

Summary
Non‐alcoholic fatty liver disease (NAFLD) is a hepatic presentation of obesity and metabolic syndrome. MicroRNA 26a (Mir‐26a) has been reported to play functions in cellular differentiation, cell growth, cell apoptosis and metastasis. A recent paper indicated that Mir‐26a regulated insulin sensitivity and metabolism of glucose and lipids. However, the role of Mir‐26a in NAFLD still needs to be investigated further. In our current study, vectors encoding pre‐Mir‐26a (LV‐26a) and an empty lentiviral vector (LV‐Con) delivered approximately 2 × 107 transforming units of recombinant lentivirus were injected into mice through the tail vein. LV‐26a‐infected mice were protected from glucose dysmetabolism and showed markedly decreased total liver weight, hepatic triglyceride deposition and serum alanine transaminase (ALT) concentration when compared with LV‐Con‐treated mice. LV‐26a‐treated mice also exhibited decreased infiltration of immune cells in the liver – something attributed to reduce infiltration of T cell receptor (TCR)‐γδ+, granulocyte‐differentiation antigen‐1 (Gr‐1)+ cells and CD11b+ cells. Next, we found that Mir‐26a inhibited the expression of interleukin (IL)−17 and IL‐6 in vivo and in vitro. Furthermore, the decreased expression of IL‐17 in the liver tissue induced by Mir‐26a was abrogated completely by IL‐6 overexpression. The decreased total liver weight, hepatic triglyceride deposition and serum ALT concentration induced by Mir‐26a was also abrogated completely by IL‐6 over‐expression. In conclusion, the Mir‐26a–IL‐6–IL‐17 axis regulates the development of NAFLD in a murine model.
Keywords: IL‐6, IL‐17, Mir‐26a, NAFLD
Introduction
Non‐alcoholic fatty liver disease (NAFLD) is a hepatic presentation of obesity and metabolic syndrome. NAFLD includes a large spectrum of hepatic pathologies that range from simple steatosis and non‐alcoholic steatohepatitis (NASH) to liver cirrhosis 1. Because of its association with obesity and insulin resistance, the prevalence of NAFLD is increasing worldwide. However, there is currently no broadly approved therapeutic strategy for treating NAFLD.
MicroRNAs (miRNAs) are expressed endogenously, small non‐coding RNAs that regulate gene expression negatively by causing degradation of target mRNAs, inhibition of the translation of these mRNAs or both 2. miRNAs play a part in crucial cellular processes such as the stress response, development, differentiation, apoptosis and proliferation 3. MicroRNA 26a (Mir‐26a) has been reported to play functions in cellular differentiation, cell growth, cell apoptosis and metastasis 4, 5. Previous studies have indicated that Mir‐26a modulated immunological functions in mouse models of experimental autoimmune encephalomyelitis (EAE), ischaemia‐reperfusion injury and transplantation 6, 7, 8. Furthermore, recent paper indicated that Mir‐26a regulated insulin sensitivity and metabolism of glucose and lipids 9. However, the role of Mir‐26a in the NAFLD still needs to be investigated further.
Obesity is associated with increased interleukin (IL)‐17 production in humans 10. IL‐17 has been demonstrated to mediate inflammatory cell‐dependent [T cell receptor (TCR)‐γδ+, granulocyte‐differentiation antigen‐1 (Gr‐1)+ cells and CD11b+ cells] hepatocellular damage 11. IL‐17 also drives reactive oxygen species (ROS) production (Nox2, p22phox, p47phox and p67phox), which can induce hepatic parenchymal cell injury 12, 13. A recent publication has indicated the function of Mir‐26a on regulatory T cells 7 but, to our knowledge, the relationship of Mir‐26a and IL‐17 has never been studied.
In our current study, we demonstrated a crucial role for Mir‐26a in attenuating the development of NAFLD. Mir‐26a over‐expressed [vectors encoding pre‐Mir‐26a infected (LV‐26a‐infected)] mice were protected from glucose dysmetabolism and showed markedly decreased total liver weight, hepatic triglyceride deposition and serum alanine transaminase (ALT) concentration when compared with LV‐Con‐treated mice. LV‐26a‐treated mice also exhibited decreased infiltration of immune cells in the liver – something attributed to reduce infiltration of TCR‐γδ+, Gr‐1+ cells and CD11b+ cells. Next, we found that Mir‐26a inhibited the expression of IL‐17 and IL‐6 in vivo and in vitro. Furthermore, the decreased expression of IL‐17 in the liver tissue induced by Mir‐26a was abrogated completely by IL‐6 overexpression. The decreased total liver weight, hepatic triglyceride deposition and serum ALT concentration induced by Mir‐26a was also abrogated completely by IL‐6 overexpression.
Materials and methods
Animals
All mice were male and from a C57BL/6 background. All protocols were approved by the Institutional Ethics Committee on Animal Use of the Hubei University of Medicine.
Vector construction and lentivirus production
A 200‐base pairs (bp) DNA fragment corresponding to pre‐Mir‐26a and its flanking sequences was amplified from mouse genomic DNA and was cloned subsequently into pLVTHM lentiviral vector 6. For Mir‐26a inhibition sequence, cDNA for IL‐6 was cloned into the same lentiviral vector. The production, purification and titration of lentivirus were performed as described by Tiscornia and colleagues 14. The packaged lentiviruses were named LV‐26a and LV‐IL‐6. The empty (untransformed) lentiviral vector LV‐Con served as control.
Protocols of LV‐26a administration
Vectors encoding pre‐Mir‐26a (LV‐26a) and an empty lentiviral vector (LV‐Con) delivering approximately 2 × 107 transforming units of recombinant lentivirus were injected into mice once through the tail vein; the efficacy of lentivirus infection was assessed 7 days later by quantitative reverse transcription polymerase chain reaction (qPCR) analysis of Mir‐26a expression in the liver of LV‐26a‐infected mice and LV‐Con‐infected mice. Lentivirus‐infected mice were fed with a high‐fat diet (HFD) or a chow diet on day 7 after virus injection.
Obesity and its downstream sequelae
Diets
Mice were fed either a HFD (Research Diets #D12492; Research Diets Inc., New Brunswick, NJ, USA) or a chow diet (chow, Lab Diet #5010; Research Diets Inc.). For all studies, animals were weighed weekly. Fresh food was provided on a weekly basis and food consumption was quantified weekly.
Glucose dysmetabolism
Following 8–12 h fasting, a single drop of blood was obtained by a tail vein nick and fasting glucose levels were quantified using a FreeStyle flash blood glucose monitoring system (Abbot Diabetes Care Inc., Alameda, CA, USA). For insulin and glucose tolerance testing, animals were fasted overnight and subsequently challenged intraperitoneally (i.p.) (10 μl/g) with recombinant human insulin dissolved in normal saline (1·5 U/kg; Novolin R; Novo Nordisk Inc., Plainsboro, NJ, USA) or with dextrose dissolved in normal saline (1 g/kg; Sigma, St Louis, MO, USA), respectively. Immediately prior to injection, and kinetically afterwards, glucose levels were quantified as above.
Liver
Hepatic triglycerides were quantified at 500–520 nm using Molecular Devices (Sunnyvale, CA, USA) vmax Kinetic Microplate Reader and SoftMax Pro version 5 software. Briefly, a preweighed quantity of frozen mouse liver tissue was mixed with 10 μl of homogenization buffer [50 mM Tris, 150 mM NaCl, 1 mM ethylenediamine tetraacetic acid (EDTA), 1 mM phenylmethane sulphonyl fluoride (PMSF)] per mg of liver tissue. A 1/8″ steel bead (McMaster‐Carr, Elmhurst, IL, USA) was added to each sample, and samples were dissociated subsequently using a TissueLyser (Qiagen, Valencia, CA, USA) at 30 Hz for 3 min. Following 30 min incubation on ice, 10 μl of each sample was diluted 1 : 10 in homogenization buffer and added to a 96‐well clear Costar flat‐bottomed plate containing 200 μl of triglyceride reagent (Pointe Scientific, Canton, MI, USA). Triglyceride standards (Pointe Scientific) were prepared according to the manufacturer's instructions.
Hepatocellular damage
ALT levels were quantified from 10 μl of mouse serum per sample in a 96‐well flat‐bottomed Costar plate (Corning, NY, USA) and a BioTek Synergy 2 Multi‐Mode Microplate Reader (BioTek, Winooski, VT, USA) with Gen5 version 2.00 software. Catatrol I and II (Catachem Inc., Oxford, CT, USA) were used as controls and were prepared according to the manufacturer's instructions. ALT buffer was prepared by mixing ALT activator reagent and ALT sample diluent reagent (Catachem Inc.) and 200 μl of buffer was added subsequently to each sample. Starting at time zero, absorbance was recorded at 340 nm and 37°C once per min for 5 min and blank value was subtracted from all samples. The ALT concentration (U/l) was calculated subsequently.
Lipid peroxidation
Levels of 4‐hydroxynonenal (4‐HNE) were quantified by enzyme‐linked immunosorbent assay (ELISA) in liver homogenates, according to the manufacturer's instructions (Cell Biolabs, San Diego, CA, USA). Protein concentrations were normalized among all groups prior to analysis.
Histology
For Red O staining, a stock solution of Oil Red O (0·5 g/100 ml) in isopropanol was prepared, stored and protected from light. Liver tissues were embedded in optimal cutting temperature gel. Air‐dried 7‐μm tissue sections were dipped in formalin, washed with Oil Red O without counterstaining with haematoxylin. Immunohistochemical staining of IL‐17 and IL‐6 were performed according to the manufacturer's instructions.
qRT–PCR
Tissue samples were homogenized in TRIzol (Invitrogen, Grand Island, NY, USA) using a TissueLyser (Qiagen) set at 30 Hz for 5 min and 1/8″ diameter stainless steel beads (McMaster‐Carr). RNA was extracted according to the manufacturer's instructions, resuspended in diethylpyrocarbonate (DEPC)‐treated water and quantified using a Nanodrop ND‐1000. One μg of RNA was treated with 1 U amplification grade DNase I (Invitrogen) and reverse‐transcribed using oligo‐dT and random hexamers in the presence of 200U Superscript II reverse transcriptase (Invitrogen). The cDNA was treated subsequently with RNAse H, diluted in DEPC‐treated water and subjected to quantitative polymerase chain reaction (qPCR) analysis using Light Cycler 480 II (Roche, Basel, Switzerland). Sybr Green I Master mix (Roche) and the following primer pairs were used: Nox2 forward CGGTGTGCAGTGCTATCATC, reverse GCTCTCCTTTCTCAGGGGTT; p67phox forward CTATCAGCTGGTTCCCACGA, reverse GCAGTGGCCTACTTCCAGAG; p47phox forward ATGACCTCAATGGCTTCACC, reverse CTATCTGGAGCCCCTTGACA; p22phox forward CCTGCSGCGATAGAGTAGGC, reverse TCATGGGGCAGATCGAGT; Mir‐26a forward AAGGAGAACCCGTAGATCCG, reverse GTGCAGGGTCCGAGGTATTC; IL‐17 forward CAGGACGCGCAAACATGA, reverse GCAACAGCATCAGAGACACAGAT; and IL‐6 forward ACTTCCATCCAGTTGCCTTCTT, reverse TCATTTCCACGATTTCCCAGA.
Flow cytometry
Single‐cell suspensions were generated from hepatic digestion as described above 3. Subsequently, flow cytometry was used to enumerate immune cell populations as described previously 7, 8, 9. Briefly, cells were treated with FACS fix buffer for 15 min (BD Biosciences). Cells were washed and co‐incubated with anti‐FcgIII/II (CD16/32; e‐Bioscience, San Diego, CA, USA) antibody for 30 min in PBS containing 0.1% bovine serum albumin (BSA) and 0·01% sodium azide. After a further wash, cells were incubated with directly conjugated monoclonal antibodies to TCR‐β‐fluorescein isothiocyanate (FITC) (H57‐597), TCR‐γδ‐allophycocyanin (APC) (GL3), CD4‐phycoerythrin (PE)‐cyanin‐7 (Cy7) (GK1·5), CD8‐Pacific Blue (53–6·7), CD11b‐peridinin chlorophyll (PerCp)‐Cy5·5 (M1/70), Gr‐1‐FITC (RB6‐8C5) and/or CD45‐PE (30‐F11) (all antibodies were from e‐Bioscience) for 30 min. Isotype control antibodies (e‐Bioscience) were used in each analysis. Data were collected and analysed using a combination of LSRII flow cytometer (BD Immunocytometry Systems) and FlowJo software (TreeStar, Inc., Ashland, OR, USA).
Western blotting
The protein levels were determined by Western blotting. Protein extracted from tissue was separated on 10% sodium dodecyl sulphate‐polyacrylamide electrophoresis gels (SDS‐PAGE) and transferred to nitrocellulose membranes (Pierce, Rockford, IL, USA). After being blocked with 5% non‐fat milk in Tris‐buffered saline (TBS) for 3 h, the membranes were incubated with the primary antibodies indicated (0·2 µg/ml) at 4°C overnight, followed by incubation with horseradish peroxidated (HRP)‐conjugated secondary antibody (1 : 5000) for 3 h. β‐actin was used as a loading control for comparison between samples.
Nucleofection
Nucleofection was performed with the mouse T cell Nucleofector® kit and Nucleofector device (Amaxa, Koelin, Germany). First, 1 × 107 naive CD4+ T cells were resuspended in 100 µl Nucleofector® solution; 2·5 µg pmaxGFP® vector or 100 pmol oligonucleotides (including pre‐Mir‐26a, pre‐Mir‐ctrl, anti‐Mir‐26a and anti‐Mir‐ctrl) were added into the solution and mixed gently. Then the mixtures were transferred gently to electroporation cuvettes and placed in the Nucleofector device. Cells were nucleofected in the X‐01 programme. Finally, transfected cells were transferred to a 12‐well plate with 1·5 ml prepared mouse T cell Nucleofector® medium in each plate and incubated in a humidified 37°C/5% CO2 incubator until analysis. The transfection efficiency, which was detected by monitoring green fluorescent protein (GFP) expression under fluorescence microscope 8 h after transfection, is approximately 55%.
CD4+ T cell activation and polarization
Four h after nucleofection, CD4+ T cells were activated by 5 µg/ml plate‐bound anti‐CD3 and 2 µg/ml soluble anti‐CD28. For propagation under T helper type 17 (Th17) conditions, 2·5 ng/ml recombinant transforming growth factor (rTGF)‐β1, 30 mg/ml rIL‐6, 10 μg/ml anti‐interferon (IFN)‐γ and 10 μg/ml anti‐IL‐4 were provided. All antibodies used were purchased from e‐Bioscience. All cytokines used were purchased from Peprotech (Rocky Hill, NJ, USA).
MiRNA target validation
A 328‐bp fragment of the IL‐6 3′ untranslated region (UTR) was amplified by PCR and cloned into the pGLO vector (Promega Corp., Madison, WI, USA), downstream of the firefly luciferase gene. This vector was named wild‐type (wt) 3′ UTR. Site‐directed mutagenesis of the Mir‐26a binding site in IL‐6 3′ UTR was performed using the Gene Tailor Site‐Directed Mutagenesis System (Invitrogen) and named mutant (mt) 3′ UTR. For reporter assays, wt or mt 30 UTR vector and the control vector pRL‐TK (Promega) were co‐transduced in human embryonic kidney (HEK)293 cells. Luciferase activity was measured 36 h after transduction using the Dual‐Luciferase Reporter Assay System 6.
Statistics
Data are presented as means ± standard deviation (s.d.). Differences were evaluated using unpaired Student's t‐test between two groups and one‐way analysis of variance (anova) for multiple comparisons, followed by a post‐hoc Student–Newmann–Keuls test when necessary. All analyses were performed using spss version 13.0 (SPSS, Chicago, IL, USA), and statistical significance was set at P < 0·05.
Results
Mir‐26a attenuates the development of NAFLD
We first explored the expression of Mir‐26a in liver from mice which were challenged in a standard model of HFD‐induced obesity – a model that induces obesity along with steatosis and hepatocellular damage by qRT–PCR. Liver tissues from mice after 20 weeks of HFD feeding showed a significantly lower expression of Mir‐26a when compared with that from mice after 20 weeks of chow feeding (Fig. 1a). Next, we investigated the potential effects of Mir‐26a on NAFLD through the construction of lentiviral vectors. Vectors encoding pre‐Mir‐26a (LV‐26a) and an empty lentiviral vector (LV‐Con) delivered approximately 2 × 107 transforming units of recombinant lentivirus to mice by injection through the tail vein. The efficacy of lentivirus infection was assessed 7 days later by qRT–PCR analysis of Mir‐26a expression in the liver of LV‐26a‐infected mice and LV‐Con‐infected mice (P < 0·05, Fig. 1b). Lentivirus‐infected mice were changed with chow or HFD feeding on day 7 after virus injection. Relative to LV‐Con‐infected mice, LV‐26a‐infected mice exhibited body weight and epididymal white adipose tissue (eWAT) weight significantly after 20 weeks of HFD feeding (Fig. 1c,d). Also, LV‐26a‐infected mice were protected from glucose dysmetabolism including fasting hyperglycaemia, glucose intolerance and insulin resistance (Fig. 1e–g). Furthermore, LV‐26a‐infected mice showed markedly decreased total liver weight, hepatic triglyceride and cholesterol deposition and serum ALT concentration when compared with LV‐Con‐treated mice (Fig. 2a–d).
Figure 1.
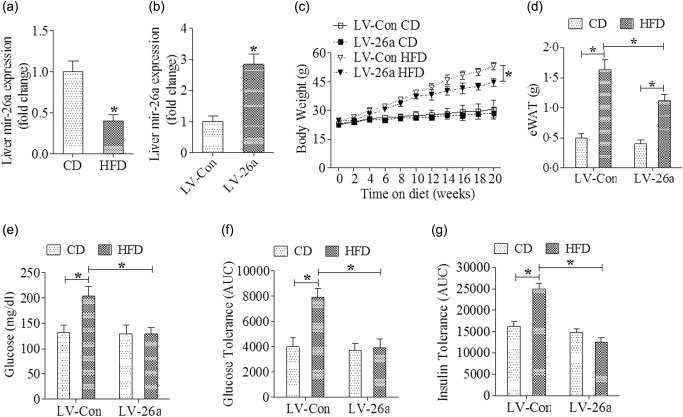
MicroRNA‐26a (Mir‐26a) attenuates high‐fat diet (HFD)‐induced obesity and protects from glucose dysmetabolism. Mice were placed on an HFD or chow diet. (a) Mir‐26a mRNA expression was analysed by reverse transcription–polymerase chain reaction (RT–PCR); (b) quantitative PCR analysis of Mir‐26a mRNA expression in the liver of mice infected with LV‐Mir‐26a or LV‐Con (day 7 after lentivirus administration); (c) indicating the body weight; (d) Indicating the epididymal white adipose tissue (eWAT) weight; (e) fasting glucose; (f) glucose tolerance test; (g) insulin tolerance test. *P < 0·05; n = 6.
Figure 2.
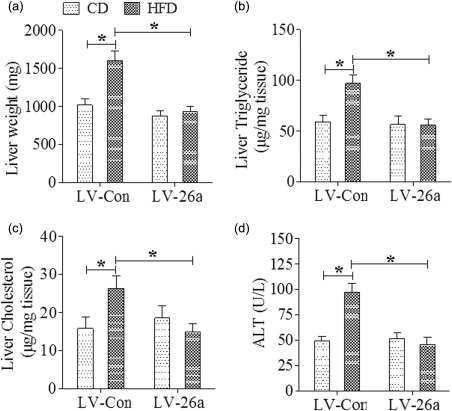
MicroRNA‐26a (Mir‐26a) attenuates hepatocellular damage during high‐fat diet (HFD)‐induced obesity. (a) Indicating total liver weight; (b) indicating hepatic triglyceride levels; (c) indicating hepatic cholesterol levels; (d) indicating serum alanine transaminase (ALT) levels. Analysis performed after 20 weeks of HFD feeding. *P < 0·05; n = 6.
Mir‐26a suppresses hepatic inflammatory responses
We next investigated the hepatic inflammatory responses. HFD consumption increased liver lipid content in LV‐Con‐infected mice, which was decreased significantly in LV‐26a‐treated mice (Fig. 3a). Similarly, after HFD challenge, LV‐26a‐treated mice exhibited decreased infiltration of immune cells in the liver – something attributed to reduce infiltration of TCR‐γδ+, Gr‐1+ cells and CD11b+ cells (Fig. 3b–g).
Figure 3.
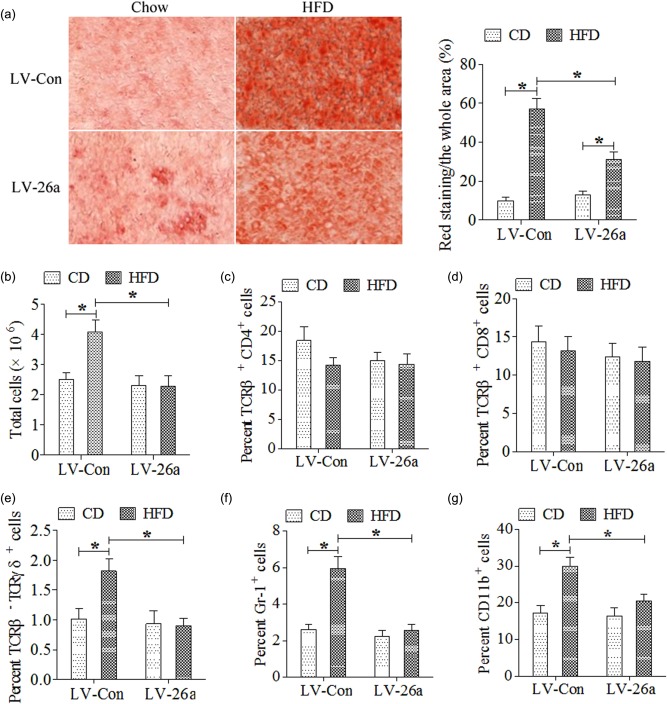
MicroRNA‐26a (Mir‐26a) decreased steatohepatitis on a high‐fat diet (HFD). (a) Representative liver histology (Oil Red O staining) after 20 weeks on chow or HFD; (b) total cells (CD45+) infiltration in liver; (c) T cell receptor (TCR)‐β+CD4+ cells infiltration in liver; (d) TCR‐β+CD8+ infiltration in liver; (e) TCR‐β–TCR‐γδ+ cells infiltration in liver; (f) CD45+ granulocyte‐differentiation antigen‐1 (Gr‐1)+ cells infiltration in liver; (g) CD45+CD11b+ cells infiltration in liver. *P < 0·05; n = 6. [Colour figure can be viewed at wileyonlinelibrary.com]
Although the molecular mechanisms that regulate hepatocellular damage during NAFLD are not well understood, the most favoured hypothesis suggests that steatosis sensitizes hepatocytes to subsequent inflammatory‐ and/or ROS‐driven oxidative stress‐mediated hepatocellular injury and fibrosis 15. Therefore, we quantified hepatic expression of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase components in LV‐Con and LV‐26a‐infected mice. As shown, LV‐26a‐infected mice on HFD had significantly decreased hepatic Nox2, p22phox, p47phox and p67phox mRNA expression, along with decreased lipid peroxidation, as quantified by hepatic 4‐HNE concentrations compared to LV‐Con‐treated mice (Fig. 4a–e).
Figure 4.
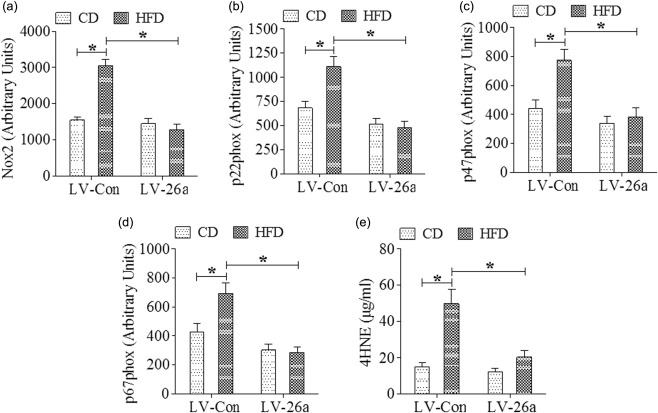
MicroRNA‐26a (Mir‐26a) decreased expression of enzymes driving hepatic reactive oxygen species (ROS) production during high‐fat diet (HFD) stress. Hepatic mRNA expression of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase components and hepatic lipid peroxidation after 20 weeks on diet, (a) Nox2, (b) p22phox, (c) p47phox, (d) p67phox, and (e) 4‐hydroxynonenal (4‐HNE). *P < 0·05; n = 6.
Mir‐26a inhibits the expression of IL‐17 and IL‐6 in vivo and in vitro
A previous paper has indicated the regulatory function of Mir‐26a on IL‐17 expression 6, and IL‐17 signalling has also been demonstrated to accelerate the progression of NAFLD in mice 15; therefore, we first investigated the effect of Mir‐26a on IL‐17 signalling expression in vivo. The results indicated that both the expression of IL‐17 and IL‐6 in liver was decreased significantly in LV‐26a‐infected mice after 20 weeks of HFD feeding when compared with that in LV‐Con‐treated mice (Fig. 5a–c).
Figure 5.
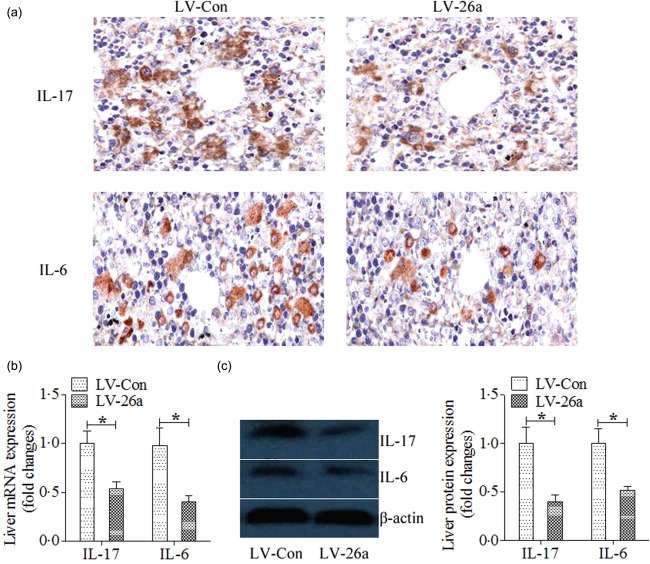
MicroRNA‐26a (Mir‐26a) inhibits the expression of interleukin (IL)−17 and IL‐6 in vivo. (a) Representative immunohistological staining of IL‐17 and IL‐6; (b) mRNA expression of IL‐17 and IL‐6 were analysed by polymerase chain reaction (PCR); (c) protein levels of IL‐17 and IL‐6 were analysed by Western blots. *P < 0·05; n = 6. [Colour figure can be viewed at wileyonlinelibrary.com]
To explore the roles of Mir‐26a in Th17 differentiation in vitro, Mir‐26a was over‐expressed by pre‐Mir‐26a and inhibited by anti‐Mir‐26a. The results showed that the frequency of Th17 was lower in CD4+ T cells which were transfected with pre‐Mir‐26a than in those which were transfected with pre‐Mir‐ctrl and anti‐Mir‐26a (Fig. 6a). In contrast, it was higher in CD4+ T cells which were transfected with anti‐Mir‐26a than in those which were transfected with anti‐Mir‐ctrl (Fig. 6a). Similarly, RAR‐related orphan receptor gamma t (ROR)‐γt expression was decreased in pre‐Mir‐26a groups compared with those in the pre‐Mir‐ctrl groups and anti‐Mir‐26a groups and was increased in the anti‐Mir‐26a groups compared with those in anti‐Mir‐ctrl groups (Fig. 6b). Both IL‐17 and IL‐6 expression in cell culture supernatant were down‐regulated significantly by pre‐Mir‐26a and were up‐regulated significantly by anti‐Mir‐26a (Fig. 6c,d).
Figure 6.

MicroRNA‐26a (Mir‐26a) inhibits the expression of interleukin (IL)−17 and IL‐6 in vitro. Pre‐Mir‐ctrl, pre‐Mir‐26a, anti‐Mir‐ctrl and anti‐Mir‐26a were transfected into CD4+ T cells, which were then activated and polarized. (a) The frequencies of T helper type 17 (Th17) cells in CD4+ T cells were determined by flow cytometry 4 days later. Th17 cells were gated with CD4+IL‐17+. The collective results of three independent experiments are shown in the histograms as mean ± standard deviation (s.d.). (b) The retinoid‐related orphan receptor gamma t (ROR‐γt) mRNA levels were analysed by quantitative polymerase chain reaction (qPCR) 3 days after transfection and activation. (c) The IL‐17 level in cell culture supernatant was detected by enzyme‐linked immunosorbent assay (ELISA) 4 days after transfection. (d) The IL‐6 level in cell culture supernatant was detected by ELISA 4 days after transfection. *P < 0·01; n = 6.
Mir‐26a–IL‐17 axis attenuates NAFLD though inhibition of IL‐6
We first investigated the role of IL‐17 in the development of NAFLD; the results showed that IL‐17 neutralization decreased total liver weight, hepatic triglyceride deposition and serum ALT concentration markedly when compared with the control group (Fig. 7a–c).
Figure 7.
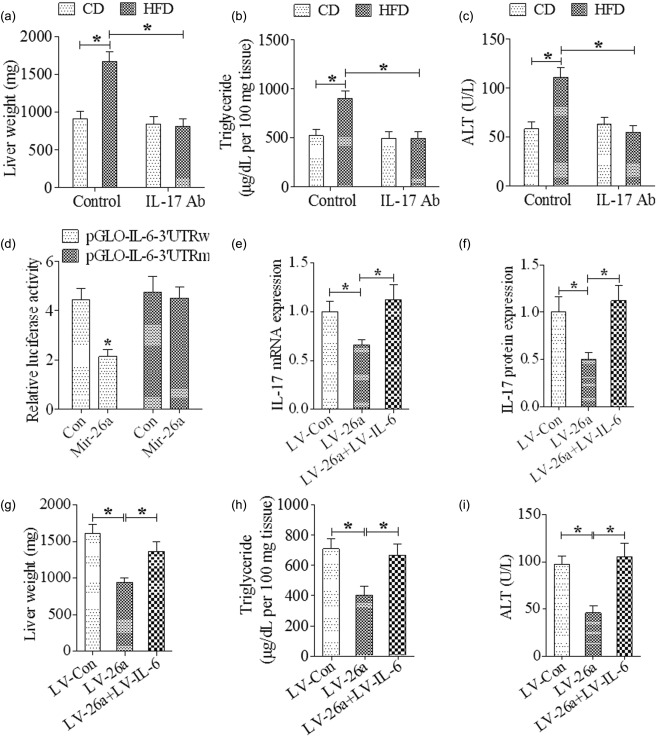
MicroRNA‐26a (Mir‐26a)–IL‐17 axis attenuates non‐alcoholic fatty liver disease (NAFLD) though inhibition of interleukin (IL)−6. (a) Indicating total liver weight after 20 weeks of high‐fat diet (HFD) feeding; (b) indicating hepatic triglyceride levels after 20 weeks of HFD feeding; (c) indicating serum alanine transaminase (ALT) levels after 20 weeks of HFD feeding; (d) luciferase activity assay of HEk293 cells at 36 h after co‐transfection with Mir‐26a and pGLO‐IL‐6‐3′ UTR or pGLO‐IL‐6‐3′ UTR mutant; (e) indicating IL‐17 mRNA level in liver after 20 weeks of HFD feeding; (f) indicating IL‐17 protein level in liver after 20 weeks of HFD feeding; (g) indicating total liver weight after 20 weeks of HFD feeding; (h) indicating hepatic triglyceride levels after 20 weeks of HFD feeding; (i) indicating serum ALT levels after 20 weeks of HFD feeding. *P < 0·01; n = 6.
Through use of the TargetScan database, it was predicted that Mir‐26a has target sites in the 3′ UTR of the IL‐6 mRNA. In order to support our data suggesting the Mir‐26a level was associated inversely with IL‐6, a luciferase reporter assay was performed to determine whether Mir‐26a could directly target the 3′ UTR of IL‐6 mRNA in HEK293 cells. The results showed that Mir‐26a decreased significantly the luciferase reporter activity of wild‐type 3′ UTR but not mutant 3′ UTR of IL‐6 (Fig. 7d).
To investigate further the role of IL‐6 in the suppressed Th17 cell expansion induced by LV‐26a, mice were co‐infected with LV‐26a and LV‐IL‐6 encoding the full‐length IL‐6 coding sequence but without the 3′ UTR. Results showed that the decreased expression of IL‐17 in the liver tissue induced by Mir‐26a was abrogated completely by IL‐6 over‐expression (Fig. 7e,f). The attenuated total liver weight, hepatic triglyceride deposition and serum ALT concentration induced by Mir‐26a were abolished by IL‐6 over‐expression (Fig. 7g–i). The decreased body weight decreased by Mir‐26a was also reversed by IL‐6 over‐expression (data not shown).
Discussion
Our current study provides strong evidence that the Mir‐26a–IL‐6–IL‐17 axis regulates the development of NAFLD in a murine model. LV‐26a‐infected mice were protected from glucose dysmetabolism and showed attenuated NAFLD. LV‐26a‐treated mice also exhibited decreased infiltration of immune cells in the liver – something attributed to reduce infiltration of TCR‐γδ+, Gr‐1+ cells and CD11b+ cells. Mir‐26a inhibits the expression of IL‐17 and IL‐6 in vivo and in vitro. Furthermore, the decreased expression of IL‐17 in the liver tissue induced by Mir‐26a was abrogated completely by IL‐6 over‐expression. The decreased total liver weight, hepatic triglyceride deposition and serum ALT concentration induced by Mir‐26a was also abrogated completely by IL‐6 over‐expression.
Imbalances of both the immune system and metabolism are involved in the development of NAFLD. However, so far the underlying mechanisms have not been elucidated completely. Both the innate immune response and the adaptive immune response participate in the aetiology of alcoholic liver disease. The innate immune system includes macrophages (Küpffer cells), dendritic cells (DC) cells, natural killer (NK) cells, T cells, neutrophils, inflammatory cytokines, acute‐phase response proteins and chemokines 16, 17. The adaptive immune response also increases the severity of NAFLD.
Recently, the Th1/Th2 paradigm has been expanded following the discovery of a distinct T helper cell subset that produces IL‐17 (Th17 cells) 18, 19, 20. Th17 cells produce IL‐17 (also referred to as IL‐17A), IL‐17F, IL‐21 and IL‐22, and mediate potent inflammatory immune responses 21. Obesity is associated with increased IL‐17 production in humans 10. Mirroring this, Th17 cell expansion is observed in obese mice 22. In response to caloric excess, mice lacking IL‐17 exhibit protection from glucose dysmetabolism, despite increased weight gain 23. The IL‐17 axis has also been linked to hepatic injury: an increased frequency of IL‐17A‐expressing cells (and increased IL‐17A expression) in diverse human liver diseases and mouse models of liver injury 24. Further, IL‐17A neutralization or genetic deletion of IL‐17A has been shown to be protective in mouse models of acute hepatitis, drug‐induced liver injury and lipopolysaccharide (LPS)‐induced liver injury during HFD stress 25, 26, 27. IL‐17RA is expressed widely in the liver and IL‐17 drives neutrophil chemokine expression by such cells. Of note, an increased hepatic neutrophil/lymphocyte ratio is predictive of NAFLD progression, and neutrophilic inflammation has been described in human NASH 28, 29. Several recent publications have demonstrated that IL‐17 signalling accelerated the progression of NAFLD 14, 27, 30. In our current study, we found that IL‐17 expression was decreased significantly in vivo and in vitro in the Mir‐26a‐teated group when compared with the control group. Also, IL‐17 neutralization decreased total liver weight, hepatic triglyceride deposition and serum ALT concentration markedly when compared with the control group. All these indicated that the inhibited IL‐17 expression contributed to the attenuated development of NAFLD induced by LV‐26a infection.
IL‐6 is a multi‐functional cytokine with a critical role in host defence, and has important features such as stimulating the hepatic acute phase response to infection and injury. Plasma IL‐6 values were increased in NASH patients in a pilot study 31. Recent studies indicated that IL‐6 contributed to the development of NAFLD 32, 33. IL‐6 has also been demonstrated to play a pivotal role in the dichotomous differentiation programmes of Th17. Under situations with low concentrations of TGF‐β, IL‐6 drives the development of Th17 cells 34. Mir‐26a is a type of highly conserved short RNA that regulates diverse cellular processes by binding to the 3′‐UTR of target messenger RNAs. In our current study, we found that over‐expression of Mir‐26a decreased the luciferase reporter activity of wild‐type 3′‐UTR but not mutant 3′‐UTR of IL‐6. Furthermore, the decreased expression of IL‐17 in the liver tissue induced by Mir‐26a was abrogated completely by IL‐6 overexpression. The decreased total liver weight, hepatic triglyceride deposition and serum ALT concentration induced by Mir‐26a was also abrogated completely by IL‐6 overexpression. All these were consistent with a previous paper demonstrating that Mir‐26a inhibited tumour growth and allograft rejection in part by suppressing IL‐6 7, 35, 36.
In summary, our data document an immune‐regulatory role for the Mir‐26a–IL‐6–IL‐17 axis in the development of NAFLD. Mir‐26a overexpression results in attenuated development of NAFLD and decreased IL‐17 expression in part by inhibition of IL‐6. Although further investigations are needed to clarify fully the precise molecular and cellular mechanism involved in the immunoregulation, Mir‐26a may be a novel therapeutic strategy to protect liver from NAFLD.
Disclosure
The authors declare that they have no competing interests.
Acknowledgement
The project was supported in part by the Bureau of Public Health of Hubei province (JX4B59).
References
- 1. Cho EH. SIRT3 as a regulator of non‐alcoholic fatty liver disease. J Lifestyle Med 2014; 4:80–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. O'Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol 2010; 10:111–22. [DOI] [PubMed] [Google Scholar]
- 3. He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 2004; 5:522–31. [DOI] [PubMed] [Google Scholar]
- 4. Zhang B, Liu XX, He JR et al Pathologically decreased miR‐26a antagonizes apoptosis and facilitates carcinogenesis by targeting MTDH and EZH2 in breast cancer. Carcinogenesis 2011; 32:2–9. [DOI] [PubMed] [Google Scholar]
- 5. Leeper NJ, Raiesdana A, Kojima Y et al MicroRNA‐26a is a novel regulator of vascular smooth muscle cell function. J Cell Physiol 2011; 226:1035–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang R, Tian A, Wang J, Shen X, Qi G, Tang Y. miR26a modulates Th17/T reg balance in the EAE model of multiple sclerosis by targeting IL6. Neuromolecular Med 2015; 17:24–34. [DOI] [PubMed] [Google Scholar]
- 7. Xie F, Chai J, Zhang Z, Hu Q, Ma T. MicroRNA 26a prolongs skin allograft survival and promotes regulatory T cells expansion in mice. Transpl Int 2015; 28:1143–51. [DOI] [PubMed] [Google Scholar]
- 8. Liang S, Wang W, Gou X. MicroRNA 26a modulates regulatory T cells expansion and attenuates renal ischemia‐reperfusion injury. Mol Immunol 2015; 65:321–7. [DOI] [PubMed] [Google Scholar]
- 9. Fu X, Dong B, Tian Y et al MicroRNA‐26a regulates insulin sensitivity and metabolism of glucose and lipids. J Clin Invest 2015; 125:2497–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sumarac‐Dumanovic M, Stevanovic D, Ljubic A et al Increased activity of interleukin‐23/interleukin‐17 proinflammatory axis in obese women. Int J Obes (Lond) 2009; 33:151–6. [DOI] [PubMed] [Google Scholar]
- 11. Kono H, Fujii H, Ogiku M et al Role of IL‐17A in neutrophil recruitment and hepatic injury after warm ischemia‐reperfusion mice. J Immunol 2011; 187:4818–25. [DOI] [PubMed] [Google Scholar]
- 12. Pietrowski E, Bender B, Huppert J, White R, Luhmann HJ, Kuhlmann CR. Pro‐inflammatory effects of interleukin‐17A on vascular smooth muscle cells involve NAD(P)H‐ oxidase derived reactive oxygen species. J Vasc Res 2011; 48:52–8. [DOI] [PubMed] [Google Scholar]
- 13. Rolo AP, Teodoro JS, Palmeira CM. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med 2012; 52:59–69. [DOI] [PubMed] [Google Scholar]
- 14. Tiscornia G, Singer O, Verma IM. Production and purification of lentiviral vectors. Nat Protoc 2006; 1:241–5. [DOI] [PubMed] [Google Scholar]
- 15. Harley IT, Stankiewicz TE, Giles DA et al IL‐17 signaling accelerates the progression of nonalcoholic fatty liver disease in mice. Hepatology 2014; 59:1830–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gao B, Jeong WI, Tian Z. Liver: an organ with predominant innate immunity. Hepatology 2008; 47:729–36. [DOI] [PubMed] [Google Scholar]
- 17. Racanelli V, Rehermann B. The liver as an immunological organ. Hepatology 2006; 43:S54–62. [DOI] [PubMed] [Google Scholar]
- 18. Langrish CL, Chen Y, Blumenschein WM et al IL‐23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 2005; 201:233–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Park H, Li Z, Yang XO et al A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 2005; 6:1133–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Harrington LE, Hatton RD, Mangan PR et al Interleukin 17‐producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 2005; 6:1123–32. [DOI] [PubMed] [Google Scholar]
- 21. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL‐17 and Th17 cells. Annu Rev Immunol 2009; 27:485–517. [DOI] [PubMed] [Google Scholar]
- 22. Ahmed M, Gaffen SL. IL‐17 in obesity and adipogenesis. Cytokine Growth Factor Rev 2010; 21:449–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zuniga LA, Shen WJ, Joyce‐Shaikh B et al IL‐17 regulates adipogenesis, glucose homeostasis, and obesity. J Immunol 2010; 185:6947–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lafdil F, Miller AM, Ki SH, Gao B. Th17 cells and their associated cytokines in liver diseases. Cell Mol Immunol 2010; 7:250–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang W, Ding X, Deng J et al Interferon‐gamma negatively regulates Th17‐mediated immunopathology during mouse hepatitis virus infection. J Mol Med (Berl) 2011; 89:399–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kobayashi M, Higuchi S, Mizuno K et al Interleukin‐17 is involved in alpha‐naphthylisothiocyanate‐induced liver injury in mice. Toxicology 2010; 275:50–7. [DOI] [PubMed] [Google Scholar]
- 27. Tang Y, Bian Z, Zhao L et al Interleukin‐17 exacerbates hepatic steatosis and inflammation in non‐alcoholic fatty liver disease. Clin Exp Immunol 2011; 166:281–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alkhouri N, Morris‐Stiff G, Campbell C et al Neutrophil to lymphocyte ratio: a new marker for predicting steatohepatitis and fibrosis in patients with nonalcoholic fatty liver disease. Liver Int 2012; 32:297–302. [DOI] [PubMed] [Google Scholar]
- 29. Rensen SS, Slaats Y, Nijhuis J et al Increased hepatic myeloperoxidase activity in obese subjects with nonalcoholic steatohepatitis. Am J Pathol 2009; 175:1473–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Giles DA, Moreno‐Fernandez ME, Divanovic S. IL‐17 axis driven inflammation in non‐alcoholic fatty liver disease progression. Curr Drug Targets 2015; 16:1315–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kugelmas M, Hill DB, Vivian B, Marsano L, McClain CJ. Cytokines and NASH: a pilot study of the effects of lifestyle modification and vitamin E. Hepatology 2003; 38:413–9. [DOI] [PubMed] [Google Scholar]
- 32. Cengiz M, Yasar DG, Ergun MA, Akyol G, Ozenirler S. The role of interleukin‐6 and interleukin‐8 gene polymorphisms in non‐alcoholic steatohepatitis. Hepat Mon 2014; 14:e24635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xin G, Qin S, Wang S, Wang X, Zhang Y, Wang J. Sex hormone affects the severity of non‐alcoholic steatohepatitis through the MyD88‐dependent IL‐6 signaling pathway. Exp Biol Med (Maywood) 2015; 240:1279–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li MO, Sanjabi S, Flavell RA. Transforming growth factor‐beta controls development, homeostasis, and tolerance of T cells by regulatory T cell‐dependent and ‐independent mechanisms. Immunity 2006; 25:455–71. [DOI] [PubMed] [Google Scholar]
- 35. Yang X, Liang L, Zhang XF et al MicroRNA‐26a suppresses tumor growth and metastasis of human hepatocellular carcinoma by targeting interleukin‐6‐Stat3 pathway. Hepatology 2013; 58:158–70. [DOI] [PubMed] [Google Scholar]
- 36. Zhang Y, Zhang B, Zhang A et al IL‐6 upregulation contributes to the reduction of miR‐26a expression in hepatocellular carcinoma cells. Braz J Med Biol Res 2013; 46:32–8. [DOI] [PMC free article] [PubMed] [Google Scholar]