

Supplemental Digital Content is Available in the Text.
Key Words: HIV-1 subtype C, first-line ART, mathematical modeling, viral dynamics, basic reproductive ratio, in vivo fitness
Abstract
Background:
Despite the high prevalence of HIV-1 subtype C (HIV-1C) worldwide, information on HIV-1C viral dynamics and response to antiretroviral therapy (ART) is limited. We sought to measure viral load decay dynamics during treatment and estimate the within-host basic reproductive ratio, R0, and the critical efficacy, εc, for successful treatment of HIV-1C infection.
Methods:
Individuals initiated on first-line ART in India and monitored for 6 months of treatment were considered. Viral load, CD4+ count, and adherence data were collected at baseline, 4, 12, 16 and 24 weeks after ART initiation. Drug resistance genotyping was performed at baseline. R0 and εc were estimated using a mathematical model.
Results:
Among 257 patients with complete data, mean baseline viral load was 5.7 log10 copies per milliliter and median CD4+ count was 165 cells per cubic millimeter. Primary drug resistance was present in 3.1% at baseline. At 6 months, 87.5% had undetectable viral load, indicating excellent response to ART despite high baseline viremia. After excluding those with transmitted resistance, suboptimal adherence and viral rebound, data from 112 patients were analyzed using a mathematical model. We estimated the median R0 to be 5.3. The corresponding εc was ∼0.8.
Conclusions:
These estimates of R0 and εc are smaller than current estimates for HIV-1B, suggesting that HIV-1C exhibits lower in vivo fitness compared with HIV-1B, which allows successful treatment despite high baseline viral loads. The lower fitness, and potentially lower virulence, together with high viral loads may underlie the heightened transmission potential of HIV-1C and its growing global spread.
INTRODUCTION
Among the most remarkable achievements in the battle against HIV has been the discovery of the effect of combination antiretroviral therapy (ART) on survival, made possible by early studies on mathematical modeling of viral load changes after ART initiation.1,2 Modeling studies have since yielded key insights into the pathogenesis of HIV, including the evolution of drug resistance and immune escape, and guided strategies of intervention.3,4 Much of this knowledge has been gleaned from studies on patients with HIV-1 subtype B (HIV-1B) infection. In the present context of the HIV epidemic, HIV-1 subtype C (HIV-1C) is the most prevalent subtype, particularly in low and middle-income countries (LMIC) like India, South Africa, and Ethiopia, and accounts for nearly 48% of all global infections.5 Whether knowledge gleaned from studies of HIV-1B infection can be applied to HIV-1C infection remains unknown. For instance, baseline viral loads in HIV-1C–infected individuals are significantly higher than in HIV-1B–infected individuals.6,7 Yet, effective first-line ART roll out in LMIC has resulted in excellent treatment response, similar to those in high-income countries.8–10 Viral dynamics modeling studies, which could potentially explain these differences and present subtype-specific guidelines for intervention, are limited within the HIV-1C setting and often restricted to ART-naive individuals.11,12
We undertook this study with the aim of determining viral dynamics in HIV-1C–infected individuals initiating first-line ART in India. By analyzing viral load measurements in these individuals at baseline and after starting ART using a mathematical model, we estimated the basic reproductive ratio, R0, for within-host HIV-1C infection. The within-host R0 is defined as the number of secondary infected cells produced by 1 infected cell in a wholly susceptible cell population.3 R0 is thus a measure of the ability of a virus to establish lasting infection among host cells and quantifies the infective and replicative fitness of the viral strain; the larger the value of R0, the more fit the strain. Successful therapy must necessarily drive R0 below 1 to ensure that replicating virus does not establish lasting infection. The minimum efficacy of therapy that achieves this target is defined as the critical efficacy, εc. More fit strains are associated with higher εc, placing more stringent requirements on therapy. Estimates of the within-host R0 and hence εc exist for HIV-1B,13–15 but are lacking for HIV-1C. This is the first study on viral decay dynamics in HIV-1C–infected subjects on treatment, with findings that have implications for refining treatment strategies in countries with predominantly HIV-1C infections.
METHODS
Study Population
HIV-infected individuals initiating first-line ART according to the Indian national guidelines16 were included in this study after informed consent. ART regimens consisted of 2 nucleoside reverse transcriptase inhibitors (zidovudine or tenofovir with lamivudine) and 1 nonnucleoside reverse transcriptase inhibitor (nevirapine or efavirenz) in the form of fixed-dose combination pills. At each visit, ART adherence was rigorously measured by means of a pill count, taking into account all drugs within the regimen in the form of dispensed pills, any lost or missed pills, pills obtained from other sources, and remaining pills. Laboratory measurements included CD4+ T cells count measured using FACSCalibur flow cytometer (BD Biosciences, San Jose, CA), and HIV-1 viral load measured by Abbott m2000rt system (Abbott Molecular Diagnostics, Wiesbaden, Germany). Patients were followed at 4, 12, 24, 36, and 48 weeks after ART initiation, until virological failure, death, or loss to follow-up. Virological failure was defined as having viral load >150 copies per milliliter on 2 consecutive measurements sampled at least 1 month apart. Individuals were classified as “responders” if their viral load was undetectable by 6 months of ART and “nonresponders” if their viral load continued to be detectable at 6 months or beyond.
Subtyping and Drug Resistance Genotyping
Subtyping was carried out using a maximum likelihood phylogenetic tree construction method as described previously.17 Drug resistance genotyping was performed using a standard procedure involving nested polymerase chain reaction to amplify the regions corresponding to 17–235 amino acids of the polymerase (pol) gene, followed by bidirectional population sequencing18 and identification of drug resistance mutations as the World Health Organization 2009 recommendations.19
Mathematical Modeling and Analysis of Patient Data
Mathematical Model
Following previous studies,20 we constructed a mathematical model to analyze viral load decline after the initiation of treatment. The model considers the time evolution of productively infected cells and long-lived infected cells under treatment that blocks de novo infection of target cells and predicts the ensuing viral load changes using the following equations20:
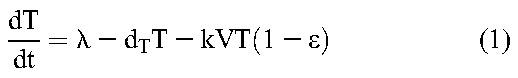 |
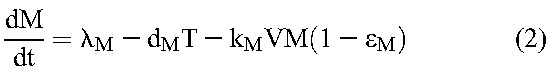 |
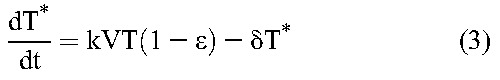 |
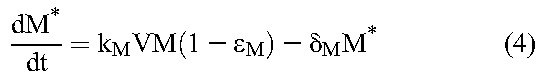 |
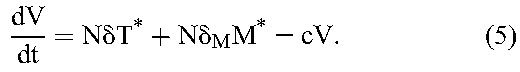 |
Here, target CD4+ T cells, T, and long-lived uninfected cells, M, are produced at rates λ and λM, die with first-order rate constants dT and dM, and are infected by free virions, V, with second-order infection rate constants k and kM, giving rise to productively infected cells, T*, and long-lived infected cells, M*, respectively. Drug action blocks these infection events with efficacy, ε and εM, respectively. Infected cells die with first-order rate constants, δ and δM, respectively, releasing virions at the burst size N per cell. Free virions are cleared with a first-order rate constant c.
Viral Load Decline During Therapy
Viral production and clearance are typically rapid compared with target cell infection and death.2,20 The pseudo-steady-state approximation, dV/dt≈0, then yields
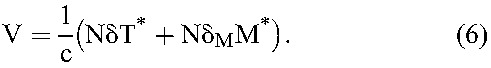 |
We obtained the dynamics of T* and M* as follows. We expect drug efficacy ε and εM to depend on adherence. For patients with optimal (>95%) adherence, we expect ε = εM≈1. (We also examined the possibility of submaximal efficacy despite optimal adherence21 and found it not to influence our key findings.) Equations 3 and 4 then yield 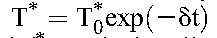 and
and 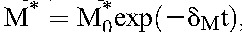 , where
, where 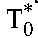 and
and 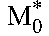 are the baseline populations of productively and long-lived infected cells, respectively. Combining the latter expressions with Equation 6 yields the predicted viral load decline in patients with optimal adherence:
are the baseline populations of productively and long-lived infected cells, respectively. Combining the latter expressions with Equation 6 yields the predicted viral load decline in patients with optimal adherence:
where 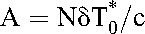 and
and 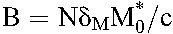 . The first and second phase slopes thus yield the death rates, δ and δM, and the preexponentials A and B the baseline populations,
. The first and second phase slopes thus yield the death rates, δ and δM, and the preexponentials A and B the baseline populations, 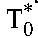 and
and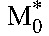 .
.
Model Fitting to Patient Data
We fit our model, Equation 7, to the viral load data from individual patients with >95% reported adherence. The first viral load measurement was 4 weeks after initiation of ART. The first phase of viral load decline, thought to last 1–2 weeks after treatment initiation,22 was thus not discernable from the available data. We therefore assumed δ, which sets the first phase slope, to be 1 per day (d−1) based on previous studies analyzing the kinetics of HIV-1B infection.23,24 Sensitivity analysis suggested that this assumption did not influence our key findings. Model fits then yielded robust estimates of the second phase slope, δM, as well as the constants A and B. We recognize that while the viral load is expected to decline in multiple phases, our measurement frequency allowed estimation of a single slope past the first phase, which we call the second phase slope. For fitting, we considered data from each patient until the time point at which the viral load was detectable or first became undetectable (<150 copies/mL). Data fitting was performed using the nonlinear regression routine LSQCURVEFIT in MATLAB.
Basic Reproductive Ratio and Critical Efficacy
To estimate the basic reproductive ratio, R0, we followed a previous derivation25 and obtained
where 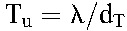 and
and 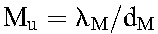 are the populations of CD4+ T cells and long-lived cells in uninfected individuals. Typically, the population of long-lived cells is small compared with CD4+ T cells,20 so that
are the populations of CD4+ T cells and long-lived cells in uninfected individuals. Typically, the population of long-lived cells is small compared with CD4+ T cells,20 so that 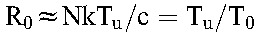 , where we recognize that
, where we recognize that 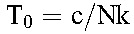 is the pool of uninfected cells in a chronically infected individual before treatment.
is the pool of uninfected cells in a chronically infected individual before treatment.
From the estimated values of the constants A and B in Equation 7 and using the viral burst size N = 50,000 virions per cell26 and the viral clearance rate c = 23/d27 of HIV-1B, we obtained estimates of the baseline populations 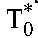 and
and 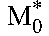 . (The estimate of R0 was insensitive to variations in N and is independent of the intracellular delay when expressed as
. (The estimate of R0 was insensitive to variations in N and is independent of the intracellular delay when expressed as 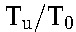 .) Subtracting
.) Subtracting 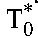 from the baseline CD4+ T-cell count yields the population of uninfected cells, T0, at baseline. (We assume that
from the baseline CD4+ T-cell count yields the population of uninfected cells, T0, at baseline. (We assume that 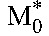 represents a population of cells distinct from CD4+ T cells, such as macrophages.20) Dividing the CD4+ T-cell count in uninfected individuals by T0 yields R0.3 When
represents a population of cells distinct from CD4+ T cells, such as macrophages.20) Dividing the CD4+ T-cell count in uninfected individuals by T0 yields R0.3 When 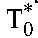 is small compared with T0, the baseline CD4+ T-cell count yields a good approximation to T0, using which in the ratio
is small compared with T0, the baseline CD4+ T-cell count yields a good approximation to T0, using which in the ratio 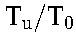 we obtain estimates of R0 from a much larger pool of patients (including those not amenable to analysis using our model above because of suboptimal adherence). We also computed the latter ratio using previously published data on HIV-1B1,2,20,28 for comparison. We examined the implications of the variation in Tu across individuals by randomly sampling Tu for each individual from its known distribution in South Indians and recalculated R0. We repeated this 100 times and determined the variation in the median R0.
we obtain estimates of R0 from a much larger pool of patients (including those not amenable to analysis using our model above because of suboptimal adherence). We also computed the latter ratio using previously published data on HIV-1B1,2,20,28 for comparison. We examined the implications of the variation in Tu across individuals by randomly sampling Tu for each individual from its known distribution in South Indians and recalculated R0. We repeated this 100 times and determined the variation in the median R0.
During treatment, following the derivation above, 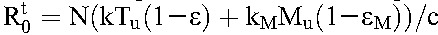 . If we assume that drug efficacy does not depend significantly on the cell type, so that
. If we assume that drug efficacy does not depend significantly on the cell type, so that 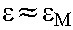 , then
, then 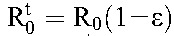 . The critical efficacy, εc, is that value of ε at which
. The critical efficacy, εc, is that value of ε at which 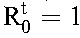 . It follows that29
. It follows that29
 |
RESULTS
Patient Characteristics and Association With Virological Response
Between 2010 and 2011, 257 individuals with HIV-1C infection initiated ART at the Infectious Diseases Clinic at St John's Medical College and were followed for the first 6 months of treatment or longer. Mean age among the 257 patients was 36.7 years (SD, 8.2), and 60% were men. Median viral load before ART initiation was 5.7 log10 copies per milliliter (interquartile range (IQR): 5.3–6.1); and median CD4+ T-cell count was 165 cells/mm3 (IQR: 99–220). Zidovudine-based ART was used in 82.1% (211/257), whereas nevirapine-based ART was initiated for 65.8% (169/257). Primary drug resistance was identified in 3.1% (8/257); 3 had nucleoside reverse transcriptase inhibitors and 5 had nonnucleoside reverse transcriptase inhibitors–related resistance. High baseline viral loads, >5 log10 copies per milliliter, were seen in 85.2% (219/257) of the individuals. After ART initiation, 56.0% (144/257) of the patients achieved viral load <150 copies per milliliter at 3 months and 87.5% (225/257) at 6 months.
Viral Load Decay During Treatment and Analysis Using Mathematical Model
For analysis of viral decay dynamics using our mathematical model, we selected patients who were likely to have been in immunological steady state at baseline, had optimal adherence, and had persistent virological suppression. From the 257 patients, we thus excluded those with primary resistance (n = 8). We also excluded patients with baseline CD4 counts <100 per microliter (n = 66), as they were more likely to have progressive disease and not be in the asymptomatic chronic steady state at treatment initiation. Furthermore, patients who showed undetectable viremia at the first available time point of 4 weeks were also not amenable to analysis and therefore excluded (n = 24). We did not consider those with viral rebound during the period analyzed (n = 24), as they were likely to have (undetected) resistance. Finally, we excluded those with suboptimal (<95%) adherence (n = 19). Viral load data from the remaining 116 patients were analyzed with our mathematical model.
A biphasic decline of the viral load was observed, which was fit well by the model (Fig. 1; also see Figures S1 and S2, Supplemental Digital Content, http://links.lww.com/QAI/A845). In patients with optimal (>95%) adherence, where the drug combination may be assumed to be maximally suppressive (ε≈1), the second phase slope yielded the loss rate of long-lived infected cells, δM = 0.030 ± 0.015 d−1, corresponding to a median lifespan of 35 days (IQR: 26–51) of these cells (see Figure S3, Supplemental Digital Content, http://links.lww.com/QAI/A845). Four patients proved to be outliers, with estimated lifespans of long-lived infected cells >180 days (see Figure S3, Supplemental Digital Content, http://links.lww.com/QAI/A845). We excluded these patients in the subsequent analysis. For the remaining 112 patients, we found δM = 0.030 ± 0.014 d−1, corresponding to a median lifespan of 35 days (IQR: 26–49) of the long-lived infected cells. We also obtained estimates of the baseline populations of productively infected (ie, infected cells that were actively producing virions) and long-lived infected cells and found that on average ∼81% of the infected cells at baseline were productively infected. Several distinct long-lived cell types are associated with the decay of viral load past the first phase.22 Our estimates of the lifespan above are thus to be viewed as weighted averages of the lifespans of these distinct cell types relevant over the 6-month study period. Furthermore, uncertainties may be introduced in our estimates from data of patients in whom viral loads became undetectable by week 12 (n = 67). Here, the last data point is set to the assay limit of detection (LOD). The true viral loads are likely to be lower than the limit of detection resulting in our second phase slopes being underestimates. Considering only patients with viral load detectable at week 12 (n = 45), we found δM = 0.034 ± 0.017 d−1, corresponding to a median lifespan of 32 days (IQR: 22–51), with ∼77% of the infected cells at baseline productively infected (see Figure S3, Supplemental Digital Content, http://links.lww.com/QAI/A845). The latter estimates were not significantly different from those above (P = 0.48 using a 2-tailed Student t test with unequal variance), justifying the use of all 112 patients together for further analysis.
FIGURE 1.
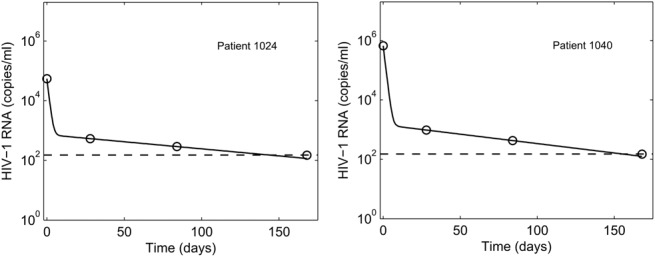
Representative fits (solid lines) of our model predictions (Equation 7) to data (symbols) from 2 patients (left and right panels) of plasma HIV-1 RNA levels as a function of time following the onset of treatment. Dashed lines represent the limit of detection (150 copies/ml). Fits to all the patients analyzed are in Figures, Supplemental Digital Content 1 and Supplemental Digital Content 2, http://links.lww.com/QAI/A845. The resulting best-fit parameter estimates are in Table, Supplemental Digital Content 6, http://links.lww.com/QAI/A845.
Basic Reproductive Ratio and Critical Efficacy
We next obtained estimates of the baseline population of uninfected CD4+ T cells by subtracting the baseline population of productively infected cells (estimated above) from the measured baseline CD4+ T-cell count. Dividing the mean CD4+ T-cell count in uninfected individuals (1039 cells/µL in south India)30 by the latter estimate yielded the median R0 to be 5.3 (IQR: 4.5–7.0) (Fig. 2). The corresponding median critical efficacy, εc, was 0.81.
FIGURE 2.
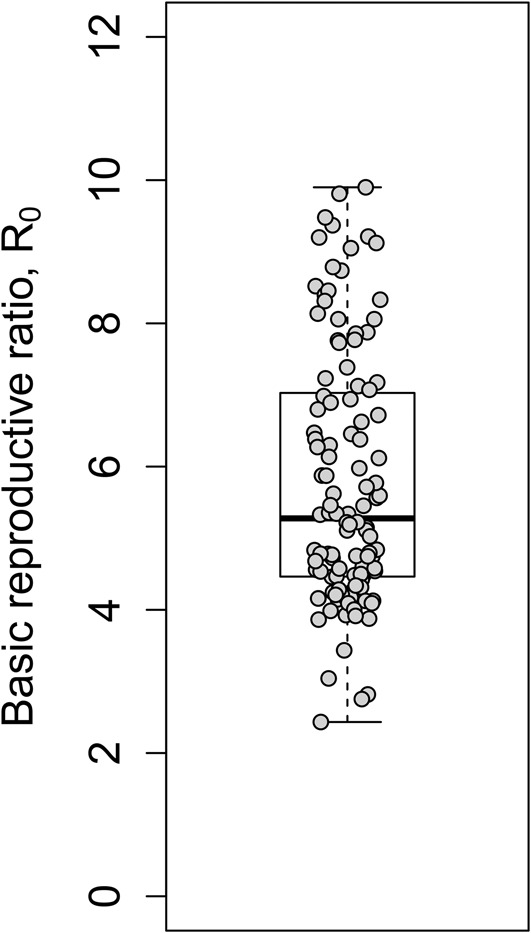
Estimates of the basic reproductive ratio obtained from analysis of viral load data of HIV-1C patients under treatment (Fig. 1; also see Table, Supplemental Digital Content 6, http://links.lww.com/QAI/A845). Symbols represent estimates for individual patients. The horizontal line within the box indicates the median, the boundaries of the box indicate the first and third quartiles, and the whiskers mark the minimum and maximum values.
Our estimate of R0 was robust to our data fitting procedure. To test the effect of the uncertainty in the estimates of the slope of the first phase of viral decline, δ (set above to 1 d−1), on R0, we repeated our analysis with δ of 0.5 and 1.5 d−1. The resulting estimates were not distinguishable from those above (median R0 of 5.3 [IQR: 4.5–7.0]; see Figure S4, Supplemental Digital Content, http://links.lww.com/QAI/A845), indicating that the estimate of R0 was robust with respect to variations in δ. Similarly, excluding patients with viral load undetectable at week 12, whose second phase slope was potentially underestimated by our fits, yielded median R0 to be 5.2 (IQR: 4.7–6.9) (n = 45), not significantly different from the estimate above (P = 0.9 using a 2-tailed Student t test with unequal variance). To account for variations in the CD4+ T-cell count in healthy individuals, we repeated our analysis by randomly choosing preinfection CD4+ T-cell counts for each patient from the known distribution in healthy South Indian adults (1039 ± 315 cells/µL).30 From 100 such repetitions, we found that the median R0 is not influenced by variations in pretreatment CD4+ T-cell counts [median of the median R0 = 5.36 (IQR: 5.22–5.49)] and is very close to the median R0 estimated above. Whereas we assumed treatment efficacy ε≈1 in patients with optimal adherence, some drug combinations may result in efficacies up to 25%–30% lower than others.21 To address the possibility of ε < 1 in the treatments used in our study, we repeated our analysis by following Wein et al31 and replaced δ and δM in Equation 7 by εδ and εMδM, respectively. With ε = εM = 0.7, marking 30% lower efficacy, we obtained estimates of R0 that were indistinguishable from those above [median 5.3 (IQR: 4.5, 7.0)]. This is because the lower ε altered estimates of δM and hence of the population of infected CD4+ T cells at baseline, 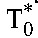 , but the latter were typically <1% of the total baseline CD4+ T-cell count and led to negligible changes in estimates of R0. Indeed, the small fraction of cells infected at baseline (∼0.3 ± 0.5%, range: 0.01%–3.3%) implied that a good approximation to R0 could be obtained by assuming that the baseline CD4+ T-cell count represents the target cell population. Data from a much larger set of patients including those with suboptimal adherence (with baseline CD4+ T-cell count >100 cells/µL and no primary resistance) (n = 183) could then be used to obtain estimates of R0. We thus estimated the median R0 to be 5.2 (IQR: 4.5–6.9) (see Figure S5, Supplemental Digital Content, http://links.lww.com/QAI/A845) (and the corresponding median εc to be 0.81), in close agreement with the estimates above (P = 0.69 using a 2-tailed Student t test with unequal variance).
, but the latter were typically <1% of the total baseline CD4+ T-cell count and led to negligible changes in estimates of R0. Indeed, the small fraction of cells infected at baseline (∼0.3 ± 0.5%, range: 0.01%–3.3%) implied that a good approximation to R0 could be obtained by assuming that the baseline CD4+ T-cell count represents the target cell population. Data from a much larger set of patients including those with suboptimal adherence (with baseline CD4+ T-cell count >100 cells/µL and no primary resistance) (n = 183) could then be used to obtain estimates of R0. We thus estimated the median R0 to be 5.2 (IQR: 4.5–6.9) (see Figure S5, Supplemental Digital Content, http://links.lww.com/QAI/A845) (and the corresponding median εc to be 0.81), in close agreement with the estimates above (P = 0.69 using a 2-tailed Student t test with unequal variance).
Estimates of R0 for HIV-1B vary from 5.7 to 19.3,13–15 with the most recent being ∼8.0.15 Our present estimate of R0 was smaller than the latter estimate for HIV-1B (P = 0.001 using a 2-tailed Student t test with unequal variance). We estimated R0 in the chronic phase of infection, whereas recent estimates for HIV-1B have been obtained in the acute phase of infection. R0 is likely to be lower in the chronic phase because of heightened immune pressure compared with the acute phase. To test whether this could underlie the difference between the estimates for HIV-1B and our present estimate, we pooled available data of baseline CD4 counts (n = 37) from several previous studies on chronic HIV-1B infection1,2,20,28 and identified the median R0 to be ∼8.6, not different from the estimate in the acute phase above (P = 0.16 using a 2-tailed Student t test with unequal variance). In the latter studies, individuals with baseline CD4 counts <100 per microliter were also included, which could lead to higher estimates of R0. (The chronic infection steady state was demonstratable even with the low CD4 counts.) We therefore included patients in our study with baseline CD4 counts <100 per microliter (yielding n = 249) and estimated the median R0 to be 6.2, which was lower than the estimate above for HIV-1B, but much less significantly so (P = 0.09 using a 1-tailed Student t test with unequal variance).
DISCUSSION
Our study reports for the first time viral decay dynamics in response to first-line ART among individuals infected with HIV-1C. The key finding is a smaller within-host basic reproductive ratio, R0, compared with HIV-1B, predicting better treatment outcomes for HIV-1C infection. We estimated the median R0 to be 5.3 (IQR: 4.5–7.0), a value that is smaller than the recent estimate of 8.0 (IQR: 4.9–11) for HIV-1B patients.15 The lower R0 suggests that the HIV-1C strain in India is less fit than the HIV-1B strain prevalent in the west.15 Correspondingly, we estimated the critical efficacy of therapy, εc, required to drive R0 below unity, to be ∼0.81 for HIV-1C, which is lower than ∼0.88 for HIV-1B. This suggests that HIV-1C infection may respond to ART more efficiently, perhaps even in the context of lower drug dosages or “suboptimal” adherence, when compared with HIV-1B. That complete virological suppression was achieved in 88% of treated individuals within 6 months of initiating ART is a testament to the excellent efficacy of the treatment regimen, especially in light of the lower εc of the HIV-1C strain prevalent in India. Similar responses to first-line ART have been reported elsewhere, both in LMIC and high-income settings.32–35 Virological response was achieved despite high viral loads at baseline, as has often been described previously with HIV-1C infection.7,18
An intriguing question that follows is how HIV-1C infection is associated with higher set-point viral loads despite its lower fitness compared with HIV-1B infection. We speculate on the answer here. Using mathematical models of viral dynamics,3 when productively infected cells dominate the infected cell populations, we recognize that 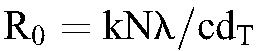 and that the set-point viral load
and that the set-point viral load 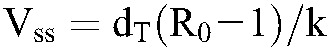 . In the latter expressions, k is the rate constant of viral infection of target cells and N the viral burst size. (Other parameters are defined in Methods.) It follows from these expressions that 2 viral strains with the same R0 can establish distinct set-point viral loads if they differ in their infection rate constants, k. (Note that dT, the natural death rate of target cells, is independent of the virus.) This decoupling of R0 and VSS provides a potential answer to the question above. Specifically, let k and N be different across the subtypes. Assuming fixed (known) values of the other parameters in the expressions above, we find that when VSS = 5.7 log10 copies per milliliter and R0 = 5.3, corresponding to the expected values for HIV-1C here, and when VSS = 4.4 log10 copies per milliliter and R0 = 8, as expected for HIV-1B,15,36 the above expressions are satisfied when k and N for the 2 subtypes obey the ratios kC/kB = 0.03 and NC/NB = 22, where the subscripts denote the subtypes. Thus, if k and N for HIV-1B are 2.4 × 10−8 mL per virion per day37 and 5 × 104 virions per cell,26 respectively, the corresponding values for HIV-1C would be 7.4 × 10−10 mL per virion per day and 1.1 × 106 virions per cell. The relative values of k and N for the 2 subtypes suggest that lower infectivity of target cells and higher virion production from infected cells could characterize HIV-1C infection compared with HIV-1B; this leads to higher set-point viral loads despite lower R0 values. Indeed, a recent study argues that the decline of CD4 counts-a marker of disease progression-depends on the viral replication capacity-a correlate of R0 -and not on the set-point viral load as long as the set-point viral load is neither too high nor too low, indicating that conditions exist wherein VSS and R0 are decoupled.38 Further evidence is presented by another recent study of hyperacute HIV-1C infection in high-risk South African women (n = 12), which found R0 in these women to be similar to that of HIV-1B infection (median ∼8) but median peak viremia to be more than 10-fold higher.39
. In the latter expressions, k is the rate constant of viral infection of target cells and N the viral burst size. (Other parameters are defined in Methods.) It follows from these expressions that 2 viral strains with the same R0 can establish distinct set-point viral loads if they differ in their infection rate constants, k. (Note that dT, the natural death rate of target cells, is independent of the virus.) This decoupling of R0 and VSS provides a potential answer to the question above. Specifically, let k and N be different across the subtypes. Assuming fixed (known) values of the other parameters in the expressions above, we find that when VSS = 5.7 log10 copies per milliliter and R0 = 5.3, corresponding to the expected values for HIV-1C here, and when VSS = 4.4 log10 copies per milliliter and R0 = 8, as expected for HIV-1B,15,36 the above expressions are satisfied when k and N for the 2 subtypes obey the ratios kC/kB = 0.03 and NC/NB = 22, where the subscripts denote the subtypes. Thus, if k and N for HIV-1B are 2.4 × 10−8 mL per virion per day37 and 5 × 104 virions per cell,26 respectively, the corresponding values for HIV-1C would be 7.4 × 10−10 mL per virion per day and 1.1 × 106 virions per cell. The relative values of k and N for the 2 subtypes suggest that lower infectivity of target cells and higher virion production from infected cells could characterize HIV-1C infection compared with HIV-1B; this leads to higher set-point viral loads despite lower R0 values. Indeed, a recent study argues that the decline of CD4 counts-a marker of disease progression-depends on the viral replication capacity-a correlate of R0 -and not on the set-point viral load as long as the set-point viral load is neither too high nor too low, indicating that conditions exist wherein VSS and R0 are decoupled.38 Further evidence is presented by another recent study of hyperacute HIV-1C infection in high-risk South African women (n = 12), which found R0 in these women to be similar to that of HIV-1B infection (median ∼8) but median peak viremia to be more than 10-fold higher.39
The origins of these potential differences between HIV-1B and C subtypes remain to be established. Recent evidence suggests that HIV-1C acquires an extra or fourth nuclear factor κB site that makes it more replication competent but less virulent.40 This latter study also found that the mean plasma viral loads, but not CD4+ T-cell counts, were significantly higher in patients with 4-nuclear factor κB–containing viral infection, suggesting that these strains had gained a potential survival advantage as a consequence of the acquired transcription factor-binding sites.40 The set-point viral load in HIV-1 infection has been argued to be evolutionarily optimized to maximize the transmission potential of the virus.36,41 The selective transmission of the less virulent 4-NF-κB strain may be driven by the high baseline viral loads, which were seen in our study as well. The ex vivo fitness of virus isolates from HIV-1B–infected individuals from Brazil and the United States were found to be higher than from HIV-1C–infected individuals from South Africa in all cell types except Langerhans cells, which are implicated in HIV-1 transmission.42 Similarly, the African subtype C strain was outcompeted by the subtype B strain in peripheral blood mononuclear cells, but was dominant to other subtypes in vaginal, cervical, penile, and rectal explants.43 HIV-1C has been argued to represent an advanced form of viral attenuation that has rendered the strain less virulent and therefore, possibly, dominant in the current global epidemic.44 The Indian HIV-1C strain is genetically distinct from the prototypical African HIV-1C strain, less neutralization sensitive45 and also less neurovirulent,46 potentially further explaining its growing prevalence and spread.
In conclusion, our study presents the first characterization of viral decay dynamics and estimates of the within-host basic reproductive ratio (R0) for individuals living with HIV-1C infection and undergoing treatment in India. Our findings of lower R0 despite higher set-point viral loads for HIV-1C compared with HIV-1B, together with the growing prevalence of HIV-1C worldwide, suggest that HIV-1C is evolving to be less virulent and yet more transmissible of the 2 strains. Unraveling the molecular underpinnings of this intriguing evolutionary possibility may provide crucial insights into virus–host interactions and the global epidemiology of infectious diseases.
Supplementary Material
Footnotes
Supported by Wellcome Trust/DBT India Alliance Senior Fellowships IA/S/13/2/501017 (AS) and IA/S/14/1/501307 (NMD).
The authors have no funding or conflicts of interest to disclose.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.jaids.com).
REFERENCES
- 1.Wei X, Ghosh SK, Taylor ME, et al. Viral dynamics in human immunodeficiency virus type 1 infection. Nature. 1995;373:117–122. [DOI] [PubMed] [Google Scholar]
- 2.Perelson AS, Neumann AU, Markowitz M, et al. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science. 1996;271:1582–1586. [DOI] [PubMed] [Google Scholar]
- 3.Nowak MA, May M. Virus Dynamics: Mathematical Principles of Immunology and Virology. New York, NY: Oxford University Press; 2000. [Google Scholar]
- 4.Padmanabhan P, Dixit N. Models of viral population dynamics. In: Current Topics in Microbiology and Immunology. Springer International Publishing Switzerland; 2015:1–26. [DOI] [PubMed] [Google Scholar]
- 5.Hemelaar J, Gouws E, Ghys PD, et al. Global trends in molecular epidemiology of HIV-1 during 2000–2007. AIDS. 2011;25:679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neogi U, Palchaudhuri R, Shet A. High viremia in HIV-1 subtype C infection and spread of the epidemic. J Inf Dis. 2013;208:866–867. [DOI] [PubMed] [Google Scholar]
- 7.Novitsky V, Ndung'u T, Wang R, et al. Extended high viremics: a substantial fraction of individuals maintain high plasma viral RNA levels after acute HIV-1 subtype C infection. AIDS. 2011;25:1515–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou J, Kumarasamy N, Ditangco R, et al. The TREAT Asia HIV Observational Database: baseline and retrospective data. J Acquir Immune Defic Syndr. 2005;38:174–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neogi U, Heylen E, Shet A, et al. Long-term efficacy of first line antiretroviral therapy in Indian HIV-1 infected patients: a longitudinal cohort study. PLoS One. 2013;8:e55421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boulle A, Schomaker M, May MT, et al. Mortality in patients with HIV-1 infection starting antiretroviral therapy in South Africa, Europe, or North America: a collaborative analysis of prospective studies. PLoS Med. 2014;11:e1001718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gray CM, Williamson C, Bredell H, et al. Viral dynamics and CD4+ T cell counts in subtype C human immunodeficiency virus type 1-infected individuals from southern Africa. AIDS Res Hum Retrovir. 2005;21:285–291. [DOI] [PubMed] [Google Scholar]
- 12.Novitsky V, Woldegabriel E, Kebaabetswe L, et al. Viral load and CD4+ T-cell dynamics in primary HIV-1 subtype C infection. J Acquir Immune Defic Syndr. 2009;50:65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Little SJ, McLean AR, Spina CA, et al. Viral dynamics of acute HIV-1 infection. J Exp Med. 1999;190:841–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stafford MA, Corey L, Cao Y, et al. Modeling plasma virus concentration during primary HIV infection. J Theor Biol. 2000;203:285–301. [DOI] [PubMed] [Google Scholar]
- 15.Ribeiro RM, Qin L, Chavez LL, et al. Estimation of the initial viral growth rate and basic reproductive number during acute HIV-1 infection. J Virol. 2010;84:6096–6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.National AIDS Control Organization. Antiretroviral Therapy Guidelines for HIV-Infected Adults and Adolescents Including Post-Exposure Prophylaxis. New Delhi, India: Ministry of Health & Family Welfare, Government of India; 2007. Available at http://www.naco.gov.in/NACO/About_NACO/Policy__Guidelines/Policies__Guidelines1/. Accessed February 5, 2016. [Google Scholar]
- 17.Neogi U, Bontell I, Shet A, et al. Molecular epidemiology of HIV-1 subtypes in India: origin and evolutionary history of the predominant subtype C. PloS One. 2012;7:e39819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neogi U, Sahoo PN, De Costa A, et al. High viremia and low level of transmitted drug resistance in anti-retroviral therapy-naive perinatally-infected children and adolescents with HIV-1 subtype C infection. BMC Infect Dis. 2012;12:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bennett DE, Camacho RJ, Otelea D, et al. Drug resistance mutations for surveillance of transmitted HIV-1 drug-resistance: 2009 update. PLoS One. 2009;4:e4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perelson AS, Essunger P, Cao Y, et al. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature. 1997;387:188–191. [DOI] [PubMed] [Google Scholar]
- 21.Louie M, Hogan C, Di Mascio M, et al. Determining the relative efficacy of highly active antiretroviral therapy. J Infect Dis. 2003;187:896–900. [DOI] [PubMed] [Google Scholar]
- 22.Coffin J, Swanstrom R. HIV pathogenesis: dynamics and genetics of viral populations and infected cells. Cold Spring Harb Perspect Med. 2013;3:a012526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Markowitz M, Louie M, Hurley A, et al. A novel antiviral intervention results in more accurate assessment of human immunodeficiency virus type 1 replication dynamics and T-cell decay in vivo. J Virol. 2003;77:5037–5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dixit NM, Markowitz M, Ho DD, et al. Estimates of intracellular delay and average drug efficacy from viral load data of HIV-infected individuals under antiretroviral therapy. Antivir Ther. 2004;9:237–246. [PubMed] [Google Scholar]
- 25.Ribeiro RM, Dixit NM, Perelson AS. Modelling the in vivo growth rate of HIV: implications for vaccination. In: Paton R, McNamara LA, eds. Multidisciplinary Approaches to Theory in Medicine. Amsterdam: Elsevier; 2006:231–246. [Google Scholar]
- 26.Chen HY, Di Mascio M, Perelson AS, et al. Determination of virus burst size in vivo using a single-cycle SIV in rhesus macaques. Proc Natl Acad Sci U S A. 2007;104:19079–19084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramratnam B, Bonhoeffer S, Binley J, et al. Rapid production and clearance of HIV-1 and hepatitis C virus assessed by large volume plasma apheresis. Lancet. 1999;354:1782–1785. [DOI] [PubMed] [Google Scholar]
- 28.Ho DD, Neumann AU, Perelson AS, et al. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373:123–126. [DOI] [PubMed] [Google Scholar]
- 29.Callaway DS, Perelson AS. HIV-1 infection and low steady state viral loads. Bull Math Biol. 2002;64:29–64. [DOI] [PubMed] [Google Scholar]
- 30.Thakar MR, Abraham PR, Arora S, et al. Establishment of reference CD4+ T cell values for adult Indian population. AIDS Res Ther. 2011;8:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wein LM, D'Amato RM, Perelson AS. Mathematical analysis of antiretroviral therapy aimed at HIV-1 eradication or maintenance of low viral loads. J Theor Biol. 1998;192:81–98. [DOI] [PubMed] [Google Scholar]
- 32.Lanoy E, May M, Mocroft A, et al. Prognosis of patients treated with cART from 36 months after initiation, according to current and previous CD4 cell count and plasma HIV-1 RNA measurements. AIDS. 2009;23:2199–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.May MT, Sterne JA, Costagliola D, et al. HIV treatment response and prognosis in Europe and North America in the first decade of highly active antiretroviral therapy: a collaborative analysis. Lancet. 2006;368:451–458. [DOI] [PubMed] [Google Scholar]
- 34.Braitstein P, Brinkhof MW, Dabis F, et al. Mortality of HIV-1-infected patients in the first year of antiretroviral therapy: comparison between low-income and high-income countries. Lancet. 2006;367:817–824. [DOI] [PubMed] [Google Scholar]
- 35.De Luca A, Marazzi MC, Mancinelli S, et al. Prognostic value of virological and immunological responses after 6 months of antiretroviral treatment in adults with HIV-1 infection in sub-Saharan Africa. J Acquir Immune Defic Syndr. 2012;59:236–244. [DOI] [PubMed] [Google Scholar]
- 36.Fraser C, Hollingsworth TD, Chapman R, et al. Variation in HIV-1 set-point viral load: epidemiological analysis and an evolutionary hypothesis. Proc Natl Acad Sci U S A. 2007;104:17441–17446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perelson AS, Kirschner DE, De Boer R. Dynamics of HIV infection of CD4+ T cells. Math Biosci. 1993;114:81–125. [DOI] [PubMed] [Google Scholar]
- 38.Claiborne DT, Prince JL, Scully E, et al. Replicative fitness of transmitted HIV-1 drives acute immune activation, proviral load in memory CD4+ T cells, and disease progression. Proc Natl Acad Sci U S A. 2015;112:E1480–E1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ndhlovu ZM, Kamya P, Mewalal N, et al. Magnitude and kinetics of CD8+ T cell activation during hyperacute HIV infection impact viral set point. Immunity. 2015;43:591–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bachu M, Yalla S, Asokan M, et al. Multiple NF-kappaB sites in HIV-1 subtype C long terminal repeat confer superior magnitude of transcription and thereby the enhanced viral predominance. J Biol Chem. 2012;287:44714–44735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fraser C, Lythgoe K, Leventhal GE, et al. Virulence and pathogenesis of HIV-1 infection: an evolutionary perspective. Science. 2014;343:1243727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ball SC, Abraha A, Collins KR, et al. Comparing the ex vivo fitness of CCR5-tropic human immunodeficiency virus type 1 isolates of subtypes B and C. J Virol. 2003;77:1021–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abraha A, Nankya IL, Gibson R, et al. CCR5-and CXCR4-tropic subtype C human immunodeficiency virus type 1 isolates have a lower level of pathogenic fitness than other dominant group M subtypes: implications for the epidemic. J Virol. 2009;83:5592–5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arien KK, Vanham G, Arts EJ. Is HIV-1 evolving to a less virulent form in humans?. Nat Rev Microbiol. 2007;5:141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hermanus T, Mkhize N, Moore P, et al. Comparison of the neutralization sensitivity of South African and Indian HIV-1 subtype C viruses to South African plasma antibodies. Retrovirology. 2012;9(suppl 2):P66. [Google Scholar]
- 46.Rao VR, Neogi U, Talboom JS, et al. Clade C HIV-1 isolates circulating in Southern Africa exhibit a greater frequency of dicysteine motif-containing Tat variants than those in Southeast Asia and cause increased neurovirulence. Retrovirology. 2013;10:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.