

Abstract
Central sleep apnoea (CSA)—the temporary absence or diminution of ventilator effort during sleep—is seen in a variety of forms including periodic breathing in infancy and healthy adults at altitude and Cheyne-Stokes respiration in heart failure. In most circumstances, the cyclic absence of effort is paradoxically a consequence of hypersensitive ventilatory chemoreflex responses to oppose changes in airflow, i.e. elevated loop gain, leading to overshoot/undershoot ventilatory oscillations. Considerable evidence illustrates overlap between CSA and obstructive sleep apnoea (OSA), including elevated loop gain in patients with OSA and the presence of pharyngeal narrowing during central apnoeas. Indeed, treatment of OSA, whether via CPAP, tracheostomy, or oral appliances, can reveal CSA, an occurrence referred to as complex sleep apnoea. Factors influencing loop gain include increased chemosensitivity (increased controller gain), reduced damping of blood gas levels (increased plant gain) and increased lung to chemoreceptor circulatory delay. Sleep-wake transitions and pharyngeal dilator muscle responses effectively raise the controller gain and therefore also contribute to total loop gain and overall instability. In some circumstances, for example apnoea of infancy and central congenital hypoventilation syndrome, central apnoeas are the consequence of ventilatory depression and defective ventilatory responses, i.e. low loop gain. The efficacy of available treatments for CSA can be explained in terms of their effects on loop gain, e.g. CPAP improves lung volume (plant gain), stimulants reduce the alveolar-inspired PCO2 difference, supplemental oxygen lowers chemosensitivity. Understanding the magnitude of loop gain and the mechanisms contributing to instability may facilitate personalised interventions for CSA.
Keywords: Central apnea, periodic breathing, ventilatory instability, loop gain
1. Introduction and Definitions
Central sleep apnoea (CSA) is characterized by the absence of airflow accompanying the cessation of ventilatory effort during sleep. In most forms, CSA is cyclic in nature manifesting as phases of hyper-ventilation alternating with apnoea: CSA can be classified into cyclic/periodic forms characterized by an oscillatory nature versus more sustained or irregular forms. CSA in periodic forms is seen commonly in preterm and term infants in the first weeks of life1, in adults sojourning to high altitude2, and in about a third of patients with heart failure3. CSA also occurs in ~5% of patients with obstructive sleep apnoea when pharyngeal patency is restored with intervention, a phenomenon termed complex sleep apnoea4–7. Periodic CSA is also seen in the form of idiopathic or primary CSA8. CSA is a common side effect of opioids9 and can be either periodic or “ataxic” in nature. Finally, we note that CSA can also occur in the form of isolated or prolonged, non-periodic central apnoeas, such as those seen with apnea of infancy/prematurity10, congenital central hypoventilation syndrome11 and respiratory muscle weakness12.
CSA is of clinical concern as it causes arterial oxygen desaturation, hypercapnia, post-apnoeic arousals from sleep, surges in ventilatory drive and negative intrathoracic pressure, sensation of dyspnoea, swings in arterial blood pressure and sympathetic excitation13–15. In patients with heart failure, CSA can promote cardiac arrhythmia, reduced cardiac function, and is strongly associated with mortality16, 17. In this review, we summarize the definitions of CSA, the mechanisms contributing to this affliction, and how it is transformed into stable breathing with treatment.
Criteria used to diagnose CSA vary somewhat depending on the patient population, the suspected aetiology and whether central hypopnoeas are scored. In adults, CSA is often defined as the presence of at least 5 central apnoeas per hour. In patients with heart failure, CSA is typically diagnosed as at least 15 events per hour with at least 50% of these being central events, but central hypopneas are included. Central hypopnoeas are generally defined as a 30–90% reduction in airflow due to a reduction in ventilatory effort; yet since effort is not directly measured (i.e. via oesophageal pressure/diaphragm EMG), non-invasive signals are used to infer the absence of pharyngeal obstruction. Signals indicative of pharyngeal obstruction include the flattening or scooping of the inspiratory flow shape, thoracoabdominal paradox (typically inward motion of the ribcage in concert with outward motion of the abdomen indicative of raised respiratory system resistance), or the presence of snoring indicating a flow-limited upper airway. It should be noted however that American Academy of Sleep Medicine scoring rules state that distinction between central and obstructive hypopnoeas is not required and can be challenging, and thus central events may be underreported18.
The duration of respiratory events is also employed in the diagnosis of CSA. In adults, as with obstructive events, apnoeas/hypopnoeas need to be at least 10 s in duration (~2–3 breaths). In preterm infants short apnoeas can yield severe desaturation or bradycardia (up to 50% reduction in saturation in 6 s), so the definition is broadened for neonates (≥20 s, or less if accompanying desaturation or bradycardia occurs), but hypopnoeas are typically ignored.
2. General Background
Introduction to ventilatory control
The primary features of the ventilatory control feedback loop that determine ventilatory effort are described as follows: Increased PCO2 and reduced PO2 are sensed at the carotid bodies located at the carotid bifurcation, making up the peripheral chemoreceptors. These chemoreceptors are well perfused and positioned to detect fast changes in PCO2/PO2 levels and are generally thought to dominate the response to transient changes in these variables. Increased PCO2 (in the form of H+) is also sensed at the medulla and pons, particularly at the retrotrapezoid nucleus in the ventrolateral medulla, making up the central chemoreceptors. The central chemoreceptors also typically dictate the baseline level of ventilatory effort. Both sets of inputs are integrated and act on the respiratory pattern generator to determine the strength and frequency of the efferent neural signals to the inspiratory muscles, namely the diaphragm and external intercostals. If the respiratory mechanics are normal, these efferent signals generate a level of inspiratory muscle pressure that yields a tidal volume excursion in direct proportion.
Traditionally, CSA has been considered a simple failure of this apparatus, described broadly as the controller, to generate ventilatory effort during sleep, akin to a severe yet temporary respiratory depression. Indeed, during normal sleep, ventilatory drive is reduced and reflex ventilatory responses to changes in PCO2 and PO2 are diminished19, 20 leading to the view that CSA is an extension of this diminution in ventilatory drive. Yet, as we discuss below, CSA in most cases is paradoxically the consequence of hypersensitivity of this chemoreceptor system.
Introduction to loop gain
To understand the negative feedback control system, we also consider the effect that ventilation has on PCO2 in the lungs and in the pulmonary venous blood leaving the lungs (arterial PCO2). An increase in arterial PCO2 will act on chemoreceptors to cause a rise in ventilation that will subsequently lead to a corrective reduction in arterial PCO2 i.e. as indicated by the metabolic hyperbola (the increase in ventilation is roughly proportional to the percent rise in PCO2). Normally, an equilibrium is achieved whereby ventilation and PCO2 levels are relatively steady. Yet on the time-scale of CSA, a fluctuation in ventilation such as a temporary hyperpnoea accompanying arousal can wash CO2 out of the lungs, leading to a temporary fall in arterial PCO2. After a circulation time, the hypocapnic arterial blood reaches the chemoreceptors, yielding a temporary reduction in ventilatory drive. But because of time delay between this disturbance and its effect on the control system, the ventilatory drive response will typically yield a ventilatory undershoot. This reflex undershoot will, in turn, raise alveolar/arterial PCO2 to elicit a delayed reflex ventilatory overshoot and so on.
The loop gain of this system, which describes the ratio of this ventilatory response (e.g. undershoot) to a prior disturbance (e.g. overshoot), ultimately determines whether the oscillation will grow into periodic central apnoeas (loop gain >1) or damp out (loop gain <1)21, 22.
It is also apparent that the ventilatory response to a disturbance has two distinct components (see Figure 1). Consider that the temporary rise in ventilation (5 L/min) washed CO2 out of the lungs such PCO2 falls by 5 mmHg (plant gain of 1 mmHg/L.min). After a lung to chemoreceptor circulatory delay, this reduction in PCO2 elicits a temporary 6 L/min reduction in ventilation (controller gain is 1.2 L/min/mmHg), such that the undershoot is larger than the initial disturbance (loop gain = 1.2) and periodic CSA will occur. CSA could be avoided if the CO2 damping was improved (lowered plant gain via increased lung volume) or if the chemoreflexes were less sensitive. Reducing circulatory delay also lowers loop gain.
Figure 1.
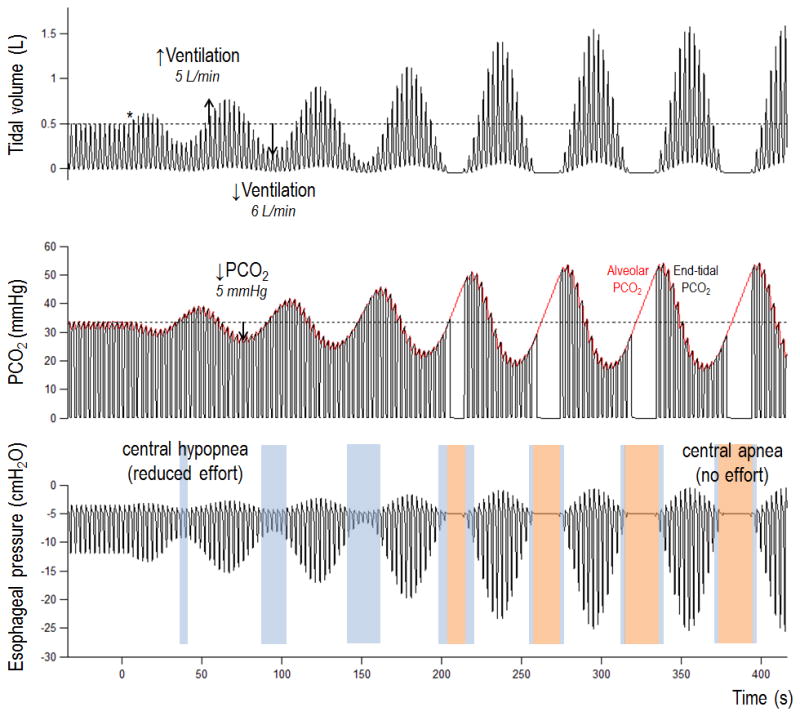
Computer simulation of CSA in heart failure illustrating the impact of loop gain >1. Loop gain was set to 1.2 at the time denoted by the asterisk. As CSA builds-up, each undershoot in ventilation is ~1.2-times larger than the prior ventilatory overshoot (see text for details). The example also illustrates the spectrum of central events, from mild hypopnoeas to more severe apnoeas (left to right).
Since controller gain describes the change in ventilation due to changes in PCO2 (or PO2), the controller gain can be modified by sleep state transitions. During the CSA cycle, as ventilatory drive rises, there is often an accompanying arousal that provides an additional increase in ventilatory drive20, 23–26 that in turn further increases the ventilatory overshoot. The effective gain relevant for the pathogenesis of CSA now becomes the chemoreflex response plus the arousal response per change in PCO2 throughout the cycle. Evidence that this effect plays a role includes: (1) CSA occurs more commonly at sleep onset or in light sleep (stage 1 non-REM) compared with during wake or deeper non-REM sleep27, 28, and (2) sedatives can improve CSA in some patients29. The increase in ventilation during arousal might relate to intrinsically greater ventilatory drive (for the same PCO2) observed during the awake state when compared to sleep, and possibly a reflex arousal ventilation, akin to a startle response26.
In principle, upper-airway effects may also promote CSA30–32. For example, changes in pharyngeal patency that occur in parallel with PCO2 will raise controller gain. In this case the overall controller gain (chemoresponsiveness) is equal to the intrinsic gain (chemosensitivity) multiplied by the effectiveness of the upper airway. In this context, some authors make a distinction between chemosensitivity (which reflects the ventilatory drive response to PCO2) and chemoresponsiveness (which reflects the change in actual ventilation in response to a PCO2 stimulus). The reason that the upper airway is considered a component of controller gain is that controller gain is essentially synonymous with chemoresponsiveness. This concept has two implications: An airway that tends to collapse as drive is reduced (i.e. via loss of muscle tone) will tend to yield a greater undershoot, thereby increasing the effective loop gain. Likewise, the same individual will exhibit a greater increase in ventilation as ventilatory drive is increased and muscle tone is re-established.
3. Pathogenesis in Patient Populations
Cheyne-Stokes respiration in congestive heart failure
Cheyne-Stokes respiration is perhaps the most widely recognised form of CSA, occurring in a substantial proportion of patients with heart failure (see example trace taken from a recent study33 in Figure 2). A reduced cardiac output and resultant increase in the circulatory delay between the lungs and chemoreceptors is believed to play an important role in the pathogenesis of CSA. Indeed, patients with a reduced cardiac output, worsened systolic function, atrial fibrillation, and prolonged lung to chemoreceptor delays are more likely to exhibit CSA34–37. Furthermore, heart failure therapies such as cardiac resynchronization and afterload reduction improve ventilatory stability38, 39. While many patients with CSA have prolonged circulatory delays, the presence of prolonged delay alone does not appear sufficient to generate CSA, highlighting the importance of increased chemosensitivity40–43.
Figure 2.

Illustrative example trace of CSA during non-rapid eye movement (NREM) sleep in a male patient with heart failure and atrial fibrillation. Note the resolution of CSA with the transition to REM.
The specific causes of increased chemosensitivity in CSA are unclear, and may differ between individuals40, 43. Elevated pulmonary capillary pressures are associated with presence of CSA and its severity, while diuresis improves CSA within individual patients34, 39, 44–47. Overnight shifts in fluid from the legs may provide another source of pulmonary congestion, with ventilatory instability more likely with increasing volume of mobilized fluid48. Left atrial distension may also drive increased chemosensitivity and CSA irrespective of pulmonary vascular congestion49. Notably, a few studies have called into question the role of pulmonary congestion in development of CSA50, 51. Recent evidence from animal models suggests abnormalities at the level of the carotid body may play an important role, leading to both enhanced chemosensitivity and sympathetic hypertonia, which might propagate CSA and worsen heart failure52, 53. Identifying the precise sources of enhanced chemosensitivity will likely provide for new therapeutic targets for CSA. For example, pharmacological reversal of the signalling mechanisms causing carotid chemoreflex hyperactivity (e.g. purinergic54) might help suppress CSA.
Idiopathic central sleep apnoea
The presence of CSA in patients without any identifiable cardiac or neurological cause is termed idiopathic CSA. The cycling period in idiopathic CSA is ~30–40 seconds and appears to be driven largely by elevated chemosensitivity to PCO240, 55. Arousals typically occur at the peak of hyperventilation and likely contribute to ventilatory overshoot, enhancing chemoresponsiveness56. Circulatory delay is by definition normal in these patients and therefore unlikely to contribute to CSA.
Periodic breathing at altitude
At high altitude, low total barometric pressure with a relatively stable fraction of oxygen results in a decreased PO2, leading to CSA of a periodic nature57, 58. Although there is variation in the altitude at which CSA will develop, CSA occurs in virtually all lowlanders at arrival to altitude59. Hypoxia promotes instability via hypoxic augmentation of the chemoreflex response to CO2 and via an increase in hypoxic chemoresponsiveness while on a steeper portion of the hypoxic ventilatory response curve60. In contrast to sojourners, highlanders are less susceptible to CSA, suggesting that genetic or adaptive factors likely play an important role in these responses57. Interestingly, the hypoxic chemosensitivity increases over days-to-weeks after arrival at altitude, facilitating an increase in ventilation and improvement in PO2, but further increasing loop gain59, 61, 62 (i.e. differences in instability are typically attributable to differences in the hypoxic ventilatory response rather than those in the magnitude of arterial hypoxemia). This instability appears to persist in lowlanders living at altitude beyond 1 year63. Adaptive increases in the hypoxic ventilatory response with acclimatization appear to improve symptoms of acute mountain sickness (via raising PO2) but come at the cost of exacerbating CSA64. CSA is also linked with hypoxemia and pulmonary hypertension accompanying chronic mountain sickness, likely acting via hypoxemic effects on chemosensitivity in such cases65.
Other factors beyond chemosensitivity may play some role in CSA at altitude. Decreases in plant gain due to hyperventilation with resulting hypocapnia, and increases in cardiac output with short circulatory delays would act as compensatory mechanisms to stabilize breathing62, 66 and thus individuals with less strong compensatory mechanisms may have more severe CSA. Subclinical pulmonary oedema appears to occur relatively frequently in sojourners and might lower lung volumes which would exacerbate CSA67. Recent research has suggested that cerebral blood flow reactivity may be important in ventilatory instability at altitude via regulation (damping) of cerebral PCO2 levels 59.
Periodic breathing in newborn infants
Periodic breathing is almost ubiquitous in term infants and those born prematurely in the first weeks of life and its high prevalence has led to the assumption that it is non-pathological68. However, in some cases, periodic breathing in preterm infants can lead to profound oxygen desaturation14 that may have serious consequences. Treatment of periodic breathing in such cases is warranted in light of the associations between reduced oxygen levels and mortality in neonatal intensive care69. Periodic breathing is rare in the first days after birth but becomes progressively more prevalent over the next 2–4 weeks before a steady decline over the first year1, 70. The increased CSA prevalence likely results from the raised hypoxic chemosensitivity that accompanies chemoreceptor “resetting” in the days after birth71, followed later by a reduced chemosensitivity with development. CSA is also thought to be due partly to hypoxemia72, 73 consequent to ventilation-perfusion heterogeneity in the developing lungs. Lower lung volumes (relative to body weight/metabolic rate) especially in preterm infants are also expected to play a role in some infants74.
Opioid-induced central sleep apnoea
Use of opioids has become a major public health issue that has garnered considerable media attention. Studies suggest that roughly a third of chronic opioid administered patients have some form of central sleep apnoea75. This breathing pattern has several important characteristics: First, opioids are sometimes associated with bradypnoea i.e. very low respiratory rates and attendant hypoventilation, hypercapnia and hypoxemia76. Second, breathing is often erratic in nature, often described as ‘ataxic’9, attributable to effects at the central respiratory pattern generator77. Third, severe CSA in opioid users often exhibits a periodic pattern remarkably similar to CSA at altitude, with a cycle period similar to idiopathic CSA (~30–40 s)76 suggesting that elevated loop gain is responsible. Detailed mechanistic studies in chronic opioid patients are relatively sparse but possible causes of elevated loop gain include: 1) an elevated alveolar PCO2 which would be expected to reduce CO2 damping (elevated plant gain), 2) severe hypoventilation and concomitant hypoxemia76 that will presumably raise hypoxic chemosensitivity, and 3) a doubling of the slope of the hypoxic ventilatory response independent of the prevailing hypoxemia78. These factors likely combine to yield an elevated loop gain and promote CSA. An increased loop gain with opioids appears paradoxical given that ventilatory drive is typically reduced, highlighting the important distinction between baseline ventilatory drive and the responsiveness to changes in drive.
Treatment of opioid induced CSA is challenging. CPAP may improve sleep apnoea in some patients, but often fails to improve it in others76, 79. Opioid effects on CSA are thought to be dose dependent such that breathing pattern may actually normalize with reduced doses80. Adaptive servo-controlled ventilation has been used effectively in small studies, but the use of this therapy clinically in this context remains to be defined 76, 79. There is a mechanistic basis for use of ventilatory stimulants (acetazolamide) or oxygen but the efficacy of such therapies is unproven. Of particular interest is a case study illustrating that acetazolamide improved opioid-induced CSA in a patient on CPAP therapy, but oxygen was ineffective81.
Overlap between and obstructive and central sleep apnoea
Obstructive sleep apnoea is a very common condition affecting roughly 10% of the US population. The details of obstructive sleep apnoea are covered elsewhere in this Review Series, but we review important concepts to give a more complete view of central apnoea pathogenesis. Obstructive sleep apnoea is known to be due to multiple underlying mechanisms: While some patients have primarily an anatomical problem, others have issues with control of upper airway dilator muscles while still others have unstable ventilatory control (high loop gain)82, 83. Some patients have multiple mechanisms underlying apnoea. In theory, treatment directed at the underlying mechanism is likely to yield improvement in apnoea using a personalized approach.
Among obstructive sleep apnoea patients with high loop gain, the question arises as to why they develop obstructive sleep apnoea rather than CSA. In reality, many patients have features of both obstructive sleep apnoea and CSA or can change features during the course of an overnight recording, emphasizing that the distinction between these two conditions can be challenging. A number of lines of evidence suggest considerable overlap between obstructive sleep apnoea and CSA:
Patients with more severe obstructive sleep apnoea have been shown to have higher loop gain than milder obstructive sleep apnoea or controls using multiple different measurement techniques82, 83. Presumably the fluctuations in output from the central pattern generator lead to upper airway collapse when output to the upper airway dilator muscles is at its nadir in those who are anatomically predisposed.
Agents which lower loop gain such as oxygen and acetazolamide improve obstructive sleep apnoea in some individuals, particularly those with high loop gain84–86.
Tracheostomy in patients with obstructive sleep apnoea can transform obstructive sleep apnoea into CSA, i.e. complex sleep apnoea (see below).
In patients with CSA, the forced oscillatory technique and direct visualization of the airway have shown evidence of upper airway narrowing/closure during central apneas and hypopneas31, 32.
Some patients have mixed apnoeas with features of both obstructive sleep apnoea and CSA e.g. patients can have minimal respiratory effort during a portion of a respiratory event but evidence of obstructive physiology during the same respiratory event. Thus, some patients are difficult to classify as strictly obstructive sleep apnoea or CSA.
REM sleep is a period of blunted chemosensitivity that often is associated with improvements in CSA including Cheyne Stokes breathing or periodic breathing at high altitude. Similarly, some obstructive sleep apnoea patients have worse breathing disturbance in NREM, presumably driven by a higher loop gain in this sleep stage.
Although obstructive sleep apnoea and CSA clearly have some similar features, this overlap is particularly evident in people with congestive heart failure. In some patients, investigators have observed an overnight conversion of obstructive sleep apnoea to CSA in conjunction with a reduction in PCO2 and an increase in circulatory delay suggesting an overnight deterioration of cardiac function and rise in chemosensitivity87. CSA can also convert to obstructive sleep apnoea with improvement in cardiac function and circulatory delay over time88, and can also revert to obstructive sleep apnoea with cardiac transplant89. CSA and obstructive sleep apnoea can both be improved with heart failure treatment in the form of cardiac resynchronization therapy90. Given the evidence of considerable overlap, the use of strict cut-offs based on percentage of central events (e.g. >50%) to define CSA may be inappropriate. We therefore favour the use of the more general term sleep apnea to encompass both central and obstructive sleep apnea manifestations of disordered ventilatory control.
Treatment emergent central sleep apnoea: “Complex sleep apnoea”
The overlap between CSA and obstructive sleep apnoea is particularly relevant for patients with obstructive sleep apnoea who exhibit CSA when the upper airway is made patent with therapies. This phenomenon has been labelled complex sleep apnoea. The conversion from obstructive sleep apnoea to CSA was reported in classic studies in the context of tracheostomy. Likewise, during CPAP titrations, removal of upper airway obstruction results in CSA in some patients. This form of CSA often resolves over time with ongoing CPAP treatment, although in individuals with higher loop gain, complex sleep apnoea may persist. Oral appliance therapy can also yield CSA in patients treated for obstructive sleep apnoea.4–7
Although unproven, a likely mechanism of complex sleep apnoea involves the relief of inspiratory flow limitation. In some patients with pharyngeal compromise, increasing ventilatory drive with increasing CO2 may not yield increased airflow due to the prevailing upper airway mechanics (i.e. ineffective muscle responses). That is, the presence of flow limitation can markedly reduce the controller gain (i.e. no rise in airflow for increasing effort). When inspiratory flow limitation is then relieved with treatment, a high chemosensitivity can be unmasked to yield CSA. In this context, application of CPAP would increase chemoresponsiveness without changing chemosensitivity per se.
Depressed ventilatory drive and chemoresponsiveness as a mechanism of CSA
In contrast to the elevated loop gain mechanism of periodic breathing, prolonged apnoeas consequent to reduced ventilatory drive and depressed chemoresponsiveness (i.e. extremely low loop gain) occur in some cases, highlighting the importance of having an intact chemoreflex control system. Infants with prolonged apnoeas (apnoea of prematurity, apnoea of infancy, and apparent life threatening events)—as opposed to periodic breathing—have been found to have a reduced chemosensitivity and a depressed ventilatory drive10 consistent with a reduced or less robust ventilatory drive response to apnoea/hypoventilation. The neonatal tendency to respond to hypoxia with further ventilatory depression (i.e. negative chemoresponsiveness) may serve to further reinforce an event once initiated. Patients with central congenital hypoventilation syndrome exhibit profound hypoventilation and hypoxemia during sleep consequent to reduced ventilatory drive and virtually non-existent sleep-related ventilatory responses to CO2 and hypoxia11. In patients with neuromuscular weakness, apnoeas may be seen particularly during REM sleep due to a combination of low chemosensitivity and severe muscle weakness during REM atonia12. While the absence of effort during these events classifies them as central, some have advocated for the terminology “pseudo-central” or “diaphragmatic” to emphasize the primary role of muscle weakness.
4. Personalising Treatments
The loop gain concept integrates several different components into a combined parameter which is helpful in determining overall stability and the likelihood of CSA. Several individual components (chemosensitivity, plant gain, and circulatory delay) interact in a multiplicative manner to yield the overall loop gain. Thus, in many cases, the improvement of any given component, strictly speaking, does not require knowledge of the particular mechanism of CSA. For example, instability caused by elevated circulatory delay could be resolved with CPAP/or lateral positioning to improve lung volume (plant gain).
However, we emphasise that identification of the underlying mechanism may help to guide therapy for a number of reasons. The isolation of an underlying abnormality is critical since such abnormalities may well be the most amenable to improvement with therapy. For instance, a normal circulatory delay may be difficult to improve, but a markedly elevated chemosensitivity may well respond to appropriate interventions (oxygen/pharmacological agents). In addition, the potential for toxicity of an intervention might be heightened if a normal value is being manipulated. For example, a patient with a normal chemosensitivity (but high plant gain and increased delays) may be more likely to exhibit hypoventilation in certain circumstances (e.g. REM) if efforts to suppress chemosensitivity are successful.
The overall loop gain can also be considered the sum of the separate feedback loops for PO2 and PCO221. Thus, identifying whether increased chemosensitivity is driven by hypoxic versus hypercapnic hyperreflexia may have important implications for therapy. For example, CSA driven by hypoxic feedback (e.g. infants/altitude) is expected to be readily resolved with supplemental oxygen administration. However, supplemental oxygen is effective in some heart failure patients with CSA but not in others91, consistent with findings that some patients with heart failure have increased responses to PO2 whereas others have increased responses to PCO243. Thus, we view a mechanistic understanding of control of breathing critical for meaningful progress towards individualized therapies to occur.
Finally, the magnitude of loop gain is also important, regardless of the particular factor destabilizing breathing. For example, it is harder to lower loop gain to below 1 and resolve CSA in a patient with a loop gain of 1.9 at baseline than if it is 1.1. Recognising the magnitude of instability may inform which treatments have the scope to resolve CSA22, 33. Clinicians could combine interventions if an individual intervention does not have sufficient potential, e.g. CPAP plus acetazolamide, or bed elevation plus oxygen. Further investigation along these lines is needed.
5. Physiological Mechanisms of Treatments
The following interventions are considered in terms of their mechanistic effects on ventilatory control:
Continuous positive airway pressure (CPAP) undoubtedly increases lung volume and consequently improves CO2 damping (reducing plant gain)22. There is little direct evidence that CPAP improves circulatory delay, but cardiac function can be improved and a preferential benefit may occur in those with increased filling pressures in whom there is a mechanistic basis for improved cardiac output20,134.
Supplemental oxygen has a profound impact on CSA in infants92 and at altitude, and improves CSA in some patients with heart failure91. Increased arterial PO2 is known to lower carotid-body chemosensitivity93. Other beneficial effects on stability are unlikely, as supplemental oxygen is expected to increase plant gain (for feedback control of PO2) and circulatory delay.
Respiratory stimulants (e.g. inhaled carbon dioxide, rebreathing, acetazolamide, theophylline) act to increase CO2 damping (reduce “plant gain”) by making alveolar PCO2 less susceptible to changes due to fluctuations in ventilation33. This phenomenon is encapsulated by a reduction in the difference between alveolar and inspired PCO2. For patients with CSA due to ventilatory depression rather than high loop gain, respiratory stimulants may act to prevent apnoea by restoring baseline ventilatory drive.
Sleeping position can have as profound an impact on CSA as CPAP. Sleeping lateral or with bed elevation can improve CSA and is likely to act in part by raising lung volume27, 94. Improvements in upper airway collapsibility may also contribute in some individuals.
Bi-level positive airway pressure with a backup rate and phrenic nerve stimulation seek to provide an additional non-chemical source of actual ventilation independent of a subject’s own ventilatory drive. The expected effect on loop gain is complex: Loop gain will be reduced by increasing ventilation and lowering the alveolar-inspired PCO2 gradient. A baseline source of ventilation will minimise the possible amplitude of hypopnoea for any reduction in ventilatory drive, thereby effectively lowering controller gain95, 96. For patients with CSA due to ventilatory depression, these interventions act to prevent apnoea by providing an additional source of ventilatory effort.
Dynamic interventions, including adaptive servo-controlled ventilation97 and dynamic inspired CO2 delivery98, seek to clamp ventilation or PCO2 levels to resolve CSA. If these interventions were completely effective, loop gain would be lowered to zero.
In treating CSA that is secondary to heart failure or opioids, we note that the primary focus must be on resolving the underlying pathophysiology causing CSA.
6. Conclusions
Central sleep apnoea, defined as the temporary absence of ventilatory effort during sleep, is seen in a variety of forms across the life span. Paradoxically, in most cases, the reduction in ventilatory effort is a consequence of hypersensitive ventilatory effort responses to changes in PCO2/PO2, i.e. elevated loop gain (overshoot/undershoot). In other patients, central apnoeas are the consequence of depressed/absent ventilatory effort responses, i.e. extremely low loop gain. Treatments for CSA can be explained in terms of effects on loop gain, e.g. CPAP improves lung volume (plant gain), stimulants reduce the alveolar-inspired CO2 difference, supplemental oxygen lowers chemosensitivity. A greater understanding of the pathophysiology in subgroups of patients may provide insight into which interventions will have the greatest beneficial impact.
Acknowledgments
Dr. Orr is PI on NIH F32 HL131306 and supported by L30 HL129451. Dr. Sands was supported by the National Health and Medical Research Council of Australia (1053201, 1038402), Menzies Foundation, American Heart Association (15SDG25890059) and American Thoracic Society Foundation, and is co-investigator on NIH R01 HL128658. Dr. Malhotra is PI on NIH R01 HL085188, K24 HL132105, and co-investigator on R21 HL121794, R01 HL119201, R01 HL081823. As an Officer of the American Thoracic Society, Dr. Malhotra has relinquished all outside personal income since 2012. ResMed, Inc. provided a philanthropic donation to the UC San Diego in support of a sleep centre.
Biography
Dr. Jeremy Orr is an Assistant Clinical Professor at the University of California San Diego. His research investigates control of breathing mechanisms in sleep disordered breathing with a particular focus on patients with pulmonary hypertension, chronic lung disease, and neuromuscular disease. Professor Atul Malhotra is the Chief of the Division of Pulmonary and Critical Care Medicine, University of California San Diego. His research interests include the pathophysiology and cardiovascular/metabolic consequences of sleep apnoea and mechanical ventilation in acute respiratory distress syndrome. Dr. Scott Sands is Instructor in Medicine at the Brigham and Women’s Hospital and Harvard Medical School. His research investigates sleep apnea pathophysiology with a focus on the development of methods for personalised treatment.
References
- 1.Glotzbach SF, Baldwin RB, Lederer NE, Tansey PA, Ariagno RL. Periodic breathing in preterm infants: incidence and characteristics. Pediatrics. 1989;84:785–92. [PubMed] [Google Scholar]
- 2.West JB, Peters RM, Aksnes G, Maret KH, Milledge JS, Schoene RB. Nocturnal periodic breathing at altitudes of 6300 and 8050 m. Journal of Applied Physiology. 1986;61:280–7. doi: 10.1152/jappl.1986.61.1.280. [DOI] [PubMed] [Google Scholar]
- 3.MacDonald M, Fang J, Pittman SD, White DP, Malhotra A. The current prevalence of sleep disordered breathing in congestive heart failure patients treated with beta-blockers. J Clin Sleep Med. 2008;4:38–42. [PMC free article] [PubMed] [Google Scholar]
- 4.Javaheri S, Smith J, Chung E. The prevalence and natural history of complex sleep apnea. J Clin Sleep Med. 2009;5:205–11. [PMC free article] [PubMed] [Google Scholar]
- 5.Onal E, Lopata M. Periodic breathing and the pathogenesis of occlusive sleep apneas. Am Rev Respir Dis. 1982;126:676–80. doi: 10.1164/arrd.1982.126.4.676. [DOI] [PubMed] [Google Scholar]
- 6.Kuzniar TJ, Kovacevic-Ristanovic R, Freedom T. Complex sleep apnea unmasked by the use of a mandibular advancement device. Sleep Breath. 2011;15:249–52. doi: 10.1007/s11325-010-0459-8. [DOI] [PubMed] [Google Scholar]
- 7.Lehman S, Antic NA, Thompson C, Catcheside PG, Mercer J, McEvoy RD. Central sleep apnea on commencement of continuous positive airway pressure in patients with a primary diagnosis of obstructive sleep apnea-hypopnea. J Clin Sleep Med. 2007;3:462–6. [PMC free article] [PubMed] [Google Scholar]
- 8.Xie A, Rutherford R, Rankin F, Wong B, Bradley TD. Hypocapnia and increased ventilatory responsiveness in patients with idiopathic central sleep apnea. American Journal of Respiratory and Critical Care Medicine. 1995;152:1950–5. doi: 10.1164/ajrccm.152.6.8520761. [DOI] [PubMed] [Google Scholar]
- 9.Farney RJ, Walker JM, Cloward TV, Rhondeau S. Sleep-disordered breathing associated with long-term opioid therapy. Chest. 2003;123:632–9. doi: 10.1378/chest.123.2.632. [DOI] [PubMed] [Google Scholar]
- 10.Gerhardt T, McCarthy J, Bancalari E. Effects of aminophylline on respiratory center and reflex activity in premature infants with apnea. Pediatric research. 1983;17:188–91. doi: 10.1203/00006450-198303000-00003. [DOI] [PubMed] [Google Scholar]
- 11.Paton JY, Swaminathan S, Sargent CW, Keens TG. Hypoxic and Hypercapnic Ventilatory Responses in Awake Children with Congenital Central Hypoventilation Syndrome. American Review of Respiratory Disease. 1989;140:368–72. doi: 10.1164/ajrccm/140.2.368. [DOI] [PubMed] [Google Scholar]
- 12.White J, Drinnan M, Smithson A, Griffiths C, Gibson G. Respiratory muscle activity and oxygenation during sleep in patients with muscle weakness. European Respiratory Journal. 1995;8:807–14. [PubMed] [Google Scholar]
- 13.van de Borne P, Oren R, Abouassaly C, Anderson E, Somers VK. Effect of Cheyne-Stokes respiration on muscle sympathetic nerve activity in severe congestive heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol. 1998;81:432–6. doi: 10.1016/s0002-9149(97)00936-3. [DOI] [PubMed] [Google Scholar]
- 14.Poets CF, Southall DP. Patterns of oxygenation during periodic breathing in preterm infants. Early Hum Dev. 1991;26:1–12. doi: 10.1016/0378-3782(91)90038-5. [DOI] [PubMed] [Google Scholar]
- 15.Naughton MT, Benard DC, Liu PP, Rutherford R, Rankin F, Bradley TD. Effects of nasal CPAP on sympathetic activity in patients with heart failure and central sleep apnea. Am J Respir Crit Care Med. 1995;152:473–9. doi: 10.1164/ajrccm.152.2.7633695. [DOI] [PubMed] [Google Scholar]
- 16.Leung RS, Diep TM, Bowman ME, Lorenzi-Filho G, Bradley TD. Provocation of ventricular ectopy by cheyne-stokes respiration in patients with heart failure. Sleep. 2004;27:1337–43. doi: 10.1093/sleep/27.7.1337. [DOI] [PubMed] [Google Scholar]
- 17.Arzt M, Schulz M, Wensel R, Montalvan S, Blumberg FC, Riegger GA, Pfeifer M. Nocturnal continuous positive airway pressure improves ventilatory efficiency during exercise in patients with chronic heart failure. Chest. 2005;127:794–802. doi: 10.1378/chest.127.3.794. [DOI] [PubMed] [Google Scholar]
- 18.Berry RB, Budhiraja R, Gottlieb DJ, Gozal D, Iber C, Kapur VK, Marcus CL, Mehra R, Parthasarathy S, Quan SF, Redline S, Strohl KP, Ward SLD, Tangredi MM. Rules for Scoring Respiratory Events in Sleep: Update of the 2007 AASM Manual for the Scoring of Sleep and Associated Events: Deliberations of the Sleep Apnea Definitions Task Force of the American Academy of Sleep Medicine. Journal of clinical sleep medicine: JCSM: official publication of the American Academy of Sleep Medicine. 2012;8:597–619. doi: 10.5664/jcsm.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Douglas NJ, White DP, Weil JV, Pickett CK, Martin RJ, Hudgel DW, Zwillich CW. Hypoxic ventilatory response decreases during sleep in normal men. Am Rev Respir Dis. 1982;125:286–9. doi: 10.1164/arrd.1982.125.3.286. [DOI] [PubMed] [Google Scholar]
- 20.Douglas NJ, White DP, Weil JV, Pickett CK, Zwillich CW. Hypercapnic ventilatory response in sleeping adults. Am Rev Respir Dis. 1982;126:758–62. doi: 10.1164/arrd.1982.126.5.758. [DOI] [PubMed] [Google Scholar]
- 21.Khoo MC, Kronauer RE, Strohl KP, Slutsky AS. Factors inducing periodic breathing in humans: a general model. J Appl Physiol. 1982;53:644–59. doi: 10.1152/jappl.1982.53.3.644. [DOI] [PubMed] [Google Scholar]
- 22.Sands SA, Edwards BA, Kee K, Turton A, Skuza EM, Roebuck T, O’Driscoll DM, Hamilton GS, Naughton MT, Berger PJ. Loop gain as a means to predict a positive airway pressure suppression of Cheyne-Stokes respiration in patients with heart failure. Am J Respir Crit Care Med. 2011;184:1067–75. doi: 10.1164/rccm.201103-0577OC. [DOI] [PubMed] [Google Scholar]
- 23.Trinder J, Ivens C, Kleiman J, Kleverlaan D, White DP. The cardiorespiratory activation response at an arousal from sleep is independent of the level of CO(2) J Sleep Res. 2006;15:174–82. doi: 10.1111/j.1365-2869.2006.00513.x. [DOI] [PubMed] [Google Scholar]
- 24.Skatrud JB, Dempsey JA. Interaction of sleep state and chemical stimuli in sustaining rhythmic ventilation. Journal of applied physiology: respiratory, environmental and exercise physiology. 1983;55:813–22. doi: 10.1152/jappl.1983.55.3.813. [DOI] [PubMed] [Google Scholar]
- 25.Mahamed S, Ali AF, Ho D, Wang B, Duffin J. The contribution of chemoreflex drives to resting breathing in man. Exp Physiol. 2001;86:109–16. doi: 10.1113/eph8602090. [DOI] [PubMed] [Google Scholar]
- 26.Horner RL, Rivera MP, Kozar LF, Phillipson EA. The ventilatory response to arousal from sleep is not fully explained by differences in CO(2) levels between sleep and wakefulness. J Physiol. 2001;534:881–90. doi: 10.1111/j.1469-7793.2001.00881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Szollosi I, Roebuck T, Thompson B, Naughton MT. Lateral sleeping position reduces severity of central sleep apnea/Cheyne-Stokes respiration. Sleep. 2006;29:1045–51. doi: 10.1093/sleep/29.8.1045. [DOI] [PubMed] [Google Scholar]
- 28.Berssenbrugge A, Dempsey J, Iber C, Skatrud J, Wilson P. Mechanisms of hypoxia-induced periodic breathing during sleep in humans. J Physiol. 1983;343:507–26. doi: 10.1113/jphysiol.1983.sp014906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quadri S, Drake C, Hudgel DW. Improvement of idiopathic central sleep apnea with zolpidem. J Clin Sleep Med. 2009;5:122–9. [PMC free article] [PubMed] [Google Scholar]
- 30.Hudgel DW, Chapman KR, Faulks C, Hendricks C. Changes in inspiratory muscle electrical activity and upper airway resistance during periodic breathing induced by hypoxia during sleep. Am Rev Respir Dis. 1987;135:899–906. doi: 10.1164/arrd.1987.135.4.899. [DOI] [PubMed] [Google Scholar]
- 31.Jobin V, Rigau J, Beauregard J, Farre R, Monserrat J, Bradley TD, Kimoff RJ. Evaluation of upper airway patency during cheyne-stokes breathing in heart failure patients. Eur Respir J. 2012 doi: 10.1183/09031936.00060311. [DOI] [PubMed] [Google Scholar]
- 32.Badr MS, Toiber F, Skatrud JB, Dempsey J. Pharyngeal narrowing/occlusion during central sleep apnea. J Appl Physiol. 1995;78:1806–15. doi: 10.1152/jappl.1995.78.5.1806. [DOI] [PubMed] [Google Scholar]
- 33.Sands SA, Edwards BA, Kee K, Stuart-Andrews CR, Skuza EM, Roebuck T, Turton A, Hamilton GS, Naughton MT, Berger PJ. Control Theory Prediction of Resolved Cheyne-Stokes Respiration in Heart Failure. European Respiratory Journal. 2016 doi: 10.1183/13993003.00615-2016. In Press. [DOI] [PubMed] [Google Scholar]
- 34.Oldenburg O, Bitter T, Wiemer M, Langer C, Horstkotte D, Piper C. Pulmonary capillary wedge pressure and pulmonary arterial pressure in heart failure patients with sleep-disordered breathing. Sleep Med. 2009;10:726–30. doi: 10.1016/j.sleep.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 35.Mortara A, Sleight P, Pinna GD, Maestri R, Capomolla S, Febo O, La Rovere MT, Cobelli F. Association between hemodynamic impairment and Cheyne-Stokes respiration and periodic breathing in chronic stable congestive heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol. 1999;84:900–4. doi: 10.1016/s0002-9149(99)00462-2. [DOI] [PubMed] [Google Scholar]
- 36.Javaheri S, Parker TJ, Liming JD, Corbett WS, Nishiyama H, Wexler L, Roselle GA. Sleep apnea in 81 ambulatory male patients with stable heart failure. Types and their prevalences, consequences, and presentations. Circulation. 1998;97:2154–9. doi: 10.1161/01.cir.97.21.2154. [DOI] [PubMed] [Google Scholar]
- 37.Orr JE, Auger WR, DeYoung PN, Kim NH, Malhotra A, Owens RL. Usefulness of Low Cardiac Index to Predict Sleep-Disordered Breathing in Chronic Thromboembolic Pulmonary Hypertension. The American journal of cardiology. 2015 doi: 10.1016/j.amjcard.2015.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sinha AM, Skobel EC, Breithardt OA, Norra C, Markus KU, Breuer C, Hanrath P, Stellbrink C. Cardiac resynchronization therapy improves central sleep apnea and Cheyne-Stokes respiration in patients with chronic heart failure. J Am Coll Cardiol. 2004;44:68–71. doi: 10.1016/j.jacc.2004.03.040. [DOI] [PubMed] [Google Scholar]
- 39.Olson TP, Frantz RP, Snyder EM, O’Malley KA, Beck KC, Johnson BD. Effects of acute changes in pulmonary wedge pressure on periodic breathing at rest in heart failure patients. Am Heart J. 2007;153:104e1–7. doi: 10.1016/j.ahj.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Solin P, Roebuck T, Johns DP, Walters EH, Naughton MT. Peripheral and central ventilatory responses in central sleep apnea with and without congestive heart failure. Am J Respir Crit Care Med. 2000;162:2194–200. doi: 10.1164/ajrccm.162.6.2002024. [DOI] [PubMed] [Google Scholar]
- 41.Xie A, Skatrud JB, Puleo DS, Rahko PS, Dempsey JA. Apnea-hypopnea threshold for CO2 in patients with congestive heart failure. Am J Respir Crit Care Med. 2002;165:1245–50. doi: 10.1164/rccm.200110-022OC. [DOI] [PubMed] [Google Scholar]
- 42.Francis DP, Willson K, Davies LC, Coats AJ, Piepoli M. Quantitative general theory for periodic breathing in chronic heart failure and its clinical implications. Circulation. 2000;102:2214–21. doi: 10.1161/01.cir.102.18.2214. [DOI] [PubMed] [Google Scholar]
- 43.Giannoni A, Emdin M, Poletti R, Bramanti F, Prontera C, Piepoli M, Passino C. Clinical significance of chemosensitivity in chronic heart failure: influence on neurohormonal derangement, Cheyne-Stokes respiration and arrhythmias. Clin Sci (Lond) 2008;114:489–97. doi: 10.1042/CS20070292. [DOI] [PubMed] [Google Scholar]
- 44.Chenuel BJ, Smith CA, Skatrud JB, Henderson KS, Dempsey JA. Increased propensity for apnea in response to acute elevations in left atrial pressure during sleep in the dog. J Appl Physiol. 2006;101:76–83. doi: 10.1152/japplphysiol.01617.2005. [DOI] [PubMed] [Google Scholar]
- 45.Calvin AD, Somers VK, Johnson BD, Scott CG, Olson LJ. Left atrial size, chemosensitivity, and central sleep apnea in heart failure. Chest. 2014;146:96–103. doi: 10.1378/chest.13-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Szollosi I, Thompson BR, Krum H, Kaye DM, Naughton MT. Impaired pulmonary diffusing capacity and hypoxia in heart failure correlates with central sleep apnea severity. Chest. 2008;134:67–72. doi: 10.1378/chest.07-1487. [DOI] [PubMed] [Google Scholar]
- 47.Solin P, Bergin P, Richardson M, Kaye DM, Walters EH, Naughton MT. Influence of pulmonary capillary wedge pressure on central apnea in heart failure. Circulation. 1999;99:1574–9. doi: 10.1161/01.cir.99.12.1574. [DOI] [PubMed] [Google Scholar]
- 48.Yumino D, Redolfi S, Ruttanaumpawan P, Su MC, Smith S, Newton GE, Mak S, Bradley TD. Nocturnal rostral fluid shift: a unifying concept for the pathogenesis of obstructive and central sleep apnea in men with heart failure. Circulation. 2010;121:1598–605. doi: 10.1161/CIRCULATIONAHA.109.902452. [DOI] [PubMed] [Google Scholar]
- 49.Lloyd TC., Jr Breathing response to lung congestion with and without left heart distension. J Appl Physiol (1985) 1988;65:131–6. doi: 10.1152/jappl.1988.65.1.131. [DOI] [PubMed] [Google Scholar]
- 50.Bitter T, Zwenke A, Dimitriadis Z, Fischbach T, Möllenberg M, Dohrmann J, Prinz C, Afsah M, Horstkotte D, Oldenburg O. Acute improvement of pulmonary hemodynamics does not alleviate Cheyne-Stokes respiration in chronic heart failure - a randomized, controlled, double-blind, crossover trial. European Respiratory Journal. 2013:42. doi: 10.1007/s11325-015-1300-1. [DOI] [PubMed] [Google Scholar]
- 51.Solin P, Snell GI, Williams TJ, Naughton MT. Central sleep apnoea in congestive heart failure despite vagal denervation after bilateral lung transplantation. Eur Respir J. 1998;12:495–8. doi: 10.1183/09031936.98.12020495. [DOI] [PubMed] [Google Scholar]
- 52.Ding Y, Li YL, Schultz HD. Role of blood flow in carotid body chemoreflex function in heart failure. J Physiol. 2011;589:245–58. doi: 10.1113/jphysiol.2010.200584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schultz HD, Marcus NJ, Del Rio R. Mechanisms of carotid body chemoreflex dysfunction during heart failure. Exp Physiol. 2015;100:124–9. doi: 10.1113/expphysiol.2014.079517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paton JF, Ramchandra R, McBryde FD, Ford AP. Carotid body involvement in a translational ovine model of renovascular hypertension: Sensitivity to purinergic P2×3 receptor antagonism with AF-130 [Abstract] Am J Respir Crit Care Med. 2016;193:A7900. [Google Scholar]
- 55.Xie A, Rutherford R, Rankin F, Wong B, Bradley TD. Hypocapnia and increased ventilatory responsiveness in patients with idiopathic central sleep apnea. Am J Respir Crit Care Med. 1995;152:1950–5. doi: 10.1164/ajrccm.152.6.8520761. [DOI] [PubMed] [Google Scholar]
- 56.Xie A, Wong B, Phillipson EA, Slutsky AS, Bradley TD. Interaction of hyperventilation and arousal in the pathogenesis of idiopathic central sleep apnea. American journal of respiratory and critical care medicine. 1994;150:489–95. doi: 10.1164/ajrccm.150.2.8049835. [DOI] [PubMed] [Google Scholar]
- 57.Lahiri S, Maret K, Sherpa MG. Dependence of high altitude sleep apnea on ventilatory sensitivity to hypoxia. Respir Physiol. 1983;52:281–301. doi: 10.1016/0034-5687(83)90086-5. [DOI] [PubMed] [Google Scholar]
- 58.Latshang TD, Lo Cascio CM, Stowhas AC, Grimm M, Stadelmann K, Tesler N, Achermann P, Huber R, Kohler M, Bloch KE. Are nocturnal breathing, sleep, and cognitive performance impaired at moderate altitude (1,630–2,590 m)? Sleep. 2013;36:1969–76. doi: 10.5665/sleep.3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Burgess KR, Lucas SJ, Shepherd K, Dawson A, Swart M, Thomas KN, Lucas RA, Donnelly J, Peebles KC, Basnyat R, Ainslie PN. Worsening of central sleep apnea at high altitude--a role for cerebrovascular function. J Appl Physiol (1985) 2013;114:1021–8. doi: 10.1152/japplphysiol.01462.2012. [DOI] [PubMed] [Google Scholar]
- 60.Lloyd BB, Jukes MG, Cunningham DJ. The relation between alveolar oxygen pressure and the respiratory response to carbon dioxide in man. Quarterly journal of experimental physiology and cognate medical sciences. 1958;43:214–27. doi: 10.1113/expphysiol.1958.sp001319. [DOI] [PubMed] [Google Scholar]
- 61.Andrews G, Ainslie PN, Shepherd K, Dawson A, Swart M, Lucas S, Burgess KR. The effect of partial acclimatization to high altitude on loop gain and central sleep apnoea severity. Respirology. 2012;17:835–40. doi: 10.1111/j.1440-1843.2012.02170.x. [DOI] [PubMed] [Google Scholar]
- 62.Bloch KE, Latshang TD, Turk AJ, Hess T, Hefti U, Merz TM, Bosch MM, Barthelmes D, Hefti JP, Maggiorini M, Schoch OD. Nocturnal periodic breathing during acclimatization at very high altitude at Mount Muztagh Ata (7,546 m) Am J Respir Crit Care Med. 2010;182:562–8. doi: 10.1164/rccm.200911-1694OC. [DOI] [PubMed] [Google Scholar]
- 63.Tellez HF, Mairesse O, Macdonald-Nethercott E, Neyt X, Meeusen R, Pattyn N. Sleep-related Periodic Breathing Does Not Acclimatize to Chronic Hypobaric Hypoxia: A 1-Year Study at High Altitude in Antarctica. American Journal of Respiratory and Critical Care Medicine. 2014;190:114–6. doi: 10.1164/rccm.201403-0598LE. [DOI] [PubMed] [Google Scholar]
- 64.Nespoulet H, Wuyam B, Tamisier R, Saunier C, Monneret D, Remy J, Chabre O, Pepin JL, Levy P. Altitude illness is related to low hypoxic chemoresponse and low oxygenation during sleep. Eur Respir J. 2012;40:673–80. doi: 10.1183/09031936.00073111. [DOI] [PubMed] [Google Scholar]
- 65.Rexhaj E, Rimoldi SF, Pratali L, Brenner R, Andries D, Soria R, Salinas C, Villena M, Romero C, Allemann Y, Lovis A, Heinzer R, Sartori C, Scherrer U. Sleep-Disordered Breathing and Vascular Function in Patients With Chronic Mountain Sickness and Healthy High-Altitude Dwellers. Chest. 2016;149:991–8. doi: 10.1378/chest.15-1450. [DOI] [PubMed] [Google Scholar]
- 66.Lahiri S. Alveolar gas pressures in man with life-time hypoxia. Respir Physiol. 1968;4:373–86. doi: 10.1016/0034-5687(68)90042-x. [DOI] [PubMed] [Google Scholar]
- 67.Pratali L, Cavana M, Sicari R, Picano E. Frequent subclinical high-altitude pulmonary edema detected by chest sonography as ultrasound lung comets in recreational climbers. Critical care medicine. 2010;38:1818–23. doi: 10.1097/CCM.0b013e3181e8ae0e. [DOI] [PubMed] [Google Scholar]
- 68.Sovik S, Lossius K. Development of ventilatory response to transient hypercapnia and hypercapnic hypoxia in term infants. Pediatr Res. 2004;55:302–9. doi: 10.1203/01.PDR.0000106316.40213.DB. [DOI] [PubMed] [Google Scholar]
- 69.Australia B-I, Tarnow-Mordi W, Stenson B, Kirby A, Juszczak E, Donoghoe M, Deshpande S, Morley C, King A, Doyle LW, Fleck BW, Davis PG, Halliday HL, Hague W, Cairns P, Darlow BA, Fielder AR, Gebski V, Marlow N, Simmer K, Tin W, Ghadge A, Williams C, Keech A, Wardle SP, Kecskes Z, Kluckow M, Gole G, Evans N, Malcolm G, Luig M, Wright I, Stack J, Tan K, Pritchard M, Gray PH, Morris S, Headley B, Dargaville P, Simes RJ, Brocklehurst P United Kingdom Collaborative G. Outcomes of Two Trials of Oxygen-Saturation Targets in Preterm Infants. N Engl J Med. 2016;374:749–60. doi: 10.1056/NEJMoa1514212. [DOI] [PubMed] [Google Scholar]
- 70.Fleming PJ, Goncalves AL, Levine MR, Woollard S. The development of stability of respiration in human infants: changes in ventilatory responses to spontaneous sighs. J Physiol. 1984;347:1–16. doi: 10.1113/jphysiol.1984.sp015049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sovik S, Lossius K, Eriksen M, Grogaard J, Walloe L. Development of oxygen sensitivity in infants of smoking and non-smoking mothers. Early Hum Dev. 1999;56:217–32. doi: 10.1016/s0378-3782(99)00048-1. [DOI] [PubMed] [Google Scholar]
- 72.Rigatto H. Periodic Breathing. In: Oommen MP, editor. Respiratory control and disorders in the newborn. Marcel Dekker Inc; New York: 2003. pp. 237–72. [Google Scholar]
- 73.Rigatto H, Brady JP. Periodic breathing and apnea in preterm infants. II. Hypoxia as a primary event. Pediatrics. 1972;50:219–28. [PubMed] [Google Scholar]
- 74.Poets CF, Rau GA, Neuber K, Gappa M, Seidenberg J. Determinants of lung volume in spontaneously breathing preterm infants. Am J Respir Crit Care Med. 1997;155:649–53. doi: 10.1164/ajrccm.155.2.9032208. [DOI] [PubMed] [Google Scholar]
- 75.Wang D, Teichtahl H, Drummer O, Goodman C, Cherry G, Cunnington D, Kronborg I. Central sleep apnea in stable methadone maintenance treatment patients. Chest. 2005;128:1348–56. doi: 10.1378/chest.128.3.1348. [DOI] [PubMed] [Google Scholar]
- 76.Farney RJ, Walker JM, Boyle KM, Cloward TV, Shilling KC. Adaptive servoventilation (ASV) in patients with sleep disordered breathing associated with chronic opioid medications for non-malignant pain. J Clin Sleep Med. 2008;4:311–9. [PMC free article] [PubMed] [Google Scholar]
- 77.Mellen NM, Janczewski WA, Bocchiaro CM, Feldman JL. Opioid-induced quantal slowing reveals dual networks for respiratory rhythm generation. Neuron. 2003;37:821–6. doi: 10.1016/s0896-6273(03)00092-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Teichtahl H, Wang D, Cunnington D, Quinnell T, Tran H, Kronborg I, Drummer OH. Ventilatory responses to hypoxia and hypercapnia in stable methadone maintenance treatment patients. Chest. 2005;128:1339–47. doi: 10.1378/chest.128.3.1339. [DOI] [PubMed] [Google Scholar]
- 79.Javaheri S, Harris N, Howard J, Chung E. Adaptive servoventilation for treatment of opioid-associated central sleep apnea. J Clin Sleep Med. 2014;10:637–43. doi: 10.5664/jcsm.3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Walker JM, Farney RJ, Rhondeau SM, Boyle KM, Valentine K, Cloward TV, Shilling KC. Chronic opioid use is a risk factor for the development of central sleep apnea and ataxic breathing. J Clin Sleep Med. 2007;3:455–61. [PMC free article] [PubMed] [Google Scholar]
- 81.Glidewell RN, Orr WC, Imes N. Acetazolamide as an adjunct to CPAP treatment: a case of complex sleep apnea in a patient on long-acting opioid therapy. J Clin Sleep Med. 2009;5:63–4. [PMC free article] [PubMed] [Google Scholar]
- 82.Younes M, Ostrowski M, Thompson W, Leslie C, Shewchuk W. Chemical control stability in patients with obstructive sleep apnea. Am J Respir Crit Care Med. 2001;163:1181–90. doi: 10.1164/ajrccm.163.5.2007013. [DOI] [PubMed] [Google Scholar]
- 83.Eckert DJ, White DP, Jordan AS, Malhotra A, Wellman A. Defining phenotypic causes of obstructive sleep apnea. Identification of novel therapeutic targets. Am J Respir Crit Care Med. 2013;188:996–1004. doi: 10.1164/rccm.201303-0448OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xie A, Teodorescu M, Pegelow DF, Teodorescu MC, Gong Y, Fedie JE, Dempsey JA. Effects of stabilizing or increasing respiratory motor outputs on obstructive sleep apnea. J Appl Physiol. 2013 doi: 10.1152/japplphysiol.00064.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wellman A, Malhotra A, Jordan AS, Stevenson KE, Gautam S, White DP. Effect of oxygen in obstructive sleep apnea: role of loop gain. Respir Physiol Neurobiol. 2008;162:144–51. doi: 10.1016/j.resp.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Edwards BA, Sands SA, Eckert DJ, White DP, Butler JP, Owens RL, Malhotra A, Wellman A. Acetazolamide improves loop gain but not the other physiological traits causing obstructive sleep apnoea. J Physiol. 2012;590:1199–211. doi: 10.1113/jphysiol.2011.223925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tkacova R, Niroumand M, Lorenzi-Filho G, Bradley TD. Overnight shift from obstructive to central apneas in patients with heart failure: role of PCO2 and circulatory delay. Circulation. 2001;103:238–43. doi: 10.1161/01.cir.103.2.238. [DOI] [PubMed] [Google Scholar]
- 88.Ryan CM, Floras JS, Logan AG, Kimoff RJ, Series F, Morrison D, Ferguson KA, Belenkie I, Pfeifer M, Fleetham J, Hanly PJ, Smilovitch M, Arzt M, Bradley TD. Shift in sleep apnoea type in heart failure patients in the CANPAP trial. Eur Respir J. 2010;35:592–7. doi: 10.1183/09031936.00070509. [DOI] [PubMed] [Google Scholar]
- 89.Mansfield DR, Solin P, Roebuck T, Bergin P, Kaye DM, Naughton MT. The effect of successful heart transplant treatment of heart failure on central sleep apnea. Chest. 2003;124:1675–81. doi: 10.1378/chest.124.5.1675. [DOI] [PubMed] [Google Scholar]
- 90.Stanchina ML, Ellison K, Malhotra A, Anderson M, Kirk M, Benser ME, Tosi C, Carlisle C, Millman RP, Buxton A. The impact of cardiac resynchronization therapy on obstructive sleep apnea in heart failure patients: a pilot study. Chest. 2007;132:433–9. doi: 10.1378/chest.06-2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Javaheri S, Ahmed M, Parker TJ, Brown CR. Effects of nasal O2 on sleep-related disordered breathing in ambulatory patients with stable heart failure. Sleep. 1999;22:1101–6. doi: 10.1093/sleep/22.8.1101. [DOI] [PubMed] [Google Scholar]
- 92.Weintraub Z, Alvaro R, Kwiatkowski K, Cates D, Rigatto H. Effects of inhaled oxygen (up to 40%) on periodic breathing and apnea in preterm infants. Journal of applied physiology. 1992;72:116–20. doi: 10.1152/jappl.1992.72.1.116. [DOI] [PubMed] [Google Scholar]
- 93.Xie A, Skatrud JB, Puleo DS, Dempsey JA. Influence of arterial O2 on the susceptibility to posthyperventilation apnea during sleep. J Appl Physiol. 2006;100:171–7. doi: 10.1152/japplphysiol.00440.2005. [DOI] [PubMed] [Google Scholar]
- 94.Joho S, Oda Y, Hirai T, Inoue H. Impact of sleeping position on central sleep apnea/Cheyne-Stokes respiration in patients with heart failure. Sleep Med. 2010;11:143–8. doi: 10.1016/j.sleep.2009.05.014. [DOI] [PubMed] [Google Scholar]
- 95.Abraham WT, Jagielski D, Oldenburg O, Augostini R, Krueger S, Kolodziej A, Gutleben KJ, Khayat R, Merliss A, Harsch MR, Holcomb RG, Javaheri S, Ponikowski P remede Pilot Study I. Phrenic nerve stimulation for the treatment of central sleep apnea. JACC Heart failure. 2015;3:360–9. doi: 10.1016/j.jchf.2014.12.013. [DOI] [PubMed] [Google Scholar]
- 96.Cowie MR, Woehrle H, Wegscheider K, Angermann C, d’Ortho M-P, Erdmann E, Levy P, Simonds AK, Somers VK, Zannad F, Teschler H. Adaptive Servo-Ventilation for Central Sleep Apnea in Systolic Heart Failure. New England Journal of Medicine. 2015;373:1095–105. doi: 10.1056/NEJMoa1506459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cowie MR, Woehrle H, Wegscheider K, Angermann C, d’Ortho MP, Erdmann E, Levy P, Simonds AK, Somers VK, Zannad F, Teschler H. Adaptive Servo-Ventilation for Central Sleep Apnea in Systolic Heart Failure. N Engl J Med. 2015;373:1095–105. doi: 10.1056/NEJMoa1506459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Giannoni A, Baruah R, Willson K, Mebrate Y, Mayet J, Emdin M, Hughes AD, Manisty CH, Francis DP. Real-time dynamic carbon dioxide administration: a novel treatment strategy for stabilization of periodic breathing with potential application to central sleep apnea. J Am Coll Cardiol. 2010;56:1832–7. doi: 10.1016/j.jacc.2010.05.053. [DOI] [PubMed] [Google Scholar]