

Abstract
Commercial priorities have been identified as negative factors in drug development. We trace the problem to inattention to sound clinical pharmacology practices. When properly applied, clinical pharmacology and associated drug development sciences can, hand in hand, facilitate success in commercial drug development.
Schneider and Lahiri, in “The Perils of Alzheimer’s Drug Development” [1], expand on concerns [2] with how sponsors and investigators potentially fail drugs by using flawed methods. In recent articles [2; 3], we described an attitude towards drug development that gives priority to the unfolding clinical pharmacology of the drug. We argued for a clinical pharmacological science able to guide investigators from step to step in development. In contrast, Schneider and Lahiri document how sponsors and investigators can respond to commercial priorities without adequate consideration of sound clinical pharmacological practices. Perhaps this occurs in part because clinical pharmacology in recent decades developed into a commercially over-influenced technology that does not take advantage of advances in other medical sciences.
To document consequences from sponsors’ misplaced priorities Schneider and Lahiri discuss Axonyx’s commercial development of phenserine, a cholinesterase inhibitor with pre- clinical evidence for potential neuroprotection consequent to its ability to lower brain amyloid-β peptide production [4, 5]. They point out that the symptomatic development of phenserine neglects the very much sought after possible disease modifying effects suggested in preclinical research with phenserine. This latter seems the scientifically and medically most consequential direction for development. With Schneider and Lahiri, we can only ascribe the decision to undertake symptomatic development to commercial priorities that displaced assumptions essential to scientifically sound and clinically more consequential development.
We propose that it is important to build on Schneider’s and Lahiri’s observations, for example, that drugs like phenserine were developed “trying to gain the financial interest of a deep-pocketed partner” [1] rather than with priority given to sound methodologies and to obtaining both clinically and scientifically consequential results. Certainly the “general lack of learning in the field” noted by Schneider and Lahiri derives in part from the majority of AD drug studies remaining unpublished because their negative outcomes do not benefit the commercial aims of sponsors [3]. We suspect as even more fundamental sources for lack of learning by sponsors and investigators underlying dispositions that allow commercial priorities to dismiss, without measured consideration, issues of clinical pharmacology likely to arise during a drug’s development. The planning and design of the commercial metrifonate clinical trials (CT) illustrate this neglect of a drug’s clinical pharmacology.
Prior to university licensing of metrifonate for commercial development, preclinical and early clinical use provided strong evidence for toxicity. We were aware that toxicity could potentially be mediated through excessive acetylcholinesterase (AChE) inhibition [6, 7], inhibition of other enzymes affected by the organophosphate class of drugs [8], by tolerance of low doses allowing too rapid dose escalation and irreversible enzyme inhibition producing cumulative drug effects [9, 10], other factors not known to us, and cautions we found in the literature.
Two pools of AChE were known in relation to the architectural structures in brain. One pool circulates freely within the cytoplasm of neurons and in the cerebrospinal-interstitial fluid of the central nervous system. A second pool consists of AChE inserted in or attached to the cell walls and concentrated on membranes within synaptic areas. AChE is synthesized in neurons and then activated as cytoplasmic AChE, fixed to the exterior cell wall and secreted into the extracellular space spontaneously and during neurotransmitter release [14]. There was evidence for different rates of turnover for the unbound soluble AChE and the membrane bound AChE. Karprzak and Salpeter[15] found long stability of about 20 days for mouse muscle synaptic AChE although Fernandez and Stiles [16] reported total recovery of synaptic activity within about 3 days after organophosphate inhibition. Rotundo and Fambrough [17] found a half-life of membrane-bound AChE in muscle cells of 50 hours and of intracellular AChE in chick muscle cells of 2–3 hours. In addition to the two active pools there was an apparently more extensive pool of newly synthesized but inactive intracellular AChE, the majority of which is rapidly degraded before it becomes active [18].
AChE was known to exist in monomeric, dimeric, tetrameric, and asymmetric forms with the more complex forms assembled from the G1 monomer. Forms have different distributions in the body [14]. Goosens et al. [19] reported no differences between the recovery rates of free G1 and membrane bound G4 forms in rat brain. Moss et al. [20] found a recovery half-time after phenylmethylsulfonyl fluoride of 11 days in whole rat brain compared to 1, 3, and 6 days for ileum, heart, and pectoralis muscle, respectively. Unni et al. [21] estimated a 2.21+/−1.22 day recovery half-time of CSF AChE in AD patients treated chronically with metrifonate. We interpreted this data to imply that membrane bound synaptic AChE recovery and possibly recovery of other enzymes important for organophosphate toxicity may have a 14 day or longer half-time recovery in spite of more rapid recovery of free CSF AChE.
To insure safety while dosing metrifonate we assumed a functional brain membrane bound G4 pool of AChE with recovery half-time of 14 days. Using the data published by Unni et al. [21] and others we calculated, for weekly and daily metrifonate administrations, the expected dose-AChE inhibition responses for RBCs, CSF, and brain. (Figure 1). We noted the risk for progression to high levels of AChE inhibition associated with daily dosing (Figure 1). We regarded these risks as important because they were made possible by properties of metrifonate: cumulative irreversible enzyme inhibition; lack of adverse effects with stepwise dosing increases; and so forth. Assuming a t1/2 recovery of 14 days, predicted brain AChE inhibitions and possible equivalent levels of inhibition of other important enzymes, such as the carboxyesterases, neurotoxin esterase, and so forth, provided strong reasons to develop a weekly dosing regimen if possible. Later we considered these factors as possible explanations for the severe adverse events experienced with the daily dosing used in Bayer’s Phase III CTs.
Figure 1.
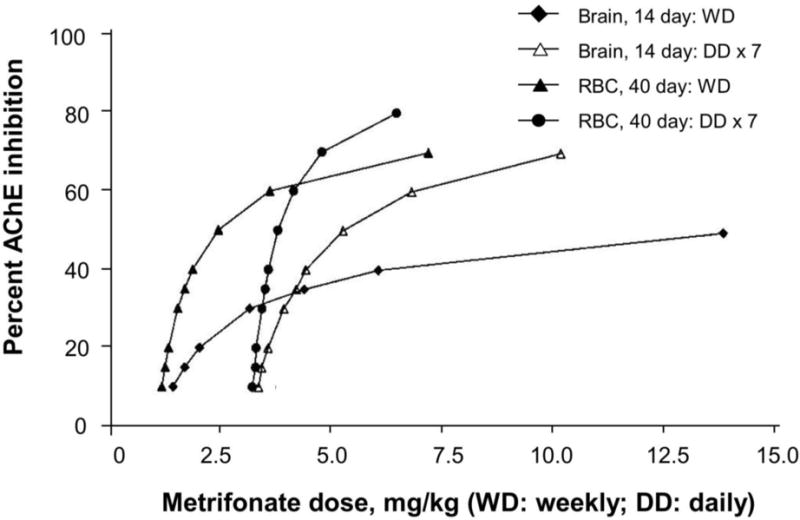
Predicted CNS membrane bound AChE inhibition after weekly (WD) and daily (DD) dosing of Alzheimer’s disease patients with metrifonate. For comparison data are provided for red blood cell (RBC) AChE inhibition after weekly and daily dosing of Alzheimer’s disease patients with metrifonate.
In response to this understanding we developed a weekly dosing schedule to provide for regeneration of enzymes irreversibly inhibited with each dosing of metrifonate. We showed for weekly dosing a U-shaped dose-response curve with maximum cognitive benefits at about 45% red blood cell (RBC) AChE inhibition [10, 11]. The decreased drug effectiveness at higher levels of inhibition fit our assumptions that receptor depolarization block might occur at high acetylcholine concentrations. After initial clinical successes using weekly dosing in two small clinical trials [12, 13], we did not pursue higher dosing or replace weekly with daily dosing. The licensee, Bayer, showed no evidence of increased efficacy with the daily dosing schedules they chose for commercial development. We regard Bayer’s decision to use daily dosing, explained at the time of licensing as required to make development profitable, as an excellent additional example of how commercial orientations facilitate but also, if unsupported by sound clinical pharmacological facts and practices, potentially undermine AD drug developments. Such decisions clearly are not made lightly by companies, whether multinational, like Bayer, or virtual, like Axonyx. In the absence of large scale grant funding for new drug developments and academic centers committed to clinical pharmacology research, commercial realities will continue to drive almost all drug development programs. We do not argue against commercial motivations for decisions, only against drug development decisions being reached without providing or taking adequate account of sound clinical pharmacological practices relevant to the decisions.
In sum, what do we recommend to render current AD drug development practices more effective? In our papers we have encouraged attitudes, assumptions, and practices that regard drug developments as informed by clinical pharmacological progressions through various animal species. The human is the most problematic species for the clinical pharmacologist to study. Consequently we argue for development in two or more non-human species of experimental conditions, assay methods, confirmations of dependent variable changes, and, as much as possible, other methods needed for a CT. This work provides the model then applied in the human species [2].
This model assumes that many critical variables will not be measurable in humans—drug concentrations at brain targets, molecular changes induced by drug in brain, and so forth. We see investigators using non-human species to establish robust patterns of change that can be applied to interpret human studies. For hypothesis testing the investigator uses changes in variables accessible in human studies to argue for parallel changes in inaccessible variables. These patterns ideally address variables able to confirm the concept of a drug’s therapeutic mechanism.
We now recognize how errors and biases can too easily lead to Type II errors in AD CTs [1–3]. Demonstrated patterns of mechanism changes induced by drug in multiple species make CTs tests of potential therapeutic mechanisms in AD and more than tests of one compound’s efficacy. Standardizations of assays and investigative methods in non-human species remove effects on CT data due to changes in methods as one goes from bench and non-humans to human trials. With stability of methods and conditions across experiments investigators anticipate consistency in humans with patterns documented in non-human animals and investigate inconsistencies before dismissing the research hypotheses.
This attitude—regarding humans as one of various animal species studied to understand the properties of drugs—and its recommended practices do not replace commercial considerations. Properly applied, clinical pharmacology and associated drug development sciences such as medical statistics facilitate sponsors realizing commercial aims for drugs they bring to development. The current failure to respect the teachings of these sciences concerns us. Expanding lists of risks to drug development witness to the need for 1) more attention to what constitutes adequately sound clinical pharmacological AD drug development practices and 2) better understanding of how clinical pharmacology integrates with commercial realities of drug development.
Acknowledgments
The authors (REB and NHG) are supported in part by the Intramural Research Program of the National Institute on Aging, NIH. The author LKU’s participation in this research was entirely completed prior to employment at Sigma-Aldrich Corporation. This research in no way reflects upon the views of Sigma-Aldrich Corporation.
Footnotes
The authors have received no other funding for this work and have no financial conflicts of interest.
References
- 1.Schneider LS, Lahiri DK. The Perils of Alzheimer’s Drug Development. Curr Alzheimer Res. 2009;6(1):77–8. doi: 10.2174/156720509787313871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Becker RE, Greig NH. Alzheimer’s disease drug development in 2008 and beyond: problems and opportunities. Curr Alzheimer Res. 2008;5(4):346–57. doi: 10.2174/156720508785132299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Becker RE, Greig NH, Giacobini E. Why do so many drugs for Alzheimer’s disease fail in development? Time for new methods and new practices? J Alzheimer Dis. 2008;15(2):303–25. doi: 10.3233/jad-2008-15213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greig NH, Sambamurti K, Yu QS, Brossi A, Bruinsma GB, Lahiri DK. An overview of phenserine tartrate, a novel acetylcholinesterase inhibitor for the treatment of Alzheimer’s disease. Curr Alzheimer Res. 2005;2(3):281–90. doi: 10.2174/1567205054367829. [DOI] [PubMed] [Google Scholar]
- 5.Klein J. Phenserine. Expert Opin Investig Drugs. 2007;16(7):1087–97. doi: 10.1517/13543784.16.7.1087. [DOI] [PubMed] [Google Scholar]
- 6.World Health Organization. Dichlorvos. Geneva: World Health Organization; 1989. http://www.inchem.org/documents/ehc/ehc/ehc79.htm. [Google Scholar]
- 7.World Health Organization. Trichlorfon. Geneva: World Health Organization; 1992. http://www.inchem.org/documents/ehc/ehc/ehc132.htm. [Google Scholar]
- 8.Taylor P. Anticholinesterase agents. In: Gilman AG, Rall TW, Nies AS, Taylor P, editors. The Pharmacological Basis of Therapeutics. Eighth Edition, New York: Pergamon Press; 1990. pp. 131–149. [Google Scholar]
- 9.Becker RE, Giacobini E. Mechanisms of Cholinesterase Inhibition in Senile Dementia of Alzheimer’s Type - Clinical, Pharmacological, and Therapeutical Aspects. Drug Dev Res. 1988;12:163–195. [Google Scholar]
- 10.Becker R, Colliver J, Elble R, Feldman E, Giacobini E, Kumar V, Markwell S, Moriearty P, Parks R, Shillcutt S, Unni L, Vicari S, Womack C, Zec R. Effects of Metifonate, A Long Acting Cholinesterase Inhibitor in Alzheimer Disease: Report of an Open Trial. Drug Dev Res. 1990;9:425–434. [Google Scholar]
- 11.Becker R, Moriearty P, Unni L. The Second Generation of Cholinesterase Inhibitors: Clinical and Pharmacological Effects. In: Becker R, Giacobini E, editors. Cholinergic Basis of Alzheimer Therapy. Birkhäuser; Cambridge, Mass: 1991. pp. 263–296. [Google Scholar]
- 12.Becker RE, Colliver JA, Markwell SJ, Moriearty PL, Unni LK, Vicari S. A Double-Blind, Placebo-Controlled Study of Metrifonate, an Acetylcholinesterase Inhibitor, for Alzheimer’s Disease. Alzheimer’s Disease and Associated Disorders. 1996;10(3):124–131. doi: 10.1097/00002093-199601030-00003. [DOI] [PubMed] [Google Scholar]
- 13.Becker RE, Colliver JA, Markwell SJ, Moriearty P, Unni LK, Varney A, Vicari S. A Six Month Study of the Effects of Metrifonate on Cognitive Decline in Alzheimer’s Disease. Alzheimer’s Disease and Associated Disorders. 1998;12(1):54–57. doi: 10.1097/00002093-199803000-00009. [DOI] [PubMed] [Google Scholar]
- 14.Schweitzer ES. Regulated and constitutive secretion of distinct molecular forms of acetylcholinesterase from PC12 cells. J Cell Sci. 1993;106:731–740. doi: 10.1242/jcs.106.3.731. [DOI] [PubMed] [Google Scholar]
- 15.Kasprzak H, Salpeter MM. Recovery of acetylcholinesterase at intact neuromuscular junctions after in vivo inactivation with di-isopropylfluorophosphate. J Neurosci. 1985;5:951–955. doi: 10.1523/JNEUROSCI.05-04-00951.1985. 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fernandez HL, Stiles JR. Intra- versus extracellular recovery of 16S acetylcholinesterase following organophosphate inactivation in the rat. Neurosci Lett. 1984;49:117–122. doi: 10.1016/0304-3940(84)90146-0. [DOI] [PubMed] [Google Scholar]
- 17.Rotundo RL, Fambough DM. Synthesis, transport and fate of acetylcholinesterase in cultured chick embryos muscle cells. Cell. 1980;22:583–594. doi: 10.1016/0092-8674(80)90368-2. [DOI] [PubMed] [Google Scholar]
- 18.Rotundo RL. Asymmetric acetylcholinesterase is assembled in the Golgi apparatus. Proc Natl Acad Sci U S A. 1984;81:479–483. doi: 10.1073/pnas.81.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goossens P, Viret J, Leterrier F. Rat brain acetylcholinesterase turnover in vivo: use of a radioactive methylphosphonothiate irreversible inhibitor. Biochem Biophys Res Commun. 1984;123:71–77. doi: 10.1016/0006-291x(84)90381-4. [DOI] [PubMed] [Google Scholar]
- 20.Moss DE, Rodriguez LA, Selim S, Ellert SO, Devine JV, Steger R. The sulfonly fluorides: CNS selective cholinesterase inhibitors with potential value in Alzheimer’s disease. In: Hutton JT, Kenny AD, editors. Senile Dementia of the Alzheimer Type. New York: Alan R. Liss; 1985. pp. 337–350. [Google Scholar]
- 21.Unni L, Vicari S, Moriearty P, Schaefer F, Becker R. The recovery of cerebrospinal fluid acetylcholinesterase activity in Alzheimer’s disease patients after treatment with metrifonate. Methods Find Clin Pharmacol. 2000;22:57–61. doi: 10.1358/mf.2000.22.1.795849. [DOI] [PubMed] [Google Scholar]