arrows on the cx43 sequence (right part of the figure). The previously determined tss is underlined. The diagram shows the relative position of the reverse primers used in the PCR amplification of the 5′-RACE products. The locations of the putative promoters P2 and P3, with respect to the previously characterized cx43 promoter (P1), are shown by bent arrows. (B) Relative luciferase (Firefly/Renilla) activity of CHO cells transiently transfected with various pGL-Basic constructs. The constructs contained either the cx43 promoter P1 or the putative promoters P2 or P3 sequences in the 5′ to 3′ or the reversed (rev) orientation. The P3(0.2) construct contained 251 nt while the P3 construct contained 1091 nt of sequence upstream of exon 1C. As controls the empty pGLBasic vector (Promega) and the C construct containing 296 nt of cx43 intron sequence immediately upstream of exon1B were used (see Materials and Methods for further details). The fold induction of each construct with respect to the empty pGLB vector is indicated. (C) Semiquantitative RT–PCR analysis of exon1C expression in various mouse tissues, which includes heart (H), uterus (U), ovary (O), brain (B), mammary gland (MG), lung (Lu) and liver (Li). The RT–PCR reactions contained one set of primers annealing to the cx43 coding region and exon1C, and a second set of primers annealing to the β-actin gene (see Materials and Methods for primer sequences). The PCR-amplified products were separated in a 2% agarose gel and visualized by ethidium bromide staining. Positions of the amplified products corresponding to mRNAs either containing (1C-1D) or skipping exon1D (1C) are indicated. Integrated density values (IDVs) of the 1C bands, as determined by the Spotdenso program (Alpha Innotech, Inc.), are shown. (D) RPA analysis of total mouse ovarian RNA (lane 3). The probe was complementary to the entire exon1C (110 nt) and the contiguous 160 nt of exon2. The proportion of exon1C (8.5%) containing transcripts was calculated as a percentage of the exon2-containing transcripts. The IDVs of the bands, as determined by the Spotdenso program, are shown. As controls the probe was hybridized to yeast RNA in the absence (lane 1) or the presence (lane 2) of RNase. The sizes of the PCR molecular weight markers (Promega) are indicated on the left-hand side. (E) RT–PCR analysis of RNA prepared from various parts of the mouse heart which included atrium (A), septum (S) and ventricle tip (V). A common antisense primer annealing to the cx43 coding region and variable forward primers specific to each of the new exons (see Materials and Methods) were used in the amplification. Products were analyzed in a 2% agarose gel and visualized by ethidium bromide staining. Subcloning and sequencing of the bands confirmed that they contained the cx43 UTR1A, 1AL, 1AS, 1BL, 1BS-1D, 1BS, 1C-1D, 1C and 1A-1E sequences, as indicated on the right-hand side of the panels. On the left, the positions of the molecular weight markers (PCR markers; Promega) are indicated.
To test whether the P2 and P3 sequences could in fact function as promoters, we subcloned ∼200 bp of the rat genomic sequence immediately upstream of either exon1B or exon1C into the MCS of the pGL2-Basic luciferase reporter plasmid (Promega). When transfected into CHO cells both P2 and P3(0.2) constructs induced luciferase activities 8- and 3-fold, respectively, as compared to the empty vector (Figure 3B). Inclusion of an additional 800 bp of upstream sequence (P3 construct in Figure 3B) increased the activity to 13-fold, which is still below the levels provided by the previously described P1 promoter (28-fold). Insertion of inverted sequences for P2 and P3 (P2rev and P3rev constructs) failed to activate the luciferase gene. This demonstrates that P2 and P3, indeed, can function as promoters. Relative promoter strength was always P1 > P3 > P2 in all cell lines tested, which included HeLa and HC-11 (data not shown).
To study whether promoter P3 is used more prominently in certain cell types, the relative abundance of the exon1C transcript was determined in seven different tissues known to express cx43. Screened tissues included heart, uterus, ovary, brain, mammary gland, lung and liver. Semiquantitative RT–PCR analysis suggested that among the tissues analyzed promoter P3 was most active in the ovary (Figure 3C). Nevertheless, RNA protection analysis showed that the percentage of transcripts produced from P3 represented only 8.5% of the total cx43 mRNA in this tissue (see Figure 3D). This correlates well with the in vitro transcription studies which classified P1 as a more active promoter than P3 in all cell lines tested (Figure 3B and data not shown). It is possible that P3 activity is regulated in response to unknown signals. Furthermore, P3 activity may be limited to specific cell types within a tissue. Thus, it may be diluted by the more commonly used P1 promoter when whole tissue is analyzed. To test whether this is the case, we decided to analyze different parts of the heart which are known to contain different cell types. We chose heart because regional differences in the regulation of cx43 expression had been shown to involve both transcriptional and post-transcriptional mechanisms (23) and also because dissection of its different parts is relatively easy. As shown in Figure 3E, promoter P2 seems to be active in atria and septum but not in the ventricle tip. In contrast, P3 functions only in ventricle but not in the atrium or the septum. Promoter P1 is active throughout the organ. Alternative splicing or exon choice also seems to be cell-type-specific since transcripts containing exon1B preferentially skip exon1D in the septum whereas in the atrium exon1D is included in about half of the transcripts. Inclusion of exon1E occurs preferentially in atrium and to some extent in the septum, but not at all in the ventricle.
Translation of cx43 is regulated by its alternative 5′-UTRs
What is the function of these alternatively spliced 5′-UTRs? It is possible that they provide a mechanism to regulate expression of cx43 post-transcriptionally either by increasing or decreasing the translational efficiency of the mRNA. To further investigate this possibility, each of the nine different 5′-UTRs was cloned immediately upstream of the luciferase coding region in the CMV promoter driven pcDNA3.1 plasmid (Invitrogen). Analysis of various cell lines transiently transfected with these constructs shows that all versions of exon1A 5′-UTRs are equally efficient in the production of protein with the exception of the exon1A–1E combination which completely blocks luciferase expression (Figure 4A). Translation of mRNA-containing exon1B as 5′-UTR was less efficient, and mRNA-containing exon1C was almost inactive. Only CHO cells showed some activity with exon1C-containing mRNA. Our results also show that mRNA containing the exon1BS–1D combination has a much lower translational activity than mRNA containing only exon1BL 5′-UTR, even though exon1BL and the exon1BS–1D combination have a similar length, indicating that it is the 3′ moiety of the leader sequence that plays a role in the regulation of translation. The same pattern of differential activity was observed when cells were transfected with in vitro transcribed mRNA containing the various 5′-UTRs linked to the luciferase coding region (Figure 4B). DNA transfection allows effects on the generation and processing of transcripts while mRNA transfection eliminates these possibilities. Thus, this result confirms that the novel 5′-UTRs indeed play a role in regulating cx43 synthesis at the post-transcriptional level. Furthermore, the different activities linked to these alternative 5′-UTRs do not appear to be a consequence of differential mRNA stability as shown in Figure 4C by semiquantitative RT–PCR. Varying the levels of total RNA used as template and reducing the number of cycles of PCR amplification did not alter the results (data not shown). Therefore, the observed differences in luciferase activities must be mainly due to differences in translation efficiencies.
Figure 4.
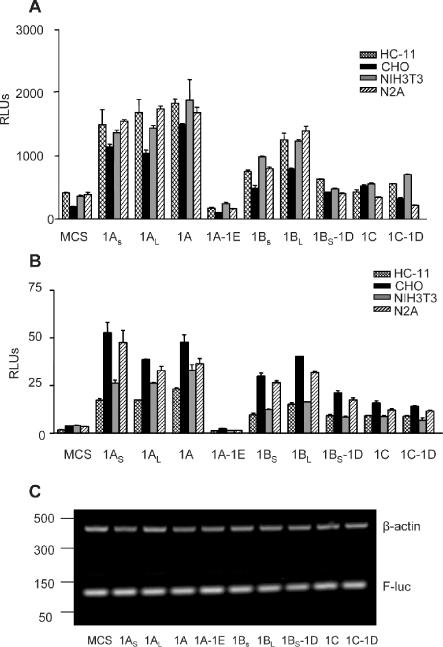
Varying translational efficiencies of the different cx43 5′-UTRs. (A) Relative luciferase (Firefly/Renilla) activities of various cell lines transiently transfected with 1.2 μg of pcDNA3.1 (Stratagene)-derived constructs containing each of the mouse cx43 5′-UTRs sequences upstream of the luciferase coding region. MCS correspond to the relative luciferase values obtained with the empty pcDNA3.1(+) vector. Cells were co-transfected with 15 ng of the pRL-TK Renilla expressing vector (Promega) to correct for variations in transfection efficiency. Values are expressed as the means ± SE of at least three replicate experiments. The cell lines included HC-11, CHO, NIH3T3 and N2A cells. (B) As in (A), but cells were co-transfected with 1 μg of in vitro transcribed, capped, chimeric cx43 5′-UTR/luciferase reporter mRNAs and 50 ng of Renilla-luciferase mRNA, as indicated. MCS corresponds to the relative luciferase values obtained from the in vitro transcribed luciferase mRNA from pSPluc+[A]60 vector. (C) Semiquatitative RT–PCR analysis of total RNA obtained from N2A cells transfected with in vitro transcribed, capped, either chimeric cx43 5′-UTR/luciferase reporter mRNAs or mRNA transcribed from the empty pSPluc+[A]60 vector (MCS), 6 h after transfection. The RT–PCR reactions contained one set of primers annealing to the Firefly-luciferase coding region (F-luc), and a second set of primers annealing to the β-actin gene (see Materials and Methods for primer sequences). The PCR-amplified products were separated in a 2% agarose gel and visualized by ethidium bromide staining. The positions of the molecular weight markers (PCR markers; Promega) are indicated on the left-hand side.
Inclusion of exon1E inhibits the translation of the cx43 transcript by introducing an additional AUG in the leader sequence
As mentioned above, inclusion of exon1E in the cx43 5′-UTR dramatically down regulates translation. This inhibitory effect could be mediated by an out-of-frame AUG triplet, located 26 nt upstream of the start codon, which is introduced into the mRNA when exon1E is included. To test whether the upstream AUG (uAUG) is responsible for this inhibitory effect, we mutated the T in the triplet to an A (1A-1EmATG). As shown in Figure 5A, mutation of this AUG significantly reversed the inhibitory effect suggesting that, at least in part, the mechanism used by the cell to downregulate translation requires this uAUG. The same effect is seen when in vitro transcribed mutant 1A-1E mRNA is used for transfection (see Figure 5B). We also noted that the secondary structure of exon1A, as determined by the mFOLD program, drastically changes when exon1E is included (Figure 5C). This profound change in the secondary structure of the leader sequence is accompanied by a decrease in its free energy indicative of a much more stable structure. This together with the additional AUG triplet may explain the strong inhibitory effect exon1E has on translation. Mutation of the uAUG triplet does not alter the secondary structure of the exon1A-1E 5′-UTR (data not shown). Since this mechanism could be utilized by the cell to downregulate production of cx43, it was of interest to find out what type of cells may be using it. For this purpose semiquantitative RT–PCR was performed on RNA from seven different tissues (Figure 5D). Again, ovary seemed to contain the highest expression levels of exon1E with respect to the other tissues analyzed. To more precisely quantify the percentage of transcripts containing exon1E in this tissue, ovarian RNA was subjected to RPA analysis. Figure 5E shows that exon1E is included only in 10% of the total cx43 message in ovary.
Figure 5.
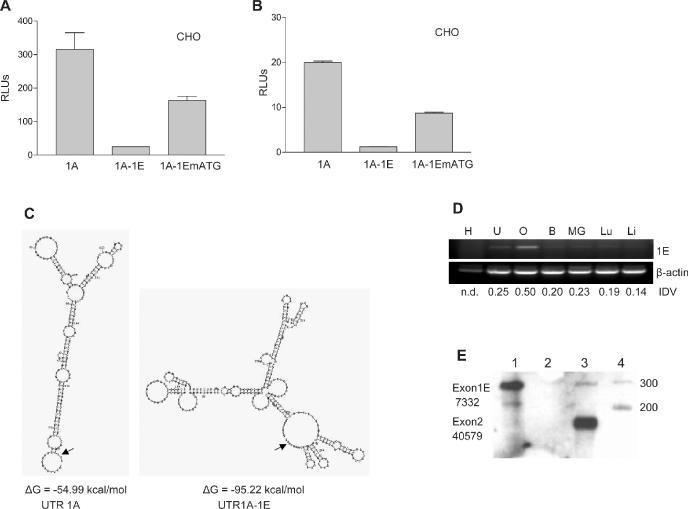
Exon1E inhibits translation of an exon1A-containing luciferase reporter mRNA. (A) Relative luciferase activity of CHO cells transfected with pcDNA3.1-derived constructs. The constructs contained exon1A cx43 5′-UTR either without (1A) or with exon1E (1A-1E) immediately upstream of the luciferase gene. Construct 1A-1EmATG is construct 1A-1E in which the upstream ATG triplet is mutated. Cells were co-transfected with 50 ng of the pRL-TK Renilla expressing vector (Promega) to correct for variations in transfection efficiency. Values are expressed as the means ± SE of at least three replicate experiments. (B) Same as in (A), but cells were transfected with in vitro transcribed, capped, chimeric cx43 5′-UTR/luciferase reporter RNAs. The 5′-UTR was exon1A or exon1A-1E either wild type or containing a mutation in the uAUG. (C) Structures of exon1A and exon1A-1E 5′-UTRs as predicted by the mFOLD program. Calculated free energies (ΔG) are indicated. The AUG translation start site is pointed by an arrow. (D) Semiquantitative RT–PCR analysis of exon1E expression in various mouse tissues, which included: heart (H), uterus (U), ovary (O), brain (B), mammary gland (MG), lung (Lu) and liver (Li). The RT–PCR reactions contained one set of primers annealing to the cx43 coding region and exon1E, and a second set of primers annealing to the β-actin gene (see Materials and Methods for primer sequences). The PCR-amplified products were separated in a 2% agarose gel and visualized by ethidium bromide staining. IDVs of the bands, as determined by the Spotdenso program (Alpha Innotech, Inc.), are shown. n.d. indicates not determined. (E) RPA analysis of 10 μg of total mouse ovarian RNA (lane 3). The probe was complementary to 110 nt of exon1E and the contiguous 160 nt of exon2. The proportion of exon1E containing transcripts (10%) was calculated as a percentage of the exon2-containing transcripts. The IDVs of the bands, as determined by the Spotdenso program, are shown. As controls, the probe was hybridized to yeast RNA in the absence (lane 1) or the presence (lane 2) of RNase. PCR molecular weight markers (lane 4) (Promega) sizes are indicated on the right-hand side.
Addition of extra 5′-UTR sequence reduces exon1A IRES activity
It had been previously shown in our laboratory that the cx43 exon1A contains IRES activity (35). Because translation efficiency was drastically reduced by the inclusion of exon1E in the transcript, we wanted to know whether it affected IRES activity. To test for IRES activity, a series of bicistronic constructs were made. They contained exon1A either with or without exon1E, or exon1AL directly upstream of the Firefly-luciferase coding sequence. In these bicistronic constructs, a Renilla luciferase gene is located upstream from the Firefly luciferase gene, separated by a prominent stem–loop to prevent ribosome scanning, as described previously (35). As indicated by the levels of relative Firefly/Renilla activities, inclusion of the exon1E sequence completely inhibited the IRES activity of exon1A (Figure 6A). As mentioned earlier, the secondary structure of the exon1A 5′-UTR not only changes quite drastically with the inclusion of exon1E but also becomes much more stable (Figure 5C).
Figure 6.
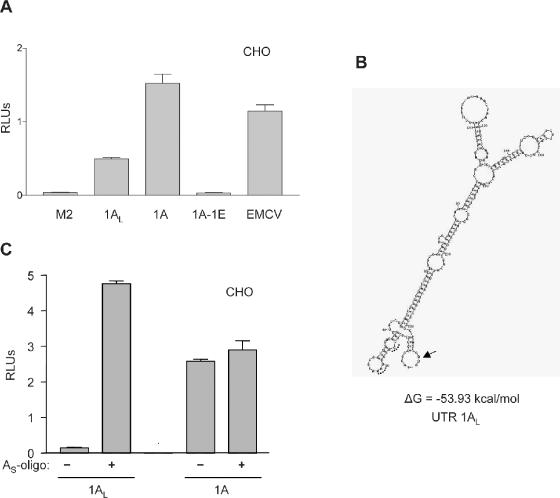
Exon1A IRES activity is inhibited by exons 1AS and 1E. (A) Relative luciferase activities of CHO cells transfected with 300 ng of SL2 bicistronic constructs. Exons 1A, 1AL or the 1A-1E combination were present immediately upstream of the Firefly-luciferase coding region. As a positive control for the IRES activity, an SL2-derived construct containing the viral EMCV element was used. The cx32 mutated 5′-UTR M2 was used as a negative control. (B) Structure of exon 1AL as predicted by the mFOLD program. The calculated free energy (ΔG) is indicated. The AUG translation start site is pointed by an arrow. Sequence annealing to oligo AS is labeled with a dotted line. (C) Relative luciferase activities of CHO cells transfected with 300 ng of SL2 bicistronic constructs containing either exon1A or exon1AL. A thioate oligo annealing to 5–27 nt of the exon1AL (oligo AS) was either included (0.5 μM) (+) or excluded (−) in the transfection mixtures and the growing medium.
Addition of the exon1AS sequence to exon1A significantly reduced its IRES activity (3-fold) both in CHO (Figure 6A) and HC-11 cells (data not shown). This result was surprising since exon1AL-containing monocistronic capped transcripts were translated with a similar efficiency (see Figure 5A and B). Still more surprising was the finding that the inclusion of the exon1AS neither altered the structure nor affected the stability of the secondary structure of the exon1A-containing 5′-UTR (cf. Figure 6B with Figure 5C). However, it was noticed that even though the general secondary structure did not change much, the region next to the start codon folded into an extra stem–loop, which could be responsible for less efficient ribosome recruitment. To test this hypothesis, the same transfection experiment was carried out in the presence of an antisense DNA complementary to nucleotides 5–27 nt from the 5′ end (oligoAS) in the 1AL 5′-UTR. If our model is correct and the accessibility of the AUG plays a role in the IRES activity, oligoAS should destabilize this extra stem–loop and therefore should recover exon1A IRES activity. As shown in Figure 6C, the presence of oligoAS not only recovered but even surpassed the levels of IRES activity of exon1A indicating that the accessibility of the region just upstream of the start codon is indeed required for exon1A-mediated IRES activity. As a negative control, oligoAS had no effect on IRES-mediated translation of the regular exon1A mRNA which lacks this sequence.
DISCUSSION
Cx43 transcription starts from three different promoters
This work establishes a new more complex structure of the cx43 gene, which includes three alternate promoters and six different exons and highlights the intricate regulation of the cx43 gene at multiple levels: transcription, pre-mRNA splicing and translation. Our first hint of the existence of new promoters (P2 and P3) came from the mapping of the transcription start sites of the newly identified exons 1B and 1C. The P2 sequence had been shown to be involved in protein binding through footprinting studies (41). It had also been proposed as a putative promoter mediating upregulation of cx43 expression through the Ras signaling pathway (40), which is known to converge with the cAMP response pathway (42). Interestingly enough, a screen of the P2 and P3 promoters for consensus transcription factor binding using the TFSearch program at http://www.bioinformatics.vg/ yielded cAMP response elements only in P2. In contrast, Sp1 and STAT factor binding sites were found in P3 but not in P2 suggesting that promoters P2 and P3 are regulated differently.
Because P1 is the most prominent promoter in all cell types tested and tissues analyzed, we cannot answer why the cx43 gene requires three alternative promoters. It is possible that the activities of the new promoters are needed by the cell for specific situations or used in a specific spatio-temporal manner, i.e. during processes such as cell differentiation and/or embryonic development. Alternatively they may be preferentially used in very scarce cell types, the activities of which would be diluted by the more commonly used P1 promoter when the whole tissue is analyzed. This latter possibility is supported by the regional differences observed in the heart. In situ hybridization studies using probes designed to anneal specifically either to exon1B or exon1C could help to narrow down the specific cell type(s) where P2 or P3 activities may be prominent. Thus, future studies on the mechanisms that regulate the transcription of the cx43 gene should include the new promoters P2 and P3. Our incomplete knowledge of cx43 gene regulation at the transcriptional level may be due to the fact that so far only P1 has been investigated.
Novel 5′-UTRs may regulate cx43 expression
In addition to transcription from three different promoters, alternative splicing provides a further level of complexity to modulate expression of cx43. The diversity found at the 5′ ends of cx43 mRNAs is likely to have an important effect on the translation of the mRNA and could possibly explain the reported discrepancies between steady-state mRNA levels and protein expression (13,17–23). In fact, even though the exon1A transcript seems to be the most abundant species in all tissues analyzed so far, we have shown that in ovary exon1A-1E and exon1C transcripts constitute ∼18.5% of the total cx43 mRNA, which is not a negligible amount. This amount may be even larger in certain ovarian cell types or in response to certain stimuli. In support of this argument, Kalma et al. (43) have recently shown that LH inhibits cx43 expression by reducing its rate of translation in the ovarian follicle. Our experiments show that these new 5′-UTR sequences lead to very different protein yields in various cell types (Figure 4A–C) indicating that they contain cis-RNA elements that respond to specific factors to modulate the translational efficiency of the transcript.
Effects on mRNA stability are unlikely because transfections with DNA constructs produced the same outcome as transfections with exogenously transcribed mRNA (cf. Figure 4A and B). Also, semiquantitative RT–PCR analysis indicated that mRNA levels remained constant throughout our experiments (Figure 4C). However, we cannot rule out the possibility that in vivo these newly identified 5′-UTRs act in combination with other sequences within the coding region or within the 3′-UTR to regulate mRNA turnover. Furthermore, they could influence the subcellular localization of the transcript which may be important for the final cx43 expression profile.
Exon1E inhibits cx43 translation
The inhibition of translation caused by inclusion of exon1E in the transcript is quite striking (Figures 4 and 5). It almost completely blocks both cap-dependent and cap-independent IRES-mediated translation of 1A-transcripts. This is not the first time that a unique splicing event within a 5′-UTR has been shown to introduce a translational control element. In fact, the presence of an additional exon (AS) included in the neuronal nitric-oxide synthase (nNOS) 5′-UTR by alternative splicing was shown to downregulate translation by stabilizing a stem–loop structure. In contrast to the cx43 exon1A-1E 5′-UTR, exonAS does not introduce an uAUG in the nNOS 5′-UTR (44).
As expected, the cx43 5′-UTRs with higher translation efficiencies, comprising exon1AS, exon1A and exon1AL, lack upstream AUG triplets, whereas those with lowest translational activities (exon1C and exon1A-1E 5′-UTRs) contain uAUGs with adequate Kozak consensus sequences. The remaining 5′-UTRs, which show intermediate translation efficiencies, contain either two or three additional uAUG, but none of them fit the Kozak consensus sequence for start of translation. The presence of uAUGs has been shown to downregulate translation mainly by ribosome stalling mechanisms. Some examples of genes regulated by upstream open reading frames (uORFs) include the AdoMetDC enzyme, the C/EBPβ transcription factor, the MDM2 oncoprotein and the Xenopus connexin41 gene (45–47). Less than 10% of eukaryotic mRNAs contain uAUGs within their transcript leader regions. They are more commonly found in genes that code for regulatory proteins. Cx43 is one of the earliest expressed connexins and has been shown to play a vital role during development (6,48). Thus, it should not be surprising to find uAUGs regulating the expression of the cx43 gene.
It has been known for years that the common exon1A cx43 mRNA is not very efficiently translated (49). Why then would a cell transcribe other mRNAs species that are even more poorly translated as it is the case of the cx43 exon1A-1E mRNA? At present we do not have a clear answer. Perhaps it provides the cell with a way to repress cx43 expression even further at times when promoter P1 cannot be shut off. Or it represents a strategy for the cell to get ready for a sudden upregulation of cx43 triggered by a factor that binds to the 5′-UTR thereby releasing the translational repression imposed by exon1E. Alternatively, exon1E's or any of the other exons' function in vivo could be dependent on trans-factors that are loaded onto the mRNA during splicing and later influence the efficiency of translation. In our transfection assays, we use processed mRNAs and therefore may miss these factors. In support of the latter argument, it has been shown that the nuclear history of a pre-mRNA can determine the translational activity of cytoplasmic mRNA (50,51).
Exon1AL represses cx43 IRES activity
Another cis-RNA element that seems to modulate translation of exon1A cx43 transcripts was found within exon1AL. In contrast to exon1E, the inhibition only affects cap-independent IRES-mediated mechanisms, it barely affects the secondary structure of the 5′-UTR and it does not introduce any uAUG (Figures 4 and 6). But somehow the sequence encompassing nucleotides 5–27 of exon 1AL from its 5′ end appears to be responsible for the inhibition since an oligonucleotide annealing to this sequence completely rescues the IRES activity (Figure 6). It is possible that the structure of the extra stem–loop introduced by the exon1AS sequence inhibits the IRES activity per se, or that the binding of an IRES repressor is obstructed by the presence of the anti-sense oligo. Whatever the mechanism might be we do not understand at present the biological significance of this cis-RNA element inhibitory of IRES activity.
Does cx43 transcription affect the downstream splicing events?
It was interesting to note that splicing of only certain exon combinations was allowed. And that exon choice seemed dependent on promoter usage and cell-type context. For example, exon1D appears linked only to exons 1BS or 1C, which are generated by transcription either from promoter P2 or P3 but it is never included in transcripts originating from promoter P1. Similarly, exon1E is only included in transcripts originating from promoter P1 but is never present in transcripts originating from either of the other two promoters. In addition, the frequency of exon skipping/inclusion and in consequence the relative abundance of 5′-UTRs containing one or two exons appears to be determined by the tissue source. This can be seen when the intensities of the faster moving bands (exon skipping) and the slower moving bands (exon inclusion) corresponding to exons 1B and 1C mRNAs are compared in Figure 1A. It is also indicated by the more frequent inclusion of exon1E in the atria, which is rarely included in the septum and which is skipped in the ventricle. On the basis of these observations, we would like to hypothesize that during splicing of the cx43 pre-mRNA exon choice is determined by the promoter at which transcription was initiated. Recently, splicing decisions have been shown to depend on the binding of transcription factors such as the estrogen receptors to synthetic promoters that contain estrogen response elements in a hormone-dependent manner, in a process termed co-transcriptional splicing (52,53). Ini, a novel transcription factor cloned in our laboratory which binds to the rat cx43 promoter and which seems to play a role during splicing in yeast (54,55) may be a good candidate to coordinate transcription and splicing and perhaps also translation of the cx43 gene.
We still do not understand why the cell requires nine different mRNA species coding for cx43. The key to the answer probably resides in the necessity for finely modulating intercellular communication in a cell type and context-dependent manner. Targeted ablation of each of the new exons in mice may lead to the generation of viable individuals with specific defects that may allow us to link the requirement of these new exons with certain physiological processes.
Acknowledgments
ACKNOWLEDGEMENTS
The authors wish to thank Dr Philip A. Marsden, Department of Medicine, University of Toronto, Canada, for the generous provision of the pSP-luc+[A]60 vector. We also thank Dr Diego G. Espinosa Heidmann, Department of Ophthalmology, University of Miami, USA, for his help in the dissection of mouse tissues. And Dr Akila Mayeda and Dr Richard Myers, Department of Biochemistry & Molecular Biology, University of Miami, USA, for critically reviewing the manuscript. This study was supported by a grant from the National Institutes of Health (HD34152) to R.W. DNA synthesis and sequence analysis was subsidized by the Sylvester Comprehensive Cancer Center through their DNA Core Lab.
DDBJ/EMBL/GenBank accession nos+ AY427554–AY427561, AY555736–AY555740
REFERENCES
- 1.Bruzzone R., White,T.W. and Paul,D.L. (1996) Connections with connexins: the molecular basis of direct intercellular signaling. Eur. J. Biochem., 23, 1–27. [DOI] [PubMed] [Google Scholar]
- 2.Bergoffen J., Scherer,S.S., Wang,S., Scott,M.O., Bone,L.J., Paul,D.L., Chen,K., Lensch,M.W., Chance,P.F. and Fischbeck,K.H. (1993) Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science, 262, 2039–2042. [DOI] [PubMed] [Google Scholar]
- 3.Abrahms C.K., Oh,S., Ri,Y. and Bargiello,T.A. (2000) Mutations in connexin 32: the molecular and biophysical bases for the X-linked form of Charcot-Marie-Tooth disease. Brain Res. Rev., 32, 203–214. [DOI] [PubMed] [Google Scholar]
- 4.Kelsell D.P., Di,W.L. and Houseman,M.J. (2001) Connexin mutations in skin disease and hearing loss. Am. J. Hum. Genet., 68, 559–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Severs N.J. (2002) Gap junction remodeling in heart failure. J. Card. Fail., 8 (Suppl. 6), S293–S299. [DOI] [PubMed] [Google Scholar]
- 6.Reaume A.G., de Sousa,P.A., Kulkarni,S., Langille,B.L., Zhu,D., Davies,T.C., Juneja,S.C., Kidder,G.M. and Rossant,J. (1995) Cardiac malformation in neonatal mice lacking connexin43. Science, 267, 1831–1834. [DOI] [PubMed] [Google Scholar]
- 7.Gutstein D.E., Morley,G.E. and Fishman,G.I. (2001) Conditional gene targeting of connexin43: exploring the consequences of gap junction remodeling in the heart. Cell Commun. Adhes., 8, 345–348. [DOI] [PubMed] [Google Scholar]
- 8.Eckardt D., Theis,M., Degen,J., Ott,T., van Rijen,H.V., Kirchhoff,S., Kim,J.S., de Bakker,J.M. and Willecke,K. (2004) Functional role of connexin43 gap junction channels in adult mouse heart assessed by inducible gene deletion. J. Mol. Cell. Cardiol., 36, 101–110. [DOI] [PubMed] [Google Scholar]
- 9.Loewenstein W.R. and Kanno,Y. (1966) Intercellular communication and the control of tissue growth: lack of communication between cancer cells. Nature, 209, 1248–1249. [DOI] [PubMed] [Google Scholar]
- 10.Trosko J.E. (2003) The role of stem cells and gap junctional intercellular communication in carcinogenesis. J. Biochem. Mol. Biol., 36, 43–48. [DOI] [PubMed] [Google Scholar]
- 11.Mesnil M. (2002) Connexins and cancer. Biol. Cell, 94, 493–500. [DOI] [PubMed] [Google Scholar]
- 12.Laird D.W., Fistouris,P., Batist,G., Alpert,L., Huynh,H.T., Carystinos,G.D. and Alaoui-Jamali,M.A. (1999) Deficiency of connexin43 gap junctions is an independent marker for breast tumors. Cancer Res., 59, 4104–4110. [PubMed] [Google Scholar]
- 13.Wiesen J.F. and Midgley,A.R.,Jr (1993) Changes in expression of connexin 43 gap junction messenger ribonucleic acid and protein during ovarian follicular growth. Endocrinology, 133, 741–746. [DOI] [PubMed] [Google Scholar]
- 14.Granot I. and Dekel,N. (1998) Cell-to-cell communication in the ovarian follicle: developmental and hormonal regulation of the expression of connexin43. Hum. Reprod., 13 (Suppl. 4), 85–97. [DOI] [PubMed] [Google Scholar]
- 15.Yu W., Dahl,G. and Werner,R. (1994) The connexin43 gene is responsive to oestrogen. Proc. R. Soc. Lond. B Biol. Sci., 255, 125–132. [DOI] [PubMed] [Google Scholar]
- 16.Granot I. and Dekel,N. (2002) The ovarian gap junction protein connexin43: regulation by gonadotropins. Trends Endocrinol. Metab., 13, 310–313. [DOI] [PubMed] [Google Scholar]
- 17.Civitelli R., Ziambaras,K., Warlow,P.M., Lecanda,F., Nelson,T., Harley,J., Atal,N., Beyer,E.C. and Steinberg,T.H. (1998) Regulation of connexin43 expression and function by prostaglandin E2 (PGE2) and parathyroid hormone (PTH) in osteoblastic cells. J. Cell. Biochem., 68, 8–21. [DOI] [PubMed] [Google Scholar]
- 18.Fishman G.I., Hertzberg,E.L., Spray,D.C. and Leinwand,L.A. (1991) Expression of connexin43 in the developing rat heart. Circ. Res., 68, 782–787. [DOI] [PubMed] [Google Scholar]
- 19.Fromaget C., el Aoumari,A., Dupont,E., Briand,J.P. and Gros,D. (1990) Changes in the expression of connexin 43, a cardiac gap junctional protein, during mouse heart development. J. Mol. Cell. Cardiol., 22, 1245–1258. [DOI] [PubMed] [Google Scholar]
- 20.Yancey S.B., Biswal,S. and Revel,J.P. (1992) Spatial and temporal patterns of distribution of the gap junction protein connexin43 during mouse gastrulation and organogenesis. Development, 114, 203–212. [DOI] [PubMed] [Google Scholar]
- 21.Ruangvoravat C.P. and Lo,C.W. (1992) Connexin 43 expression in the mouse embryo: localization of transcripts within developmentally significant domains. Dev. Dyn., 194, 261–281. [DOI] [PubMed] [Google Scholar]
- 22.Dahl E., Winterhager,E., Traub,O. and Willecke,K. (1995) Expression of gap junction genes, connexin40 and connexin43, during fetal mouse development. Anat. Embryol., 191, 267–278. [DOI] [PubMed] [Google Scholar]
- 23.Van Kempen M.J., Vermeulen,J.L., Moorman,A.F., Gros,D., Paul,D.L. and Lamers,W.H. (1996) Developmental changes of connexin40 and connexin43 mRNA distribution patterns in the rat heart. Cardiovasc. Res., 32, 886–900. [DOI] [PubMed] [Google Scholar]
- 24.Perez-Armendariz E.M., Saez,J.C., Bravo-Moreno,J.F., Lopez-Olmos,V., Enders,G.C. and Villalpando,I. (2003) Connexin43 is expressed in mouse fetal ovary. Anat. Rec., 271A, 360–367. [DOI] [PubMed] [Google Scholar]
- 25.Sullivan R., Ruangvoravat,C., Joo,D., Morgan,J., Wang,B.L., Wang,X.K. and Lo,C.W. (1993) Structure, sequence and expression of the mouse Cx43 gene encoding connexin 43. Gene, 130, 191–199. [DOI] [PubMed] [Google Scholar]
- 26.Sohl G. and Willecke,K. (2003) An update on connexin genes and their nomenclature in mouse and man. Cell Commun. Adhes., 10, 173–180. [DOI] [PubMed] [Google Scholar]
- 27.Neuhaus I.M., Dahl,G. and Werner,R. (1995) Use of alternate promoters for tissue-specific expression of the gene coding for connexin32. Gene, 158, 257–262. [DOI] [PubMed] [Google Scholar]
- 28.Sohl G., Gillen,C., Bosse,F., Gleichmann,M., Muller,H.W. and Willecke,K. (1996) A second alternative transcript of the gap junction gene connexin32 is expressed in murine Schwann cells and modulated in injured sciatic nerve. Eur. J. Cell Biol., 69, 267–275. [PubMed] [Google Scholar]
- 29.Sohl G., Theis,M., Hallas,G., Brambach,S., Dahl,E., Kidder,G. and Willecke,K. (2001) A new alternatively spliced transcript of the mouse connexin32 gene is expressed in embryonic stem cells, oocytes, and liver. Exp. Cell Res., 266, 177–186. [DOI] [PubMed] [Google Scholar]
- 30.Jacob A. and Beyer,E.C. (2001) Mouse connexin 45: genomic cloning and exon usage. DNA Cell Biol., 20, 11–19. [DOI] [PubMed] [Google Scholar]
- 31.Dupays L., Mazurais,D., Rucker-Martin,C., Calmels,T., Bernot,D., Cronier,L., Malassine,A., Gros,D. and Theveniau-Ruissy,M. (2003) Genomic organization and alternative transcripts of the human Connexin40 gene. Gene, 305, 79–90. [DOI] [PubMed] [Google Scholar]
- 32.Condorelli D.F., Parenti,R., Spinella,F., Trovato Salinaro,A., Belluardo,N., Cardile,V. and Cicirata,F. (1998) Cloning of a new gap junction gene (Cx36) highly expressed in mammalian brain neurons. Eur. J. Neurosci., 10, 1202–1208. [DOI] [PubMed] [Google Scholar]
- 33.Sohl G., Degen,J., Teubner,B. and Willecke,K. (1998) The murine gap junction gene connexin36 is highly expressed in mouse retina and regulated during brain development. FEBS Lett., 428, 27–31. [DOI] [PubMed] [Google Scholar]
- 34.Hudder A. and Werner,R. (2000) Analysis of a Charcot-Marie-Tooth disease mutation reveals an essential internal ribosome entry site element in the connexin-32 gene. J. Biol. Chem., 275, 34586–34591. [DOI] [PubMed] [Google Scholar]
- 35.Schiavi A., Hudder,A. and Werner,R. (1999) Connexin43 mRNA contains a functional internal ribosome entry site. FEBS Lett., 464, 118–122. [DOI] [PubMed] [Google Scholar]
- 36.Werner R. (2000) IRES elements in connexin genes: a hypothesis explaining the need for connexins to be regulated at the translational level. Life, 50, 173–176. [DOI] [PubMed] [Google Scholar]
- 37.Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 38.Zuker M. (2000) Calculating nucleic acid secondary structure. Curr. Opin. Struct. Biol., 10, 303–310. [DOI] [PubMed] [Google Scholar]
- 39.Zuker M. (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res., 31, 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carystinos G.D., Kandouz,M., Alaoui-Jamali,M.A. and Batist,G. (2003) Unexpected induction of the human connexin 43 promoter by the ras signaling pathway is mediated by a novel putative promoter sequence. Mol. Pharmacol., 63, 821–831. [DOI] [PubMed] [Google Scholar]
- 41.Echetebu C.O., Ali,M., Izban,M.G., MacKay,L. and Garfield,R.E. (1999) Localization of regulatory protein binding sites in the proximal region of human myometrial connexin 43 gene. Mol. Hum. Reprod., 5, 757–766. [DOI] [PubMed] [Google Scholar]
- 42.Richards J.S. (2001) New signaling pathways for hormones and cyclic adenosine 3′,5′-monophosphate action in endocrine cells. Mol. Endocrinol., 15, 209–218. [DOI] [PubMed] [Google Scholar]
- 43.Kalma Y., Granot,I., Galiani,D., Barash,A. and Dekel,N. (2004) Luteinizing hormone-induced connexin 43 down-regulation: inhibition of translation. Endocrinology, 145, 1617–1624. [DOI] [PubMed] [Google Scholar]
- 44.Newton D.C., Bevan,S.C., Choi,S., Robb,G.B., Millar,A., Wang,Y. and Marsden,P.A. (2003) Translational regulation of human neuronal nitric-oxide synthase by an alternatively spliced 5′-untranslated region leader exon. J. Biol. Chem., 278, 636–644. [DOI] [PubMed] [Google Scholar]
- 45.Meijer H.A. and Thomas,A.A. (2003) Ribosomes stalling on uORF1 in the Xenopus Cx41 5′ UTR inhibit downstream translation initiation. Nucleic Acids Res., 31, 3174–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meijer H.A. and Thomas,A.A. (2002) Control of eukaryotic protein synthesis by upstream open reading frames in the 5′-untranslated region of an mRNA. Biochem. J., 367, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morris D.R. and Geballe,A.P. (2000) Upstream open reading frames as regulators of mRNA translation. Mol. Cell. Biol., 20, 8635–8642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yancey S.B., Biswal,S. and Revel,J.P. (1992) Spatial and temporal patterns of distribution of the gap junction protein connexin43 during mouse gastrulation and organogenesis. Development, 114, 203–212. [DOI] [PubMed] [Google Scholar]
- 49.Werner R., Levine,E., Rabadan-Diehl,C. and Dahl,G. (1989) Formation of hybrid cell–cell channels. Proc. Natl Acad. Sci. USA, 86, 5380–5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Matsumoto K., Wassarman,K.M. and Wolffe,A.P. (1998) Nuclear history of a pre-mRNA determines the translational activity of cytoplasmic mRNA. EMBO J., 17, 2107–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nott A., Le Hir,H. and Moore,M.J. (2004) Splicing enhances translation in mammalian cells: an additional function of the exon junction complex. Genes Dev., 18, 210–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim V.N. and Dreyfus,G. (2001) Nuclear mRNA binding proteins couple pre-mRNA splicing and post-splicing events. Mol. Cells, 12, 1–10. [PubMed] [Google Scholar]
- 53.Auboeuf D., Honig,A., Berget,S.M. and O'Malley,B.W. (2002) Coordinate regulation of transcription and splicing by steroid receptor coregulators. Science, 298, 416–419. [DOI] [PubMed] [Google Scholar]
- 54.Oltra E., Pfeifer,I. and Werner,R. (2003) Ini, a small nuclear protein that enhances the response of the connexin43 gene to estrogen. Endocrinology, 144, 3148–3158. [DOI] [PubMed] [Google Scholar]
- 55.Oltra E., Verde,F., Werner,R. and D'Urso,G. (2004) A novel RING-finger-like protein Ini1 is essential for cell cycle progression in fission yeast. J. Cell. Sci., 117, 967–974. [DOI] [PubMed] [Google Scholar]