

Abstract
Metal sulfide minerals are assumed to form naturally at ambient conditions via reaction of a metallic element with (poly)sulfide ions, usually produced by microbes in oxygen-depleted environments. Recently, the formation of mercury sulfide (β-HgS) directly from linear Hg(II)-thiolate complexes (Hg(SR)2) in natural organic matter and in cysteine solutions was demonstrated under aerated conditions. Here, a detailed description of this non-sulfidic reaction is provided by computations at a high level of molecular-orbital theory. The HgS stoichiometry is obtained through the cleavage of the S-C bond in one thiolate, transfer of the resulting alkyl group (R’) to another thiolate, and subsequent elimination of a sulfur atom from the second thiolate as a thioether (RSR’). Repetition of this mechanism leads to the formation of RS-(HgS)n-R chains which may self-assemble in parallel arrays to form cinnabar (α-HgS), or more commonly, quickly condense to four-coordinate metacinnabar (β-HgS). The mechanistic pathway is thermodynamically favorable and its predicted kinetics agrees with experiment. The results provide robust theoretical support for the abiotic natural formation of nanoparticulate HgS under oxic conditions and in the absence of a catalyst, and suggest a new route for the (bio)synthesis of HgS nanoparticles with improved technological properties.
Nucleation of metal sulfide solids typically occurs when solubility is exceeded by elevated concentration of reduced sulfur, metal cation, or both components1,2. In environmental aquatic systems, metal ions are commonly complexed with natural organic matter or inorganic anions, including sulfide, and free sulfide ions (S(-II)) produced mainly by dissimilatory sulfate reducing microbes3,4 are considered necessary for solid nucleation. Sulfide can also be generated in the laboratory from intracellular cysteine by photosynthetic aerobic microorganisms5,6 and from decomposition of sulfur compounds, such as thioglycolic acid, thioglycerol, dithiocarbamate, thioacetamide, and cystine, by hydrothermal, solvothermal, and biomimetic synthesis routes, sonochemical reaction, microwave irradiation, and hydrolysis7,8,9,10,11,12,13,14,15,16,17,18.
Recently it was shown that sulfide ions were not required to form a metal sulfide solid19. Metacinnabar (β-HgS) precipitated directly from linear Hg-thiolate complexes (Hg(SR)2) in natural organic matter (NOM) and from Hg-dicysteinate complexes (Hg(Cys)2) in aerated and deaerated aqueous solutions in the dark without a catalyzing agent. These results are relevant to soil and aquatic systems, especially in cases where organo-sulfide is the dominant sulfide source. The reaction was rather slow and took several days for Hg(II) complexed to NOM at a concentration of 30–200 mg of Hg/kg of NOM dry weight (ppm). A global reaction pathway was proposed that has similarities to one suggested for β-HgS precipitation in sodium hydrosulfide (NaHS) solution20,21. In its reaction with NaHS, Hg(II) initially forms an unstable low coordination chain-type complex (–S-Hg-S-Hg-S-) that rapidly transforms to a four-coordinate mercury sulfide with the short range ordering of β-HgS. The disordered β-HgS nanostructures eventually yield β-HgS crystals. In the case of thiolate as the source of reduced sulfur, the starting reactant is the linear Hg(SR)2 complex (RS-Hg-SR), which is the most stable coordination of mononuclear Hg with thiolate ligands at neutral and acidic pH22,23. Because β-HgS nanostructures appear rapidly once –S-Hg-S-Hg-S- chains are formed in sulfidic solution20, we infer that formation of the chain structure limits the rate of formation of β-HgS from Hg(SR)2. The pathway proposed19 for chain formation in natural organic matter is the cleavage of the S-R bond according to the reaction:
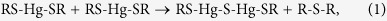 |
followed by growth of the chain through the addition of new Hg(SR)2 complexes:
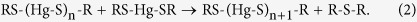 |
Given that β-HgS also was obtained from Hg-dicysteinate complexes (Hg(Cys)2)19, the R group in natural organic matter can be an alkyl ligand of the general form CH2-R’. Thus, reaction (1) involves the cleavage of a S-C bond in an R’-CH2-S-Hg-S-CH2-R’ entity. It can be described as a transfer of an alkyl group between two aliphatic thiolates (SR−) followed by dissociation of the resultant R-S-R thioether and bonding of Hg to the exposed S. Elimination of one sulfur from two Hg(SR)2 complexes decreases the S to Hg ratio from 4:2 to 3:2 in the mercury product and triggers the formation of HgS when the reaction is repeated as in (2).
Here, we present quantum chemical calculations of the structure and energetics of the transition state in reaction (1) that support our previous experimental results. The results show that the proposed dealkylation of the Hg(SR)2 complex is thermodynamically allowed and has an activation free-energy barrier consistent with the kinetics of formation of β-HgS in natural organic matter. We also discuss how cinnabar (α-HgS) and metacinnabar are formed by the proposed reaction mechanism directly from Hg-thiolate complexes in the absence of any catalyst or external reagent.
Results
Structural mechanism of dealkylation
According to (1), the free reactants (FRs) are two linear Hg-thiolate complexes of formula RS-Hg-SR. The R group was represented in the computational work by a methyl group (CH3). This simplification has been validated previously on stability calculations of Hg(II) complexes with thiolate and thioether ligands22,24, and is also justified by the independence of the dissociation energy of the R−SH bond with respect to the nature of the R radical25. The direct transfer of an alkyl group between the two Hg-thiolate complexes may be regarded as a nucleophilic substitution with two sulfur atoms as nucleophilic centers. Thus, the three directly interacting atoms, that is, the donor sulfur (Sd), the acceptor sulfur (Sa), and the C atom of the CH3 group, should be collinear in the transition state (TS) to provide an adequate overlap of orbitals (Fig. 1). Based on the equivalence of the four S atoms, the transition state has a configurational degeneracy of eight. It decays to an intermediate product (IP) in which the Sd atom is placed nearly equidistant with respect to the two CH3 groups carried by Sa. An internal rearrangement of the system leads to a more stable configuration for the product complex (PC), in which the Sd atom bonds to the Hg atom of the acceptor complex (Hga) to form the mercury sulfide dimer Hg2S3(CH3)2. The scan of the potential energy surface with respect to the Sd-Hga distance shows that this rearrangement occurs with practically no energy barrier (Supplementary Fig. S1). Simultaneously, the CH3-Sa-CH3 group (thiodimethane) moves away to a Hga-Sa distance of 3.52 Å. It can further dissociate from the mercury sulfide dimer, leaving the two as free products (FPs).
Figure 1. Mechanistic pathway of formation of a Hg(II) sulfide dimer by dealkylation38 of two Hg-thiolate complexes.

Gibbs free energy diagram (at 298 K and 1 atm) of the cleavage of the S-C bond by an alkyl group transfer between two linear Hg-thiolate complexes, and optimized structures for the reaction pathway with four explicit water molecules (not shown for clarity). The height of the activation-energy barrier for the alkyl group transfer relative to the free reactant state decreases from 39.1 kcal mol−1 to 36.2 kcal mol−1 with two explicit water molecules, to 34.7 kcal mol−1 with four, and to 31.9 kcal mol−1 with seven. The final value, corrected for overestimation of the solvation entropy in the continuum solvation models is 22 kcal mol−1. The same correction applies to the IP and PC states (corrected levels not shown). FR = free reactants; TS = transition state; IP = intermediate product; PC = product complex; FP = free products. Bond lengths are in angstroms. Dark red, Hg; yellow, thiolate sulfur SR− and sulfide sulfur HgSHg; orange, thioether sulfur RSR; dark gray, C; light gray, H. Cartesian coordinates are given in the Supplementary Materials.
Energetics of dealkylation
The transition state has an activation free-energy barrier of 39.1 kcal mol−1 without water molecules in the reaction core. Better estimates of the free energy are obtained when explicit water molecules are added to Hg(II) complexes to account for strong short-range hydrogen bonding interactions between the anion (here CH3S−) and the solvent26. The length of the Sd…H hydrogen bonds effectively decreased from 2.37 Å in the free reactants to 2.24 Å in the transition-state structure when two water molecules were placed near the Sd atom, thus confirming the importance of solute-solvent covalent interactions26,27 (see Supplementary Materials). Overall, the activation energy decreased to 36.2 kcal mol−1 with two explicit water molecules, 34.7 kcal mol−1 with four, and 31.9 kcal mol−1 with seven (Fig. 1 and Supplementary Fig. S2). In the model with seven water molecules, the specific interactions between the three reactive ligands, Sa, Sd. and CH3, and the solvent are integrally taken into account since all the related hydrogen bonds are formed.
The energy barrier of 31.9 kcal mol−1 is lowered to about 22 kcal mol−1 after correcting for improper evaluation of the solvation entropy in the continuum solvent models28,29,30,31 (see Supplementary Materials). The same Gibbs free energy correction applies to the intermediate product (IP) and to the product complex (PC). To compare with experiment, the range of reaction times reported for the formation of β-HgS in natural organic matter and from Hg(Cys)219 indicates an energy barrier on the order of 24 kcal mol−1, as estimated from Eyring’s formula32 for the reaction rate constant. The predicted value is close enough to the experimental value to validate the proposed reaction mechanism.
One might expect the transfer of a methyl group between two identical atoms (Sd and Sa) to be reversible. The back transfer of the methyl group here is unlikely because of the internal rearrangement of the system leading to the product-complex state. This state is more stable than the free-reactant state by −8.0 kcal mol−1, and further decays to the free product state which is −10.3 kcal mol−1 lower in energy than the free reactants (as calculated with 4 H2O, Fig. 1). Although the Hg atoms are not directly involved in the nucleophilic substitution, they play a key role in the product rearrangement through the intermolecular Hg-S forces and the attractive intramolecular short-range Hg-Hg interactions of van der Waals type33,34,35.
An alternative to the dealkylation reaction is the insertion of the Hg atom from one linear Hg-thiolate complex between the S and C atoms of the S-C bond from the other complex, as observed in coordination complexes with Co(III) and W(III)36,37. Calculations performed for two possible reaction pathways each gives a high Gibbs free energy for the intermediate product (see Supplementary Materials).
Formation of HgS
The -S-(Hg-S)n-Hg-S- chain formed by repetition of the dealkylation mechanism has a specific conformation (Fig. 2a). Because the sulfur ligands are linearly coordinated to Hg, the chain conformation is completely determined by the Hg-S-Hg angle and the S-S-S-S dihedral angle defined by four successive S atoms. The first angle ranges from 89.5° to 95.6° and the second from −86.7 to −101.4° in the optimized Hg6S7(CH3)2 model shown in Figure 2a. This conformation is close to that in cinnabar (α-HgS), which has infinite chains throughout its structure with a Hg-S-Hg angle of 104.7° and a dihedral S-S-S-S angle of −98.5°38 (Fig. 2b,c). A primitive α-HgS nanostructure, as observed experimentally in aqueous solution with sodium hydrosulfide (NaHS) before the subsequent formation of β-HgS20, is obtained by optimizing the geometry of three HS-(Hg-S)3-Hg-SH chains in aqueous solution (Fig. 2d). Once formed, the zigzag -S-(Hg-S)n-Hg-S- chains self-assemble to make the trimer 3[Hg4S5H2], which is geometrically comparable to three neighboring Hg4S5 units in cinnabar. The coordination around the Hg atoms in α-HgS is “2 + 4”, with two short intra-chain Hg-S bonds 2.37 Å in length and four long inter-chain Hg-S bonds of 3.10–3.29 Å38. Similarly, the cohesion of the HS-(Hg-S)3-Hg-SH aggregate is realized by inter-chain Hg-S bonds ranging from 3.15 Å to 3.37 Å. In aqueous solution with sodium hydrosulfide (NaHS), the early-formed 2 + n (n < 4) coordination is unstable and quickly evolves to a 4 coordination (i.e., tetrahedral) with the local ordering of metacinnabar (β-HgS)20. The same transformation is assumed to occur in natural organic matter because only nanoparticulate β-HgS is detected19. The 2 + n to 4 transition is however difficult to model because β-HgS is thermodynamically metastable at room temperature21,39,40.
Figure 2. Formation of cinnabar by association of -S-(Hg-S)n-Hg-S- chains.
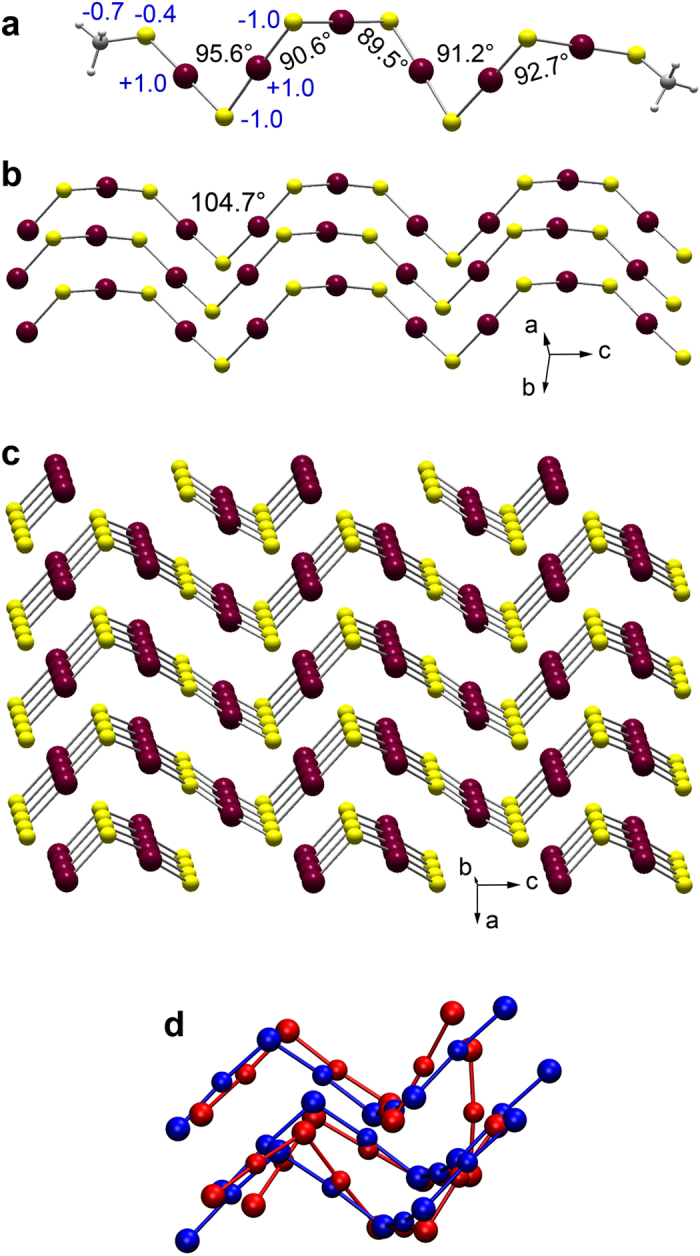
(a) Hg6S7(CH3)2 model optimized in aqueous solution with the CPCM model. Hg-S-Hg angles are in black. Atomic charges, in units of elementary charge e and calculated by natural population analysis (NPA)61, are in blue. Hg (dark red) has a natural charge of +1.0 e, sulfide S (yellow) of -1.0 e, thiol S (yellow) of −0.4 e, C (dark gray) of −0.7 e, and H (light gray) of +0.2 e (not represented). (b) Three parallel -S-(Hg-S)n-Hg-S- chains in cinnabar38. (c) Cinnabar as an assemblage of replicated chains. d) Best superposition of the trimer 3[Hg4S5H2] optimized in aqueous solution (red, H atoms not shown) and three fragments of adjacent Hg4S5 chains from the structure of cinnabar (blue).
We have proposed a new mechanism for cleavage of the S-C bond of thiolate in the presence of Hg(II), based on the transfer of one alkyl group (R) between two linear Hg-thiolate complexes (Hg(SR)2), and elimination of a sulfur atom by formation of a thioether (RSR). This reaction initially produces a mercury sulfide dimer and subsequently mercury sulfide clusters if replicated. The mechanism provides robust theoretical support for the experimental nucleation of nanoparticulate metacinnabar from Hg(II)-thiolate complexes in natural organic matter and from Hg-dicysteinate complexes19. It also offers an explanation for the occurrence of metacinnabar under oxic conditions in soils19,41,42, for what has been termed ‘old’ soil mercury, i.e., mercury deposited from the atmosphere that becomes relatively recalcitrant within weeks to months43,44,45, and for metal sulfides associated with dissolved natural organic matter in river water46. The nucleation of HgS particles from Hg-thiolate complexes is significantly slower, therefore yields less defective structures than with free sulfides20 because the sulfur release is controlled by a non-negligible energy barrier. This could lead to interesting effects on the size, shape, and crystallinity of metacinnabar nanocrystals and improved control over (bio)synthesis, structures, properties, and functionality of this technologically important material10,13,15,18.
Method
Calculations were performed with GAUSSIAN 0947 using a computational method validated previously on Hg-thiolate complexes22. All calculations were performed in aqueous solutions using the supermolecule-continuum solvent model, as developed in the framework of the conductor-like polarizable continuum model CPCM48, which allows explicit water molecules in contact with the reactants in a continuum bulk solvent. The geometry optimizations were performed using the second-order Møller-Plesset perturbation theory (MP2)49, and single point energies were evaluated using the hybrid method Integrated Molecular Orbital and Molecular Orbital (IMOMO)50, ONIOM version51,52. The IMOMO method combines calculations of energies at two levels of theory: a higher one applied to a limited part of the system (called the “model system”, here the Hg-thiolate complexes without explicit water molecules) and a lower one applied to the whole system (called the “real system”) which includes water molecules. The model system was treated at the coupled cluster level of theory with single and double substitutions and corrections for triple substitutions (CCSD(T))53,54,55,56 and the real system was treated at the MP2 level. The C, H, and O centers were treated using the aug-cc-pVDZ basis set57 while the S centers were represented at the aug-cc-pVTZ level58. The mercury atom was treated using the Stuttgart-Dresden-Bonn quasirelativistic pseudopotentials (SDD)59 for the core electrons and the associated valence basis set (describing 20 valence electrons of Hg). Two polarization functions of f type taken from ref.60 were added in order to ameliorate the Hg basis set. Other computational details are given in the Supplementary Materials.
Additional Information
How to cite this article: Enescu, M. et al. Nucleation of mercury sulfide by dealkylation. Sci. Rep. 6, 39359; doi: 10.1038/srep39359 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
Support was provided by the French National Research Agency (ANR) under grant ANR-12-BS06-0008-01 (Mercurius Project). The Mesocenter from Franche-Comté and the Froggy platform of the CIMENT Infrastructure in Grenoble (ANR Grant ANR-10-EQPX- 29-01) provided computing resources.
Footnotes
Author Contributions M.E. and A.M. designed the research and performed calculations. M.E., A.M. and K.L.N. discussed results and wrote the manuscript.
References
- Wilkin R. T. & Barnes H. L. Formation processes of framboidal pyrite. Geochim. Cosmochim. Acta 61, 323–339 (1997). [Google Scholar]
- Rickard D. & Luther G. W. Chemistry of iron sulfides. Chem. Rev. 107, 514–562 (2007). [DOI] [PubMed] [Google Scholar]
- Moreau J. W. et al. Extracellular proteins limit the dispersal of biogenic nanoparticles. Science 316, 1600–1603 (2007). [DOI] [PubMed] [Google Scholar]
- Labrenz M. et al. Formation of sphalerite (ZnS) deposits in natural biofilms of sulfate-reducing bacteria. Science 290, 1744–1747 (2000). [DOI] [PubMed] [Google Scholar]
- Lefebvre D. D., Kelly D. & Budd K. Biotransformation of Hg(II) by cyanobacteria. Appl. Environ. Microbiol. 73, 243–249 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards C. D., Beatty J. C., Loiselle J. B. R., Vlassov K. A. & Lefebvre D. D. Aerobic transformation of zinc into metal sulfide by photosynthetic microorganisms. Appl. Microbiol. Biotechnol. 97, 3613–3623 (2013). [DOI] [PubMed] [Google Scholar]
- Breen M. L., Dinsmore A. D., Pink R. H., Qadri S. B. & Ratna B. R. Sonochemically produced ZnS-coated polystyrene core-shell particles for use in photonic crystals. Langmuir 17, 903–907 (2001). [Google Scholar]
- Zhu J. J., Zhou M. G., Xu J. Z. & Liao X. H. Preparation of CdS and ZnS nanoparticles using microwave irradiation. Mater. Lett. 47, 25–29 (2001). [Google Scholar]
- Wang H., Zhang H. R. & Zhu J. J. A microwave assisted heating method for the rapid synthesis of sphalerite-type mercury sulfide nanocrystals with different sizes. J. Cryst. Growth 233, 829–836 (2001). [Google Scholar]
- Higginson K. A. et al. Synthesis and characterization of colloidal β-HgS quantum dots. J. Phys. Chem. B 106, 9982–9985 (2002). [Google Scholar]
- Wang H. & Zhu J. J. A sonochemical method for the selective synthesis of α-HgS and β-HgS nanoparticles. Ultrason. Sonochem. 11, 293–300 (2004). [DOI] [PubMed] [Google Scholar]
- Qin D. Z. et al. Biomimetic synthesis of HgS nanoparticles in the bovine serum albumin solution. J. Nanopart. Res. 10, 559–566 (2008). [Google Scholar]
- Onwudiwe D. C. & Ajibade P. A. ZnS, CdS and HgS nanoparticles via alkyl-phenyl dithiocarbamate complexes as single source precursors. Int. J. Mol. Sci. 12, 5538–5551 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haveli S. D. et al. Hair fiber as a nanoreactor in controlled synthesis of fluorescent gold nanoparticles. Nano Lett. 12, 6212–6217 (2012). [DOI] [PubMed] [Google Scholar]
- Liu X. et al. Antibody-biotemplated HgS nanoparticles: Extremely sensitive labels for atomic fluorescence spectrometric immunoassay. Analyst 137, 1473–1480 (2012). [DOI] [PubMed] [Google Scholar]
- Zhang L. et al. Synthesis of HgS nanocrystals in the Lysozyme aqueous solution through biomimetic method. Appl. Surf. Sci. 258, 8185–8191 (2012). [Google Scholar]
- Patriarche G., Walter P., Van Eslandre E., Ayache J. & Castaing J. Characteristics of HgS nanoparticles formed in hair by a chemical reaction. Phil. Mag. 93, 137–151 (2013). [Google Scholar]
- Judy-Azar A. R. & Mohebbi S. An easy route to synthesize superfine meta cinnabar (β-HgS) semiconductor nanoparticles and their optical properties. Mater. Lett. 106, 233–237 (2013). [Google Scholar]
- Manceau A. et al. Formation of mercury sulfide from Hg(II)-thiolate complexes in natural organic matter. Environ. Sci. Technol. 49, 9787−9796 (2015). [DOI] [PubMed] [Google Scholar]
- Charnock J. M. et al. The structural evolution of mercury sulfide precipitate: an XAS and XRD study. Am. Miner. 88, 1197–1203 (2003). [Google Scholar]
- Bell A. M. T., Pattrick R. A. D. & Vaughan D. J. Structural evolution of aqueous mercury sulphide precipitates: energy-dispersive X-ray diffraction studies. Miner. Mag. 74, 85–96 (2010). [Google Scholar]
- Enescu M. & Manceau A. High-level ab initio calculation of the stability of mercury–thiolate complexes. Theor. Chem. Acc. 133, n° 1457 (2014). [Google Scholar]
- Mah V. & Jalilehvand F. Glutathione complex formation with mercury(II) in aqueous solution at physiological pH. Chem. Res. Toxicol. 23, 1815–1823 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manceau A. et al. Structure, bonding, and stability of mercury complexes with thiolate and thioether ligands from high-resolution XANES spectroscopy and first-principles calculations. Inorg. Chem. 54, 11776−11791 (2015). [DOI] [PubMed] [Google Scholar]
- Mackle H. The thermochemistry of sulfur-containing molecules and radicals-II The dissociation energies of bonds involving sulphur: the heats of formation of sulphur-containing radicals. Tetrahedron 19, 1159–1170 (1963). [Google Scholar]
- Afaneh A. T., Schreckenbach G. & Wang F. Y. Theoretical study of the formation of mercury (Hg2+) complexes in solution using an explicit solvation shell in implicit solvent calculations. J. Phys. Chem. B 118, 11271–11283 (2014). [DOI] [PubMed] [Google Scholar]
- Reed A. E., Curtiss L. A. & Weinhold F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 88, 899–926 (1988). [Google Scholar]
- Giesen D. J., Cramer C. J. & Truhlar D. G. Entropic contributions to free-energies of solvation. J. Phys. Chem. 98, 4141–4147 (1994). [Google Scholar]
- Yu Y. B., Privalov P. L. & Hodges R. S. Contribution of translational and rotational motions to molecular association in aqueous solution. Biophys. J. 81, 1632–1642 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertz D. H. Relationship between the gas-phase entropies of molecules and their entropies of solvation in water and 1-octanol. J. Am. Chem. Soc. 102, 5316–5322 (1980). [Google Scholar]
- Lau J. K. C. & Deubel D. V. Hydrolysis of the anticancer drug cisplatin: Pitfalls in the interpretation of quantum chemical calculations. J. Chem. Theor. Comput. 2, 103–106 (2006). [DOI] [PubMed] [Google Scholar]
- Cardey B. & Enescu M. A computational study of thiolate and selenolate oxidation by hydrogen peroxide. Chemphyschem 6, 1175–1180 (2005). [DOI] [PubMed] [Google Scholar]
- Schwerdtfeger P., Li J. & Pyykko P. The polarizability of Hg and the ground-state interaction potential of Hg2. Theor. Chim. Acta 87, 313–320 (1994). [Google Scholar]
- Pyykko P. Strong closed-shell interactions in inorganic chemistry. Chem. Rev. 97, 597–636 (1997). [DOI] [PubMed] [Google Scholar]
- Pyykko P. & Straka M. Ab initio studies of the dimers (HgH2)2 and (HgMe2)2. Metallophilic attraction and the van der Waals radii of mercury. Phys. Chem. Chem. Phys. 2, 2489–2493 (2000). [Google Scholar]
- Rajsekhar G., Rao C. P., Saarenketo P. K., Kolehmainen E. & Rissanen K. C.-S. bond cleavage by cobalt: synthesis, characterization and crystal structure determination of 1,2-di-(o-salicylaldiminophenylthio)ethane and its Co(III) product with C-S bond cleaved fragments. Inorg. Chem. Comm. 5, 649–652 (2002). [Google Scholar]
- Boorman P. M., Gao X. L. & Parvez M. C-S bond-cleavage in reactions of thiolate nucleophiles with bridging thioethers in anionic ditungsten(III) complexes. J. Chem.l Soc. - Dalton Trans., 25–31 (1992). [Google Scholar]
- Auvray P. & Genet F. Affinement de la structure cristalline du cinabre α-HgS. Bull. Soc. Fr. Mineral. Cristallogr. 96, 218–219 (1973). [Google Scholar]
- Potter R. W. & Barnes H. L. Phase relations in binary Hg-S. Am. Miner. 63, 1143–1152 (1978). [Google Scholar]
- Cardona M. et al. Electronic, vibrational, and thermodynamic properties of metacinnabar β-HgS, HgSe, and HgTe. Phys. Rev. B 80, 195204 (2009). [Google Scholar]
- Harris L. A. et al. Imaging and microanalyses of mercury in flood plain soils of East Fork Poplar Creek. Water Air Soil Poll. 86, 51–69 (1996). [Google Scholar]
- Barnett M. O. et al. Formation of mercuric sulfide in soil. Environ. Sci. Technol. 31, 3037–3043 (1997). [Google Scholar]
- Hintelmann H. et al. Reactivity and mobility of new and old mercury deposition in a Boreal forest ecosystem during the first year of the METAALICUS study. Environ. Sci. Technol. 36, 5034–5040 (2002). [DOI] [PubMed] [Google Scholar]
- Demers J. D., Blum J. D. & Zak D. R. Mercury isotopes in a forested ecosystem: Implications for air-surface exchange dynamics and the global mercury cycle. Global Geochem. Cycles 27, 222–238 (2013). [Google Scholar]
- Obrist D., Pokharel A. K. & Moore C. Vertical profile measurements of soil air suggest immobilization of gaseous elemental mercury in mineral soil. Environ. Sci. Technol. 48, 2242–2252 (2014). [DOI] [PubMed] [Google Scholar]
- Rozan T. F., Lassman M. E., Ridge D. P. & Luther G. W. Evidence for iron, copper and zinc complexation as multinuclear sulphide clusters in oxic rivers. Nature 406, 879–882 (2000). [DOI] [PubMed] [Google Scholar]
- Frisch M. J. et al. Gaussian 09, Revision A.1., (Gaussian, Inc., Wallingford CT, 2009). [Google Scholar]
- Cossi M., Rega N., Scalmani G. & Barone V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 24, 669–681 (2003). [DOI] [PubMed] [Google Scholar]
- Møller C. & Plesset M. S. Note on an approximation treatment for many-electron systems. Phys. Rev. 46, 618–622 (1934). [Google Scholar]
- Humbel S., Sieber S. & Morokuma K. The IMOMO method: Integration of different levels of molecular orbital approximations for geometry optimization of large systems: Test for n-butane conformation and S(N)2 reaction: RCl + Cl. J. Chem. Phys. 105, 1959–1967 (1996). [Google Scholar]
- Svensson M. et al. ONIOM: A multilayered integrated MO + MM method for geometry optimizations and single point energy predictions. A test for Diels-Alder reactions and Pt(P(t-Bu)3)2 + H2 oxidative addition. J. Phys. Chem. 100, 19357–19363 (1996). [Google Scholar]
- Dapprich S., Komaromi I., Byun K. S., Morokuma K. & Frisch M. J. A new ONIOM implementation in Gaussian98. Part I. The calculation of energies, gradients, vibrational frequencies and electric field derivatives. J. Mol. Str. - Theochem. 461, 1–21 (1999). [Google Scholar]
- Scuseria G. E., Janssen C. L. & Schaefer H. F. An efficient reformulation of the closed-shell coupled cluster single and double excitation (CCSD) equations. J. Chem. Phys. 89, 7382–7387 (1988). [Google Scholar]
- Pople J. A., Headgordon M. & Raghavachari K. Quadratic configuration-interaction - A general technique for determining electron correlation energies. J. Chem. Phys. 87, 5968–5975 (1987). [Google Scholar]
- Raghavachari K., Trucks G. W., Pople J. A. & Head-Gordon M. A fifth-order perturbation comparison of electron correlation theories. Chem. Phys. Lett. 157, 479–483 (1989). [Google Scholar]
- Watts J. D., Gauss J. & Bartlett R. J. Coupled-cluster methods with noniterative triple excitations for restricted open-shell Hartree-Fock and other general single determinant reference functions - Energies and analytical gradients. J. Chem. Phys. 98, 8718–8733 (1993). [Google Scholar]
- Dunning T. H. Gaussian-basis sets for use in correlated molecular calculations. 1. The atoms boron through neon and hydrogen. J. Chem. Phys. 90, 1007–1023 (1989). [Google Scholar]
- Woon D. E. & Dunning T. H. Gaussian-basis sets for use in correlated molecular calculations. 3. The atoms aluminum through argon. J. Chem. Phys. 98, 1358–1371 (1993). [Google Scholar]
- Andrae D., Haussermann U., Dolg M., Stoll H. & Preuss H. Energy-adjusted ab initio pseudopotentials for the second and third row transition-elements. Theor. Chim. Acta 77, 123–141 (1990). [Google Scholar]
- Martin J. M. L. & Sundermann A. Correlation consistent valence basis sets for use with the Stuttgart-Dresden-Bonn relativistic effective core potentials: The atoms Ga-Kr and In-Xe. J. Chem. Phys. 114, 3408–3420 (2001). [Google Scholar]
- Glendening E. D., Landis C. R. & Weinhold F. NBO 6.0: Natural bond orbital analysis program. J. Comput. Chem. 34, 1429–1437 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.