

To the Editor
Biochemical, epidemiologic, clinical, and genetic research over several decades has shown that any increment in fetal hemoglobin (HbF) reduces the clinical severity of sickle cell anemia, with significantly improved survival in U.S. patients with HbF levels above the 75% percentile (8.6%) or with an absolute HbF ≥ 0.5 g/dl with hydroxyurea (HU) treatment (1–2). While having 100% F-cells results in a benign condition in compound heterozygotes for HbS and hereditary persistence of HbF (HPFH), a level of 70–75% F-cells has been observed in the milder haplotypes, such as the Arabian-Indian haplotype (3–5). Perhaps the most important protector of the sickle erythrocyte from deoxy HbS polymer induced injury is the concentration of HbF/F-cell. A recent analysis of a population of African patients found low concentrations of HbF/F-cells in sickle cell patients in Tanzania, supporting the importance of this parameter. (6) The amount of HbF/F-cell required to entirely prevent HbS polymerization was recently proposed as a therapeutic target. (1)
To investigate the impact of HU on HbF expression parameters other than total HbF in adult patients, we analyzed F-cells and HbF/F-cell in 56 adult sickle cell disease patients attending a sickle cell clinic for routine care, of whom 33 (60%) were taking hydroxyurea at modest stable doses of 1000–1500 mg/day. Subjects were 20–65 years of age, with median age 31 years; 45% were females. Patients with an acute illness or transfusion within 4 weeks were not included. Proportions of F-cells and mean fluorescent intensity (MFI) of F-cells were analyzed from heparinized peripheral blood by flow cytometry. Cells were stained with a specific HbF antibody (Becton–Dickinson). F-cells and mean fluorescence intensity of positive cells was determined using Cell Quest software and used as an estimate of HbF/F-cell. HbF was analyzed by HPLC (Variant).
The mean HbF in HU-treated subjects was 8.8 % compared to 5.0 % in untreated subjects (Figure 1A), a level nearly identical to that observed in the Multi-Center Study of Hydroxyurea that led to its FDA approval. Mean % F-cells was 34% in HU-treated subjects compared to 22.9 % in untreated subjects, (p=0.01, t-test), shown in Figure 1B. Fourteen of the 33 (42%) of HU-treated subjects demonstrated F-cell proportions ≥ 40%. Mean fluorescent intensity of F-cells in untreated patients compared to HU-treated patients was 37 vs 48 fluorescence units, respectively, shown in Figure 1B (p = 0.01). Several recently identified targeted HbF therapeutic inducing agents which act through differing mechanisms to increase fetal globin mRNA, HbF, and F-cells in vitro and in vivo, including sodium 2,2 dimethylbutyrate (ST20), benserazide (BEN), and the LSD-1 inhibitor RN-1 were evaluated for effects on HbF expression in erythroid progenitor cells cultured from at least 10 sickle cell patients (3). All therapeutic candidates significantly induced fetal globin mRNA levels by 2.5–10-fold above untreated control cells from the same patients (Figure 1C); mean increases above control were 2.5 to 2.8-fold with HU, RN-1, or ST20, (all, p<0.01), 5.8-fold with BEN, p<0.001); and 7-fold with combined treatment with BEN and HU (p=0.01), analyzed by a nonparametric test.
Figure 1.
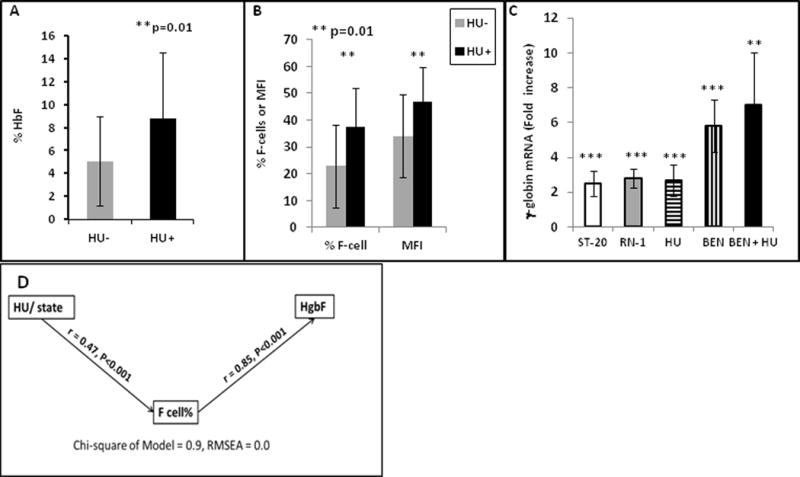
A–B. Mean levels of HbF, F-cells, or MFI in sickle cell patients who were taking HU (dark bars) were significantly higher in treated than in untreated patients. C. Fetal (γ)-globin mRNA in erythroid progenitors cultured from sickle cell patients is significantly increased (*) with added therapeutics compared to untreated controls from the same patient, from 2.5 to 2.8-fold with ST20, RN-1, or HU, 5.8-fold with BEN (p< 0.001), and 7-fold with BEN + HU (n=10, p = 0.011). Standard deviation is shown by the vertical bars. D. Diagram of the Pathway analysis, demonstrating that the main effect of Hydroxyurea (HU) is due to increases in % F-cells, which in turn contribute to total HbF.
Therapeutic targets for amelioration of clinical severity of sickle cell disease have been proposed as 20–30% HbF, 70–75% F-cells, and 10 pg HbF/cell, twice the threshold of 4–6 pg/cell which is the minimum previously detectable in flow cytometry assays (1). F-cells undergo selective survival and have longer lifespans than non-F cells (3–5). We used a pathway analysis to deconstruct the total effects of hydroxyurea as either direct (HbF) or indirect (mediated by F-cell percentage). Pathway analysis tests a hypothetical pathway from predictors to responses against observed data using multiple regression equations. Standardized regression coefficients are computed for each relationship, adjusted for the other relationships, and shown next to each line connecting predictors to responses, and is shown for the patient data in Figure 1D. This analysis indicates that HU contributes first to higher proportions of F-cells (r=0.47, p< 0.001), and secondly to the amount of HbF (r=0.85, p<0.01), whereas, in contrast, a direct effect of HU to HbF was not statistically significant (r = 0.09, P = 0.3). In this analysis, 82% of the total effect of HU on HbF is an indirect effect mediated by F-cells. These data suggest that addition of a second, or perhaps multiple, HbF inducers may produce higher concentrations of HbF content in erythroid cells which differentiate with, or are primed by, HU. The findings here particularly suggest that addition of benserazide as a second therapeutic with HU may induce HbF expression closer to therapeutic targets proposed. As individual patients have highly variable baseline HbF expression patterns, monitoring these parameters may guide treatments to ameliorate clinical severity and indicate when multiple therapies are warranted.
Acknowledgments
This work was supported by NIH Grants 1P50 HL-118006, R01 DK-52962, R41 HL-108516, and R42 HL-110727. The content is solely the responsibility of the authors and does not necessarily represent the official view of the National Institutes of Health.
Footnotes
Author Contributions
Y. Dai, J. Sangerman, A. D. Faller, D. Maharaj and X. Niu performed assays; A. Rock, O. Owoyemi, and P. Oneal obtained clinical correlations; S. Perrine, M. Nouraie, M. Steinberg, analyzed results and wrote the paper. M. Nouraie performed statistical analyses. S. Nekhai, S. Cui, and R. Taylor reviewed the manuscript.
Conflict of Interest: S. Perrine: Inventor on patents related to this work
References
- 1.Steinberg MH, Chui DHK, Dover GJ, Sebastiani P, Alsultan A. Fetal hemoglobin in sickle cell anemia: a glass half full? Blood. 2014;123:481–485. doi: 10.1182/blood-2013-09-528067. [DOI] [PubMed] [Google Scholar]
- 2.Steinberg MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up. Am J Hematol. 2010;85:403–408. doi: 10.1002/ajh.21699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perrine SP, Pace BS, Faller DV. Targeted fetal hemoglobin induction for treatment of beta hemoglobinopathies. Hematol Oncol Clin N Amer. 2014;28:233–248. doi: 10.1016/j.hoc.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 4.Boyer SH, Dover GJ, Serjeant GR, et al. Production of F-cells in sickle cell anemia: regulation by a genetic locus or loci separate from the beta globin cluster. Blood. 1984;64:1053–1058. [PubMed] [Google Scholar]
- 5.Franco RS, Yasin Z, Palascak MB, Ciraolo P, et al. The effect of fetal hemoglobin on the survival characteristics of sickle cells. Blood. 2006;108:1073–6. doi: 10.1182/blood-2005-09-008318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Urio F, Lyimo M, Mtatiro SN, Cox SE, Mmbando BO, Makani J. High prevalence of individuals with low concentration of fetal hemoglobin in F-cells in sickle cell anemia in Tanzania. Am J Hematol. 2016;91(8):E323–324. doi: 10.1002/ajh.24390. [DOI] [PMC free article] [PubMed] [Google Scholar]