


Abstract
Poly(ADP-ribose) (PAR) is a posttranslational modification predominantly synthesized by PAR polymerase-1 (PARP-1) in genome maintenance. PARP-1 detects DNA damage, and damage detection is coupled to a massive increase PAR production, primarily attached to PARP-1 (automodification). Automodified PARP-1 then recruits repair factors to DNA damage sites. PARP-1 automodification eventually leads to release from DNA damage thus turning off catalytic activity, although the effects of PAR on PARP-1 structure are poorly understood. The multiple domains of PARP-1 are organized upon detecting DNA damage, creating a network of domain contacts that imposes a major conformational transition in the catalytic domain that increases PAR production. Presented here are two novel fluorescent sensors that monitor the global and local structural transitions of PARP-1 that are associated with DNA damage detection and catalytic activation. These sensors display real-time monitoring of PARP-1 structural transitions upon DNA damage detection, and their reversal upon PARP-1 automodification. The fluorescent sensors are further used to investigate intramolecular and intermolecular PARP-1 activation, followed by the observation that intramolecular activation of PARP-1 is the predominant response to DNA strand breaks in cells. These results provide a unique perspective on the interplay between PARP-1 DNA damage recognition, allosteric regulation, and catalytic activity.
INTRODUCTION
PARP-1 (also known as ARTD1) is the founding member of the PARP family, consisting of 17 human proteins related by the structurally conserved ADP-ribosyltransferase (ART) fold (1). PARP-1 is a multi-domain enzyme with important roles in cellular processes such as transcription, cell fate determination and DNA repair (2). The catalytic activity of PARP-1 converts nicotinamide adenine dinucleotide (NAD+) into long and branched chains of PAR covalently attached to nuclear proteins through heteromodification and primarily to itself through automodification (3). In response to DNA damage, PARP-1 rapidly recruits DNA repair and chromatin remodeling factors to compromised genomic sites through coordination of its DNA damage detection and PAR catalysis activities (4). This response is tightly regulated through automodification, resulting in the eventual release of PARP-1 from sites of damage and subsequent decline in catalytic activity (5). The important role of PARP-1 in the DNA damage response has provided several therapeutic implications for inhibition of PARP-1, and clinical PARP inhibitors have matured into a new approach of targeted therapy against DNA repair deficient tumors, exemplified by the recent FDA approval of Olaparib (Lynparza™, AstraZeneca) (6).
PARP-1 has six distinct folded domains, and structures have been obtained for each individual domain and some combinations of domains (Figure 1A) (7). The carboxyl-terminal catalytic domain (CAT) combines the signature ADP-ribosyltransferase fold (ART) with an alpha-helical sub-domain (HD) that regulates catalytic activation (8,9). A tryptophan-glycine-arginine (WGR) domain is important for DNA binding and allosteric activation (9) and is a regulatory feature of the DNA damage response PARPs 1, 2 and 3 (8). A BRCT (BRCA1 C-terminus) fold is located within the region known as the automodification domain of PARP-1. Two homologous N-terminal zinc fingers, Zn1 and Zn2, bind to the ends of damage DNA (10), and a structurally distinct third zinc finger, Zn3, has important roles in communicating DNA damage recognition to the catalytic domain (9,11).The global architecture of PARP-1 domains in the absence of DNA damage is unknown, although they are thought to resemble a ‘beads-on-a-string’ like architecture (Figure 1A) (12,13). In the presence of DNA damage, PARP-1 domains collapse together on the ends of DNA strand breaks (9,14), and this organization of domains suggests a global structural compaction that occurs as part of the DNA damage recognition mechanism (Figure 1B). PARP-1 automodification results in varying lengths of linear and branched chains of PAR covalently attached to PARP-1 (15–18), and this event is strongly correlated with release from DNA damage (19,20). The long-standing model for PARP-1 release describes the growing steric bulk and negative charge of PAR as the forces responsible for decreased affinity for DNA damage (2,21). However, PARP-1 domain disassembly during DNA damage-dependent automodification is not understood.
Figure 1.

PARP-1 structure and conformational changes in response to DNA damage. (A) The architecture of PARP-1 is composed of six domains from N- to C-terminus: zinc finger 1 (Zn1; PDB-ID:3ODA), zinc finger 2 (Zn2; PDB-ID:3ODE), zinc finger 3 (Zn3; PDB-ID:2JVN), BRCA-1 C-terminus fold (BRCT; PDB-ID:2COK), Tryptophan-Glycine-Arginine domain (WGR; PDB-ID:2CR9) and catalytic domain (CAT; PDB-ID:1A26), which consists of an alpha-helical subdomain (HD) and ADP-ribosyltransferase fold (ART). In the absence of DNA damage, the overall structure resembles a ‘beads-on-a-string’ assembly. To visualize this concept, unstructured linker regions between domains are depicted as beads representing each residue as a circle. (B) In the presence of DNA damage, essential PARP-1 domains (PDB-ID: 4DQY) collapse together to form a network of DNA-protein and protein-protein interactions. This structural collapse bridges communication between DNA damage detection domains and the CAT domain, facilitating DNA damage-dependent catalytic activation. A global structural change results from the overall rearrangement of domains on DNA damage, which is predicted to place the termini in proximity. The domain–domain contacts resulting from DNA damage recognition causes a destabilization in the HD that leads to catalytic activation. A notable structural change observed in the crystal structure displaced residues L698 and L701 from the hydrophobic interior of the HD (inset).
The complex of PARP-1 essential domains (Zn1, Zn3, WGR and CAT) bound to a DNA double-strand break depicted an allosteric mechanism for coordinating PARP-1 DNA damage detection and activation of the catalytic domain (9). In the crystal structure, the assembly of PARP-1 domains on DNA damage supported a major structural transition in the HD. Most prominently, two leucine residues were displaced from the HD hydrophobic core (Figure 1B). A correlation between PARP-1 activation and de-stabilizing HD mutants supported that the observed structural transition was relevant to the PARP-1 activation mechanism; however, the mechanism by which the HD regulated the catalytic domain was not clear. A recent study of PARP-1 dynamics using hydrogen/deuterium exchange with mass spectrometry revealed that specific regions of the HD that form stable helices in the absence of DNA transitioned to an unfolded state upon PARP-1 binding to DNA damage (22). The structural transition required a key contact formed between PARP-1 domains when bound to DNA, and the transition occurred in the absence of NAD+ (22). The same study showed that the HD is an autoinhibitory domain that must be severely distorted to relieve inhibition and allow NAD+ to have full access to the ART fold (22). Notably, the reversibility of this major structural transition has not been explored.
The crystal structure of PARP-1 essential domains bound to a DNA double-strand break and the NMR/crystallographic model for PARP-1 binding to DNA single-strand breaks indicate a monomeric intramolecular mechanism of PARP-1 DNA damage detection and catalytic activation (9,14). A number of structural and biophysical reports align with the model of monomeric intramolecular DNA recognition and catalytic activation for PARP-1 (9,12,14,23,24); however, an intermolecular mechanism of PARP-1 activation has been invoked to explain certain biochemical (25) and DNA binding (26) analyses. The crystal structure of the Zn1-Zn2 fragment of PARP-1 bound to a DNA duplex with a single 5′-nucleotide overhang presented a complex that was only achievable through separate polypeptides, leading to the proposal of an intermolecular mechanism for binding DNA damage (27), although it is not known how this complex could lead to catalytic activation. PARP-1 activation in response to DNA damage can be completely inactivated through mutations to the catalytic domain or to the regulatory domains. Mixtures of different mutants can restore DNA-dependent activation (11,15), and mixtures of different PARP-1 domain combinations can also reconstitute DNA-dependent activity (9). These reports indicate that PARP-1 domains can indeed come together as separate polypeptides at least at a biochemical level, suggesting that intermolecular activation mechanisms are possible. Importantly, a direct comparison of the efficiency of PARP-1 activation via an intramolecular versus an intermolecular mechanism has not been made. Furthermore, there has been no examination of a viable intermolecular mechanism of activation in cell-based assays.
In an effort to better understand PARP-1 activation in response to DNA damage, we have developed fluorescent sensors to detect the structural changes that occur during DNA damage recognition and activation, and the subsequent reversal of these structural changes upon PARP-1 automodification. These innovative detection systems represent the first real-time monitoring of global structural compaction and allosteric activation status of PARP-1. In addition, our results provide biochemical evidence of a modest level of PARP-1 intermolecular activation, in contrast to an efficient level of intramolecular activation that we also observe to be the predominant activation mechanism in cells in response to DNA strand breaks. Together with recent structural analysis, our data provides further support for the monomeric, intramolecular activation mechanism of PARP-1.
MATERIALS AND METHODS
Gene cloning and mutagenesis
To construct the intramolecular FRET sensor, a pmCherry-N1 vector was cut at Bgl2 and Age1 restriction sites to introduce the custom sequence TCATATGTCGAATTCTGCAGTCAGGCTCGAGCGA, which contains Nde1 and Xho1 cut sites. This product was then cut at Nhe1 and Bgl2 restriction sites to insert eGFP from a digested pEGFP-C1 vector. Next, the bacterial expression vector pET28 was digested at Nde1 and Xho1 to introduce the sequence AGCTAGCATGACTGGTGGACAAGCTTGCGGCCGCA, which contains Nhe1 and Not1 cleavage sites while knocking out the Nde1 and Xho1 sites. The fluorescent protein construct was then cut at Nhe1 and Not1 and the designed eGFP-mCherry was ligated into the modified pET28 vector. PARP-1 constructs were then ligated between the two fluorescent proteins through Nde1 and Xho1 restriction sites.
To construct the N-terminal PARP-1 fragment for sortaseA coupling, residues LPETGGGRR replaced residues 376–377 in a pET24 vector containing PARP-1 residues 1–377 by amplifying with the primers 5′-AGCACATCGTGAGCAAATCGGCGGTGGTGGTGGTTCTGCTTCAGCAGATAAGCCATTATC and 5′- TCTGCGACCACCACCGGTTTCCGGCAGCGAGGCTGTGGAGGGCGGAGGCG (Supplementary Figure S1A). For the C-terminal fragment, a pET28 vector coding for full-length PARP-1 (residues 1 to 1014) with an N-terminal SUMO-like tag (SMT) was used as template for QuikChange mutagenesis with primers 5′- AGCACATCGTGAGCAAATCGGCGGTGGTGGTGGTTCTGCTTCAGCAGATAAGCCATTATC and 5′- ACCACCACCACCGCCGATTTGCTCACGATGTGCTTCAATAATATCGTTATCCTCC in order to delete residues 1–382 of full-length PARP-1 and replace those residues with three glycine residues, thus producing SMT-GGG-383-1014 PARP-1.
Mutagenesis was performed using the QuickChange Protocol (Stratagene).
Expression and purification of proteins
PARP-1 constructs were expressed in Escherichia coli strain Rosetta2 cells with an N- or C-terminal His6-tag and purified by Ni(II)-NTA affinity chromatography, followed by heparin affinity chromatography, and finally size exclusion chromatography (SEC) in 25 mM Hepes at pH 8.0, 150 mM NaCl, 0.1 mM TCEP and 1 mM EDTA as previously published (28). Ni(II)-NTA elutions of SMT-GGG-383-1014 PARP-1 constructs were mixed with 200 μg of SUMO protease ULP-1 for 1 h at 4°C, thus removing SMT and leaving an N-terminus with three consecutive glycines (GGG-383-1014). After protease digestion, the sample was diluted to 50 mM imidazole and passed over a freshly charged Ni(II)-NTA column to remove unprocessed proteins and ULP-1. The elution from this purification was diluted to 250 mM NaCl and purified by heparin affinity chromatography.
Sortase coupling
A 0.5 mL mixture of GGG-383-1014 (250 μM), 1-375-LPETG (300 μM), and Sortase A (5.4 μM) in Sortase A buffer (50 mM Tris pH 8.0, 50 μM ZnSO4, 1 mM DTT, 10 mM CaCl2, and 200 mM NaCl) was placed in a Slide-A-lyzer dialysis cassette (3.5K) and dialyzed at 4°C overnight in Sortase A buffer while stirring (Supplementary Figure S1B). The following day, the mixture was diluted in Ni(II) column buffer and passed over a Ni(II)-NTA affinity column and washed to remove unreacted components. The flow-through was diluted to 250 mM NaCl and purified over a heparin column with a 470–600 mM NaCl gradient.
Fluorescent labeling
The C-terminal PARP-1 construct GGG-383-1014 was diluted to 50 μM in 25 mM Hepes pH 8.0, 150 mM NaCl, 0.1 mM TCEP including 100 μM of the cysteine-reactive fluorescent label. This mixture was left to react at 4°C overnight protected from light. On the following day the mixture was concentrated and washed in a spin column to remove unreacted dye, and then passed onto a heparin column, washed with 50 mM Tris pH 7.0, 250 mM NaCl, and 0.1 mM TCEP, and eluted with 50 mM TRIS pH 7.0, 1 M NaCl and 0.1 mM TCEP.
HD destabilization assay
Labeled PARP-1 was placed in a 384-well plate (50 μl/well) at 1 μM with 1 μM DNA (unless specified otherwise) in 25 mM Hepes pH 8.0, 32.5 mM NaCl, 60 mM KCl, 8 mM MgCl2, 50 μg/ml BSA, 0.02 mM TCEP, 4% glycerol and 11.4 mM β-mercapto-ethanol (BME). Measurements were taken using a TECAN Infinite M-200 pro plate reader, with excitation at 395 nm and emission at 460 nm and 535 nm. DNA release measurements were taken immediately following addition of 2 mM NAD+. For controls with no DNA, the DNA annealing buffer was added instead.
PARP-1 FRET assay
PARP-1 was placed in a 384-well plate (50 μl/well) at 1 μM with 1 μM DNA (unless specified otherwise) in 25 mM Hepes pH 8.0, 32.5 mM NaCl, 60 mM KCl, 8 mM MgCl2, 50 μg/ml BSA, 0.02 mM TCEP, 4% glycerol and 11.4 mM BME. Measurements were taken using a TECAN Infiniti M-200 plate reader, with excitation at 420 nm and emission at 512 and 612 nm.
DNA-dependent SDS-PAGE automodification assay
PARP-1 automodification reactions were performed at RT in 25 mM Hepes pH 8.0, 32.5 mM NaCl, 60 mM KCl, 8 mM MgCl2, 50 μg/ml BSA, 0.02 mM TCEP, 4% glycerol and 11.4 mM BME using 1 μM of PARP-1 and 1 μM of DNA. At each timepoint the reaction was quenched with 0.1 M EDTA in SDS-loading buffer, resolved on a 12% SDS-PAGE, and treated with Imperial protein stain (Pierce). In specifically described cases, UV visualization was used in place of protein stain.
DNA-dependent colorimetric automodification assay
Catalytic activity measurements were carried out in Ni(II)-NTA-His prime 96-well plates (5Prime) using an equimolar ratio of PARP-1/WT to DNA (50 nM), followed by addition of 400 μM NAD+ (1% biotinylated NAD; Trevigen), and detection by Strep-HRP (Pierce) using TMB-ELISA (Thermo-Scientific) as the colorimetric substrate (28).
DNA binding assay
PARP-1 was mixed with equimolar DNA at 1 μM in 50 mM Tris pH 7.0, 125 mM NaCl, 0.1 mM TCEP, 5% glycerol and 0.1 mM EDTA for 30 min at RT. Mobility shift assays were carried out in 0.9% agarose in 1× TG running buffer, and gel electrophoresis was conducted for 3 h with 100 V at 4°C. Images of gels were acquired under UV light.
Immunofluorescence microscopy
PARP-1 knockout mouse embryo fibroblasts cells (MEFs) (29) were cultured in DMEM containing 1% penicillin/streptomycin supplemented with 10% fetal bovine serum (FBS). Cells were grown on glass coverslips in 6-well plates and transfected with Fugene® 6 (Promega) using 1.5 μg DNA. Cells were treated with 10 mM H2O2 for 10 min at 24 h post-transfection, then fixed with ice-cold methanol for 2 min, washed with 2 ml PBS, and then blocked for 20 min in blocking buffer (25 mM Tris pH 7.5, 150 mM NaCl, 1% Triton X-100 (TBST), 2.5% blocking reagent). The primary antibody used to detect PAR formation (diluted 1:100 in TBST buffer) was anti-PADPR (Abcam) mouse monoclonal antibody [10H]. The secondary antibody (diluted 1:1000 in TBST buffer/1% blocking) used was an anti-mouse antibody conjugated to Atto-647 (Active-Motif). Cells were mounted in ProLong® Diamond Antifade Mountant with DAPI (Life Technologies). Spectral imaging (12-bit, 1024 × 1024) was performed with a Zeiss LSM510 Meta confocal laser scanning microscope using a 40× 1.3 NA oil immersion DIC Plan-Neofluar objective lens. Expressed eGFP and mCherry sensors were excited with a 405, 488, 543, 643 nm Argon ion laser line and emitted light was collected for DAPI (470 long-pass filter), eGFP (505-530 band-pass), mCherry (615–668) and Atto-647 (700–754), respectively. Images of fixed cells were processed in ImageJ.
Live cell microscopy and laser irradiation
MEFs were sensitized for microirradiation 16 h post-transfection with 1 μM BrdU for 24 h at 37°C, 5% CO2 before addition of Hoechst stain (10 μg/ml). Live cell experiments were carried out using a Zeiss LSM-510 Meta Confocal laser scanning microscope equipped with a 405 nm diode laser (set to 50% power) focused through 63×/1.4 NA oil immersion lens to locally irradiate nuclear, pre-selected sites for five iterations. Images were recorded by excitation with a 488 and 543 nm argon laser.
RESULTS
A novel sensor to monitor PARP-1 allosteric activation
To monitor the PARP-1 allosteric activation mechanism, cysteine residues were engineered into the HD domain to covalently bind small molecule fluorescent sensors. Cysteine residues were positioned near regions expected to undergo major structural transitions upon recognition of DNA damage (9,22). Modified HD residues were site-selectively labeled using the cysteine-reactive probe acrylodan (6-acryloyl-2-dimethylaminonaphthalene) (Figure 2A). Covalently attached to a cysteine residue, acrylodan exhibits a signal intensity and a peak wavelength of fluorescence emission that depends on the local chemical environment of the probe (30). In order to site-specifically label PARP-1, solvent accessible non-conserved cysteine residues were mutated to alanine; however, multiple cysteine residues in the N-terminal zinc fingers are functionally important and could not be mutated. Despite being involved in zinc coordination, these cysteine residues were susceptible to off-target reactivity with acrylodan and thus precluded selective labeling of the full-length PARP-1 (not shown). To overcome this challenge, we produced a C-terminal fragment of PARP-1 (residues 383–1014) that naturally lacked functionally important cysteines and permitted selective labeling at defined positions after two solvent accessible cysteine residues were mutated to alanine (C845A, C908A). The selectively labeled and purified C-terminal fragment was joined to the N-terminal fragment of PARP-1 (residues 1–375) using the transpeptidase sortase A (31), and then purified to yield the expected full-length PARP-1 polypeptide (Supplementary Figure S1C, lanes 1–4, and Materials and Methods). Importantly, sortase-joined full-length PARP-1 and variants thereof retained DNA binding and catalytic activity comparable to that of native PARP-1 (Supplementary Figure S1D and E).
Figure 2.

A PARP-1 sensor to monitor HD destabilization. (A) The small molecule acrylodan reacts specifically with cysteine residues forming a covalent attachment. Upon reaction, this molecule has varied fluorescent peak emission dependent upon local solvent polarity. (B) Structure of PARP-1 essential domains bound to DNA damage (PDB code 4DQY; gray), highlighting and comparing activated HD (PDB code 4DQY; blue) with non-activated HD (PDB code 1A26; red). The major observable difference in the crystal structure lies within the ‘leucine switch’, exemplified by L698 and L701 transitioning out of the HD hydrophic interior. The mutated residue K700C was selectively labeled with acrylodan (magenta sphere), allowing for real-time detection of HD destabilization. (C) Emission profile of acrylodan-labeled PARP-1 in the absence and presence of dsDNA. A construct lacking the engineered cysteine at position 700 (WT_2XC, two native cysteines removed; see Methods) displayed very low fluorescence intensity, indicating that the K700C mutant was selectively labeled. (D) Acrylodan-PARP-1 was used to show specificity for DNA structure by comparing changes in the emission ratio (λ460/λ535) of dsDNA, nickDNA, gapDNA, and poly(dT). For the no DNA control, only the DNA annealing buffer was added. Data points were conducted in triplicates ± S.D.
Comparison of PARP-1 CAT structures in the absence and presence of DNA damage-dependent regulatory domains revealed residues lining the hydrophobic core of the HD (L698 and L701) that switch to the exterior upon activation by double-stranded DNA (dsDNA), a mimic of a double-strand break (Figure 2B) (9). Based on this observation, we introduced cysteine residues at five positions surrounding this key conformational change between α-helices B and C (‘leucine switch’), followed by acrylodan labeling and sortase-mediated assembly of full-length PARP-1. In the absence of dsDNA, most labeled positions revealed a red-shifted emission peak indicating a strong solvent exposure profile as expected from labeling surface accessible cysteine residues (Supplementary Figure S2A) (32). The addition of dsDNA caused a blue shift in the acrylodan-labeled PARP-1 emission peak, which is indicative of a change to a more hydrophobic probe environment. The exposure of hydrophobic residues that occurs upon DNA damage-dependent activation is consistent with the blue-shift observed for most of the variants of PARP-1 labeled at different positions. The only exception was position 760 that detected a more hydrophobic environment initially, and this is likely a consequence of the distance from position 760 to the center of the HD structural transition. Alternative solvatochromic fluorophores such as monobromobimane and IANBD were tested at the same and additional positions. Although the spectral profiles of these sensors paralleled observations with acrylodan, the signal intensities and spectral shifts were weaker (data not shown). The acrylodan label at position K700C was selected for further analysis due to its major spectral shift (28 nm) and two-fold intensity change upon addition of dsDNA (Figure 2C). In determining the specificity of this method for monitoring PARP-1 activation, dsDNA was compared to mimics of single-strand break damage within a DNA duplex, nickedDNA and gapDNA, and a single strand of DNA with no duplex structure, poly(dT) (Supplementary Figure S2B). Single-strand break DNA damage potently activates PARP-1, but a single strand of DNA, such as poly(dT), does not (8). As expected, dsDNA, nickedDNA, and gapDNA all created a spectral shift (reported as a change in the λ460/λ535 ratio) at equimolar amounts of DNA, while poly(dT) had no effect (Figure 2D). These data further support the model that PARP-1 catalytic activation by specific DNA structures proceeds through HD structural changes, and identify a new way to monitor PARP-1 allosteric activation independent from PAR catalytic production.
A novel sensor to detect the global structural compaction of PARP-1
While HD structural changes are the final step of PARP-1 catalytic activation, a DNA recognition response of N-terminal domains precedes and initiates the allosteric activation mechanism. The organization of PARP-1 domains onto DNA strand breaks places the polypeptide termini into close proximity (9), whereas in the absence of DNA damage PARP-1 domains are not organized and are expected to exist in a more open and flexible configuration. We reasoned that the expected structural compaction of PARP-1 domains could be monitored using fluorescent resonance energy transfer (FRET) (33). FRET is the natural phenomenon of energy transfer from a fluorophore in the excited state (donor) to a fluorophore in the ground state (acceptor) (34). The efficiency of this transfer requires spectral overlap between donor emission and acceptor excitation, and is based on the relative orientation and distance between the two fluorophores. Thus, detection of differences in energy transfer efficiency of FRET pairs fused to PARP-1 termini can serve as an indication of global conformational changes.
Full-length PARP-1 was tagged with an N-terminal green fluorescent protein (eGFP) and with a C-terminal red fluorescent protein (mCherry) (Figure 3A). The doubly tagged construct of PARP-1 displayed good spectral separation with low excitation overlap between the fluorophores (Supplementary Figure S3A). Despite the low fluorescence quantum yield of mCherry, we were able to obtain measurable FRET changes consistent with other studies using this pair of fluorophores (33). Furthermore, PARP-1 bearing these tags retained robust DNA binding and catalytic activity (Supplementary Figure S3B and C, respectively), consistent with the cell-based analysis of PARP-1 DNA damage detection using fusions with eGFP (35).
Figure 3.

A FRET-based assay to detect PARP-1 global compaction. (A) A model representing dually-tagged PARP-1 with the eGFP and mCherry FRET pair, predicting a global conformational change of PARP-1 that results in a proximity of the N- and C-termini that is close enough for FRET detection. (B) Depiction of constructs used as FRET sensors. DEVD represents the cut site for caspase-3 (CAS-3). (C) The emission ratio of λ612/λ512 was used to evaluate sensors in the absence (control) and presence of caspase-3 after a 2 h incubation at 37°C. (D) The specificity of the PARP-1/WT sensor was compared to the peptide sensor by titrating 0.2 μM increments of dsDNA, poly(dT), or no DNA into 1 μM PARP-1 sensor. For the no DNA control, only DNA annealing buffer was added. The emission ratio (λ612/λ512) for each construct was normalized to zero in the absence of DNA, and the percentage change of the FRET ratio is displayed. Data points were conducted in triplicates ± S.D.
To set a range for the observed FRET response, we created a control protein construct in which the two fluorescent proteins were connected by a short linker polypeptide rather than PARP-1, thus holding the fluorescent proteins close together and providing a high efficiency FRET signal. A cleavage site for the protease caspase-3 (DEVD) was also engineered into the short linker polypeptide so that caspase-3 treatment would fully separate the fluorescent proteins and thereby provide a threshold for a ‘no FRET’ signal (Figure 3B). PARP-1 naturally contains a caspase-3 cleavage site located in a linker region that connects the DNA binding zinc fingers (Zn1, Zn2) to the rest of the protein. A tagged PARP-1 CAT domain construct was also used as a control since it lacks the caspase-3 cleavage site. FRET-pair tagged PARP-1 in the absence of DNA had a low but detectable FRET signal in comparison to the control protein. Treatment with caspase-3 reduced the low FRET signal of PARP-1 alone (Figure 3C). These data indicate that in the absence of DNA damage the domains of PARP-1 are held in a configuration that holds the terminal ends of the polypeptide at the limit of a detectable FRET signal, consistent with a flexible and extended conformation of PARP-1. In the presence of dsDNA, there was a stable increase in the FRET signal (Figure 3D), in line with a DNA-dependent collapse and organization of PARP-1 domains that increases the proximity of the N- and C-termini. As a control, the FRET signal of the fluorescent proteins connected by a short polypeptide did not change in the presence of dsDNA. Upon addition of poly(dT) to PARP-1, there was no appreciable change in the FRET signal of either PARP-1 or the control peptide. Overall, these results support the model of PARP-1 structural compaction upon detection of damaged duplex DNA structures.
Essential domains are required for fluorescence-based detection of PARP-1 structural changes
PARP-1 strictly requires four domains (Zn1, Zn3, WGR and CAT) for DNA damage-dependent catalytic activation, with the Zn1, Zn3 and WGR domains making direct contacts with DNA. Although not strictly required for DNA damage-dependent activation, the Zn2 domain has robust DNA binding affinity (10,24) and contributes to activation and interaction with single-strand break damage (14,36). To distinguish which domains are contributing to the FRET signal associated with structural compaction, PARP-1 mutants with a loss of function in each essential DNA binding domain were analyzed (Figure 4A, left panel). Point mutations disrupting the DNA binding function of the Zn1 domain (R34A) or the WGR domain (W589A) completely prevented the DNA damage-dependent FRET change, even though these PARP-1 mutants still interact with DNA due to the presence of a functional Zn2 domain (37). A PARP-1 mutant that deleted the Zn2 domain still provided a robust FRET signal, supporting studies that indicate that the Zn2 is not strictly required for DNA damage-dependent activation (9). Interestingly, when both non-essential domains (Zn2 and BRCT) were deleted individually or in tandem, there were no significant differences in FRET efficiency compared to full-length PARP-1 in the absence or presence of DNA damage (Supplementary Figure S3D). Accordingly, these observations agree with a global structural compaction that is mediated by the essential domains and their contacts with DNA.
Figure 4.

Effects of DNA binding and allosteric regulation disruptions on PARP-1 structural changes. (A) The full-length PARP-1/WT FRET sensor was compared to mutants that disrupt the DNA binding function of the Zn1 (R34A), Zn2 (ΔZn2) or WGR (W589A) domains (left), or mutants that disrupt domain–domain interactions important for DNA damage-dependent catalytic activation, by targeting the Zn1-Zn3 interface (W246A), Zn3-WGR-CAT interface (W318R), or Zn1-WGR interface (R591A) (right). (B) A fluorescence emission profile for acrylodan-labeled -PARP-1 in the absence and presence of equimolar dsDNA (1 μM) (left). Deletion of the DNA binding domains (ΔZn1Zn2–Zn3, residues 1–382) and domain contact mutant (W318R) are compared to the functional PARP-1/WT sensor. The emission ratios (λ460/λ535) of these profiles are plotted (right). Data points were conducted in triplicates ± S.D.
Similar to global compaction, the local changes in HD structure upon PARP-1 activation coincide with a proper alignment of the essential domains on DNA damage. Correspondingly, in the absence of the N-terminal DNA binding domains—Zn1, Zn2 and Zn3—we observed no change in the signal of acrylodan-labeled PARP-1 (Δ1–382) in the presence of DNA damage (Figure 4B, middle panel). Collectively, these observations support a model of PARP-1 DNA damage detection that leads to both global structural compaction and a local HD structural change.
Domain–domain contacts primarily influence PARP-1 allostery rather than compaction
Following DNA damage recognition, a network of interactions between PARP-1 domains is formed (9,37). Point mutations at domain contact points efficiently shut down PARP-1 DNA damage-dependent catalytic activity; however, it was not clear to what extent these mutations might influence the DNA damage-induced PARP-1 compaction described above. We analyzed three domain contact mutations introduced into the FRET-pair PARP-1 construct. WGR residue R591 mediates a key interaction between the Zn1 and CAT domains. The PARP-1 mutant R591A mutant showed a measureable deficiency in FRET signal (Figure 4A, right panel), indicating that the ability of PARP-1 to organize on DNA damage has been compromised, likely at the stage of positioning the WGR and CAT domains. PARP-1 mutant W246A, which disrupts the interface between Zn1 and Zn3, exhibited no apparent effect on the FRET signal (Figure 4A, right panel). PARP-1 mutant W318R disrupts an interface formed between the Zn3, WGR, and CAT domains, and also showed no apparent deficiency in the FRET signal that indicates PARP-1 compaction (Figure 4A, right panel). The severe catalytic deficiencies of W246A and W318R therefore do not arise from a major deficiency in assembly on damaged DNA, but are rather due to a lack of efficient allosteric communication. Consistent with this interpretation, HXMS analysis of PARP-1 W318R indicated protein-DNA contacts similar to those observed for WT PARP-1, but demonstrated a complete lack of HD structural changes associated with activation (22). Furthermore, the W318R mutation does not influence PARP-1 localization to DNA damage sites in cells, despite preventing PAR production in response to DNA damage (37). We introduced the W318R mutation into the acrylodan-labeled PARP-1 construct and confirmed that the mutation completely prevents HD structural changes associated with activation (Figure 4B, bottom panel), further demonstrating that this assay reports on the allosteric activation mechanism of PARP-1.
DNA damage-induced structural changes are reversible in the presence of NAD+
PARP-1 domain assembly on DNA damage and subsequent HD destabilization prime the CAT domain to process NAD+ into large, highly negatively charged PAR. PAR is mostly found covalently attached to PARP-1 itself, an event that correlates with reduced affinity for DNA damage and catalytic inactivation (21). It is generally held that steric clashes and charge repulsion lead to PARP-1 dissociation; however the effects on PARP-1 structure due to automodification are not well understood. Most notably, the reversibility of the HD structural transition was not known. Using the fluorescent PARP-1 sensors described above, we tested the reversibility of DNA damage-dependent structural changes by adding NAD+ and monitoring PARP-1 over a sufficient time course to reach a near maximum level of PAR production (Figure 5A). The HD destabilization observed with acrylodan-labeled PARP-1 in the presence of dsDNA was reversed following the addition of NAD+, reaching 65% reversion after incubation with NAD+ for 10 min. For comparison, PAR formation reached ∼80% of the maximum observed level (taken as 60 min) after 10 min (Figure 5A). The reversal of HD destabilization was inhibited in the presence of the PARP inhibitor olaparib (Figure 5B), indicating that CAT domain production of PAR is required for the structural reversal of HD destabilization. Complete inhibition required an olaparib concentration higher than that required to shut down PARP-1 activity in assays that detect PAR production, indicating that the HD destabilization assay is sensitive to low levels of PARP-1 activation that are not detectable in other assays. Indeed, a modest level of reversal is observed at 60 min in the presence of olaparib (Figure 5B), even though there is no detectable PAR formed at this time point in the corresponding activity assay (Figure 5A). Addition of NAD+ in the absence of the dsDNA template had no effect on the spectroscopic parameters of acrylodan-labeled PARP-1 (Supplementary Figure S4), further supporting the specificity of the NAD+ induced event.
Figure 5.
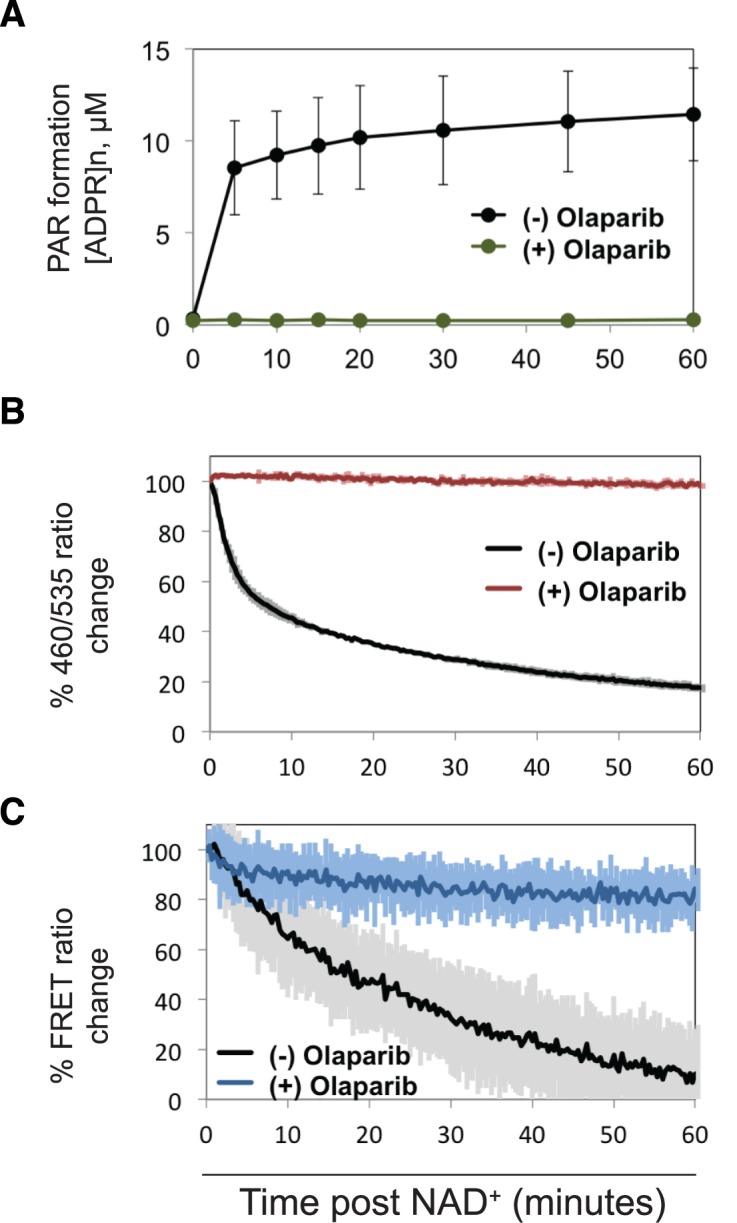
Reversal of DNA-dependent PARP-1 structural changes upon automodification. (A) Catalytic activity of PARP-1/WT was monitored over a time course long enough to reach a maximum of PAR formation. The inhibitor olaparib was used at 100 μM (0.1% DMSO) to prevent formation of PAR. Activity was measured using the PARP-1 colorimetric assay. (B) The change in HD destabilization was detected by recording the emission ratio (λ460/λ535) of acrylodan-PARP-1 mutant K700C over an hour after the addition of NAD+. (C) The change in FRET was detected by recording the emission ratio (λ612/λ512) of eGFP-PARP-1-mCherry over an hour after addition of NAD+. In each case 2 mM final NAD+ was added to an equimolar ratio of PARP-1 to DNA (1 μM each), and the 0 and 100% normalization was set by recording the values in the absence and presence of DNA damage. For the no inhibitor control, 0.1% DMSO was added instead. Data points were conducted in triplicates ± S.D.
The global structural compaction observed using FRET pair tagged PARP-1 in the presence of DNA damage was also reversed following the addition of NAD+ (Figure 5C). At the 15-min time point, PARP-1 compaction was 50% reversed. The reversal of DNA-dependent structural compaction was inhibited by olaparib, consistent with PAR production leading to the reversal of PARP-1 domain assembly on DNA damage.
Intramolecular PARP-1 activation is biochemically more robust than intermolecular activation
Recent structural studies have indicated an intramolecular mechanism of DNA damage-dependent PARP-1 activation, in which a single monomeric PARP-1 polypeptide achieves the assembly of domains on DNA damage required for activation (9,14). However, there are biochemical experiments in which two different, inactive variants of PARP-1 are mixed to restore DNA damage-dependent activity (11,15). Furthermore, fragments of PARP-1 mixed together can reconstitute DNA damage-dependent activity, and the detailed structural analysis of interactions between PARP-1 domains has resulted from studies using fragments of PARP-1 (9,10,14,38,39). Collectively, previous studies have indicated a propensity for PARP-1 domains to assemble from separate polypeptides, which has confounded the model of a monomeric intramolecular activation mechanism. We used the acrylodan-labeled PARP-1 system to evaluate the relative efficiency of intramolecular activation compared to intermolecular activation, where HD destabilization served as the readout for activation. As shown earlier, the mutation W318R in the Zn3 domain disrupts allosteric communication and thus prevents a signal of HD destabilization by the allosteric activation sensor (Figure 4B). When PARP-1/WT with no label was added to acrylodan-labeled PARP-1/W318R in the presence of dsDNA, HD destabilization of PARP-1/W318R was observed (Figure 6A), indicating that the unlabelled WT protein was capable of restoring function to the deficient PARP-1/W318R mutant. Notably, the strength of the signal did not reach the level of that observed for acrylodan-labeled PARP-1/WT on it own, indicating that the intermolecular combination does not work as efficiently as the intramolecular assembly of PARP-1 domains. Similarly, a catalytic active site mutant (E988A) was capable of partially restoring HD destabilization of PARP-1/W318R, and the intermolecular combination again did not achieve the activation level of the WT sensor alone.
Figure 6.

Inefficient intermolecular activation of PARP-1. Mixtures of PARP-1 fragments and mutants were analyzed in the presence of DNA damage to observe the efficiency of intermolecular activation. (A) Both WT and W318R versions of the acrylodan-labeled PARP-1 mutant K700C (0.5 μM) were mixed with unlabelled PARP-1/WT and mutants (0.5 μM) in the presence of dsDNA (1 μM). Detection was determined from the emission ratio (λ460/λ535) that reveals the extent of HD destabilization. (B) A schematic representation of intramolecular and intermolecular activation mechanisms. (C) The acrylodan-labeled PARP-1/WT and W318R mutant (0.5 μM) were mixed with dsDNA (1 μM) in the presence and absence of unlabeled PARP-1/WT (0.5 μM). Acrylodan-labeled W318R mutant was also mixed with unlabeled PARP-1 mutants. Mixtures were reacted with NAD+ for the specified amount of time and then resolved on 10% SDS-PAGE and visualized under UV light so that only the fluorescent acrylodan-labeled PARP-1 is visible. Data points were conducted in triplicates ± S.D.
We reasoned that the monomeric interaction of PARP-1 with DNA damage and the resulting multi-domain contacts with DNA create a stable complex that makes the observed intermolecular complementation an inefficient process, since there is steric frustration between the two polypeptides as they compete for domain interfaces and contact points. Notably, we have shown that the W318R mutant binds to DNA with robust affinity (37) and forms the compacted conformation on DNA damage (Figure 4A); thus W318R still forms a stable monomeric complex with DNA damage that is largely resistant to intermolecular contacts due to steric constraints. To test our line of reasoning, we deleted domains of unlabelled PARP-1 in this intermolecular complementation experiment and assessed whether this reduced steric constraints and improved the efficiency of HD destabilization. A CAT domain deletion supported HD destabilization at a similar level, and a WGR-CAT deletion supported a higher efficiency of HD destabilization (Figure 6A). Since the WGR domain is a critical component of PARP-1 interaction with DNA damage and domain–domain contacts, deletion of this region has likely made these binding sites more available for intermolecular complementation by removing steric hindrance (Figure 6B).
In correspondence with the HD destabilization signal, we observed PAR production with the intermolecular mixtures of PARP-1 using an SDS-PAGE activity assay that monitors automodification as an upward shift in electrophoretic migration. By using UV light detection of the gel, acrylodan-labeled PARP-1 can be selectively monitored (Figure 6C). As expected, acrylodan-labeled PARP-1/W318R alone was not automodified; however, a shift in migration was observed with W318R in the presence of PARP-1/WT, E988A or the C-terminal truncation mutants (Figure 6C). The shifts observed under these intermolecular conditions were not as efficient as those observed using WT protein alone, requiring longer incubation times to achieve even a modest level of activation, consistent with an inefficient process. It is also notable that even though the WGR-CAT deletion mutant was able to more efficiently support HD destabilization (Figure 6A), the intermolecular complementation did not translate into an increase in apparent PAR production (Figure 6C). This difference likely results from the fact that an intermolecular combination of domains is not able to optimally position PARP-1 for the automodification reaction (Figure 6B), as occurs in the intramolecular activation mechanism (9,14,22). Collectively, these results support biochemical transactivation as the domains of PARP-1 can ‘mix-and-match’ to a certain degree and thus support intermolecular activation. However, intermolecular activation is not an efficient process as steric interference between the domains of the two PARP-1 polypeptides leads to suboptimal automodification when compare to intramolecular activation.
PARP-1 intermolecular activation is not an effective response to DNA damage in cells
Although biochemical mixing of recombinant proteins can lead to a certain level of PARP-1 intermolecular complementation, there are no reported experiments that have tested the relevance of intermolecular PARP-1 activation in cells. If intermolecular activation is a relevant mechanism in cells, we hypothesized that two distinct PARP-1 mutants that are both deficient in DNA damage-dependent catalytic activation on their own should be capable of producing PAR when combined, as observed biochemically. Using mouse embryonic fibroblasts derived from the PARP-1 knockout mouse, we introduced human PARP-1 protein through cDNA transfection to assess the ability of two inactive PARP-1 molecules to support PAR production in response to DNA damage caused by H2O2 treatment. The PARP-1−/− cells contain functional PARP-2; however the amount of PAR produced by PARP-2 in response to DNA damage is minimal compared to that of PARP-1. An eGFP-tagged PARP-1/W318R mutant and an mCherry-tagged PARP-1/E988A mutant were analyzed, as well as the WT versions of the fluorescent fusion proteins. The purified fusion proteins are inactive on their own, but demonstrate automodification activity when mixed together biochemically (Supplementary Figure S5A). In cells, each of the mutant proteins efficiently localized to sites of laser-induced DNA damage (Supplementary Figure S5B), consistent with biochemical analysis that indicates that the mutations do not influence DNA binding affinity. On their own, the two PARP-1 mutants were unable to produce PAR in cells in response to H2O2 (Figure 7A–B), also consistent with biochemical analysis of their catalytic activity. However, the WT versions of the PARP-1 fusions with eGFP and mCherry robustly produced PAR in response to DNA damage, both on their own and in combination (Figure 7A). In contrast, cells that expressed both of the mutant proteins were not able to produce PAR in response to DNA damage (Figure 7A), indicating that intermolecular complementation does not occur in a cellular context. Upon observation of several replicate cells (Figure 7B), we consistently observed robust PAR production activation with WT PARP-1, but no PAR production of statistical significance in cells carrying the two mutant proteins. Taken together with the inefficient complementation observed using the HD allosteric activation sensor, we conclude that PARP-1 intermolecular activation is not relevant to the robust PAR production of PARP-1 observed in the cellular response to DNA damage.
Figure 7.

Cell-based analysis reveals no evidence of intermolecular PARP-1 activation. PARP-1−/− MEFs were transfected with PARP-1/WT, W318R, or E988A carrying either an eGFP or mCherry tag to show expression and nuclear localization. (A) Cells were treated with 10 mM H2O2 in serum-free media or mock-treated (no treatment, no H2O2) for 10 min before being washed with PBS and fixed with methanol and immunostained with mouse anti-PAR monoclonal antibody (10H) and Atto647N (STED) goat anti-mouse IgG antibody for analysis by immunofluorescence. Images are representative of at least 5 images taken at 40× magnification. Excitation of Atto647N was kept constant across every image, and the signal was adjusted by 1.5× across every image post-acquisition. (B) Fluorescent intensity of nuclear PAR staining across multiple cells. Individual cell values are depicted and the average signal and S.D. are shown. Statistical significance was determined using a Student's t test. ****P < 0.0001; n.s., no significance.
DISCUSSION
Here, we have developed two new fluorescence-based sensors that are capable of detecting global structural compaction and HD destabilization as indicators of the collapsed DNA damage bound status, and the catalytic activation status of PARP-1, respectively. In agreement with previous reports (9,10,15), experiments performed with the newly developed sensors indicate that the Zn1, Zn3, WGR and CAT domains are the minimally required components of DNA damage-dependent catalytic activation. Furthermore, our results support studies that identify allosteric regulation as a critical component for DNA damage-dependent catalytic activation (9,22,37) by showing that HD destabilization is sensitive to disruption of this network of interactions between PARP-1 domains (Figure 4). Interestingly, it was found that disruption of domain–domain interactions essential for allosteric regulation had only a slight effect on the global compaction of domains on DNA damage. This indicates that the functional DNA binding surfaces of essential domains are still capable of forming a collapsed structure on DNA, and it is the disrupted coordination of domain contacts that prevents efficient allosteric activation. The identification of novel compounds that inhibit allosteric regulation of PARP-1 may therefore serve as very effective ‘trapping’ agents (20).
Recent structural and biophysical analyses, including X-ray crystallography, solution NMR, small-angle X-ray scattering, and analytical ultracentrifugation, have indicated that a single PARP-1 polypeptide can assume the activated conformation on single-strand and double-strand break DNA damage (9,12–14,24). These studies thus support a monomeric PARP-1 interaction with DNA damage and an intramolecular activation mechanism. A crystal structure of the Zn1–Zn2 fragment of PARP-1 on duplex DNA suggested dimer formation between two PARP-1 polypeptides (27). However, the Zn1 domain bound in an unexpected conformation relative to other studies (9,10,14) and blocked the WGR binding site on DNA (9); thus the relevance of the crystal structure to a PARP-1 activation mechanism needs further development. The bulk of PARP-1 structural and biophysical analysis thus support an intramolecular activation mechanism; however, there are biochemical reports of two PARP-1 proteins with different inactivating mutations being able to support DNA damage-dependent activation when mixed together (11,15), which has been difficult to reconcile with an intramolecular activation mechanism.
In this study, we provide evidence that the intermolecular mechanism of PARP-1 activation is not efficient biochemically (Figure 6), and is not supported in a cell-based model (Figure 7). We reproduced the restoration of DNA damage-dependent activity by combining the inactive E988A and W318R mutants in a biochemical assay (Figure 6 and Supplementary Figure S5A). However, the fluorescence-based assay of HD destabilization (Figure 6A), as well as the PARP-1 activity assay (Figure 6C), both indicated that the efficiency of intermolecular activation was not as robust as intramolecular activation. Most notably, the co-expression of these two mutants did not produce PAR in cells in response to DNA damage over background levels, indicating that intermolecular activation cannot explain the robust PAR production of PARP-1 in the DNA damage response.
PARP-1 has a strong preference for automodification (modification on PARP-1 itself) over heteromodification (modification of other nuclear proteins). The PARP-1 conformation during the intramolecular activation mechanism provides a rationale for preferential in cis modification (9,14), and recent biochemical analysis has indicated that in cis modification is indeed possible (14). A compelling feature of intramolecular activation and in cis modification is that the same PARP-1 molecule that detects DNA damage becomes the platform for PAR-mediated recruitment of repair factors, and at some level of automodification the PARP-1 molecule will release itself from the damage. It is important to emphasize that heteromodification of other nuclear proteins such as histones is still possible with the intramolecular activation mechanism of PARP-1. In other words, intramolecular activation can lead to both in cis automodication of PARP-1, and in trans heteromodification of other proteins. The distribution between automodification and heteromodification will likely be determined by the conformation of PARP-1 and the cellular context, including local chromatin structure and potential binding partners/targets for modification. Interestingly, a recent study identified a PARP-1 binding partner HPF-1 that influences the distribution of PARP-1 automodification versus histone heteromodification during the DNA damage response (40).
Our fluorescence-based sensors indicated that the DNA-damage induced structural changes of PARP-1 reversed back to the non-activated conformation following the addition of NAD+. This relates to earlier studies of PARP-1, where it had been observed that PAR glycohydrolase (PARG) restored enzymatic activity and DNA binding affinity of PARP-1 (21). As predicted, the time course of the structural reversal coincided with an increase in PAR production (Figure 5). The PARP inhibitor olaparib prevented PAR production and the reversal of both HD destabilization and global compaction. A high concentration of inhibitor was required to completely prevent structural reversal (100 μM), as 10 μM olaparib still led to a slow reversal of structural change (data not shown). At this lower concentration, detection of PAR formation was strongly inhibited, which emphasizes the sensitivity of our HD allosteric sensor, and provides insights into the PARP inhibitor concentrations required to fully shut down PARP-1 activity.
In moving forward, identification of biologically relevant automodification sites and their consequential effects on structural integrity will greatly benefit our understanding of PARP-1 regulation. The functional differences between mono-ADP-ribosylation compared to poly-ADP-ribosylation in cells will also be important, as the enzyme that removes PAR modification, PARG, leaves a mono-ADP-ribose residue and the enzyme that removes the final mono-ADP-ribose residue, terminal (ADP-ribose) glycohydrolase (TARG), may have important implications as therapeutic targets (41,42). Presumably, inhibition of PARG could result in an increased pool of automodified, inactive PARP-1. Current PARP-1 inhibitors that target the catalytic active site represent an important class of targeted oncology agents that have progressed through FDA approval against BRCA deficient ovarian cancer. Alternative routes to pharmacological manipulation of PARP-1 activity may therefore have important therapeutic outcomes. Use of the presented HD allosteric sensor thus has further implications as a screening assay to identify small molecules that disrupt PARP-1 allosteric activation.
Supplementary Material
Acknowledgments
We thank Drs S. Eustermann and D. Neuhaus (MRC Laboratory of Molecular Biology) for a generous gift of wild-type Staph Aureus sortase A enzyme. We thank Dr Teresa Fernandes-Alnemri (Thomas Jefferson University) for the generous gift of Caspase-3 enzyme. We thank A. Sverzhinsky and M.F. Langelier for feedback and discussions on the manuscript.
Footnotes
Present address: Jamin D. Steffen, JBS Science, Inc., 3805 Old Easton Road, Doylestown, PA 18902.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [GM087282 to J.M.P. and CA174221 to J.S.]. Funding for open access charge: National Institutes of Health [GM087282].
Conflict of interest statement. None declared.
REFERENCES
- 1.Hottiger M.O., Hassa P.O., Luscher B., Schuler H., Koch-Nolte F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010;35:208–219. doi: 10.1016/j.tibs.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 2.D'Amours D., Desnoyers S., D'Silva I., Poirier G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999;342:249–268. [PMC free article] [PubMed] [Google Scholar]
- 3.Ogata N., Ueda K., Kawaichi M., Hayaishi O. Poly(ADP-ribose) synthetase, a main acceptor of poly(ADP-ribose) in isolated nuclei. J. Biol. Chem. 1981;256:4135–4137. [PubMed] [Google Scholar]
- 4.Hassa P.O., Haenni S.S., Elser M., Hottiger M.O. Nuclear ADP-ribosylation reactions in mammalian cells: Where are we today and where are we going. Microbiol. Mol. Biol. Rev. 2006;70:789–829. doi: 10.1128/MMBR.00040-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferro A.M., Olivera B.M. Poly(ADP-ribosylation) in vitro. reaction parameters and enzyme mechanism. J. Biol. Chem. 1982;257:7808–7813. [PubMed] [Google Scholar]
- 6.Kim G., Ison G., McKee A.E., Zhang H., Tang S., Gwise T., Sridhara R., Lee E., Tzou A., Philip R., et al. FDA approval summary: Olaparib monotherapy in patients with deleterious germline BRCA-mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin. Cancer Res. 2015;21:4257–4261. doi: 10.1158/1078-0432.CCR-15-0887. [DOI] [PubMed] [Google Scholar]
- 7.Karlberg T., Langelier M.F., Pascal J.M., Schuler H. Structural biology of the writers, readers, and erasers in mono- and poly(ADP-ribose) mediated signaling. Mol. Aspects Med. 2013;34:1088–1108. doi: 10.1016/j.mam.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Langelier M.F., Riccio A.A., Pascal J.M. PARP-2 and PARP-3 are selectively activated by 5′ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic Acids Res. 2014;42:7762–7775. doi: 10.1093/nar/gku474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langelier M.F., Planck J.L., Roy S., Pascal J.M. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science. 2012;336:728–732. doi: 10.1126/science.1216338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Langelier M.F., Planck J.L., Roy S., Pascal J.M. Crystal structures of poly(ADP-ribose) polymerase-1 (PARP-1) zinc fingers bound to DNA: structural and functional insights into DNA-dependent PARP-1 activity. J. Biol. Chem. 2011;286:10690–10701. doi: 10.1074/jbc.M110.202507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Langelier M.F., Ruhl D.D., Planck J.L., Kraus W.L., Pascal J.M. The Zn3 domain of human poly(ADP-ribose) polymerase-1 (PARP-1) functions in both DNA-dependent poly(ADP-ribose) synthesis activity and chromatin compaction. J. Biol. Chem. 2010;285:18877–18887. doi: 10.1074/jbc.M110.105668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lilyestrom W., van der Woerd M.J., Clark N., Luger K. Structural and biophysical studies of human PARP-1 in complex with damaged DNA. J. Mol. Biol. 2010;395:983–994. doi: 10.1016/j.jmb.2009.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mansoorabadi S.O., Wu M., Tao Z., Gao P., Pingali S.V., Guo L., Liu H.W. Conformational activation of poly(ADP-ribose) polymerase-1 upon DNA binding revealed by small-angle X-ray scattering. Biochemistry. 2014;53:1779–1788. doi: 10.1021/bi401439n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eustermann S., Wu W.F., Langelier M.F., Yang J.C., Easton L.E., Riccio A.A., Pascal J.M., Neuhaus D. Structural basis of detection and signaling of DNA single-strand breaks by human PARP-1. Mol. Cell. 2015;60:742–754. doi: 10.1016/j.molcel.2015.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Altmeyer M., Messner S., Hassa P.O., Fey M., Hottiger M.O. Molecular mechanism of poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues as ADP-ribose acceptor sites. Nucleic Acids Res. 2009;37:3723–3738. doi: 10.1093/nar/gkp229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tao Z., Gao P., Liu H.W. Identification of the ADP-ribosylation sites in the PARP-1 automodification domain: analysis and implications. J. Am. Chem. Soc. 2009;131:14258–14260. doi: 10.1021/ja906135d. [DOI] [PubMed] [Google Scholar]
- 17.Chapman J.D., Gagne J.P., Poirier G.G., Goodlett D.R. Mapping PARP-1 auto-ADP-ribosylation sites by liquid chromatography-tandem mass spectrometry. J. Proteome Res. 2013;14:1868–1880. doi: 10.1021/pr301219h. [DOI] [PubMed] [Google Scholar]
- 18.Gagne J.P., Ethier C., Defoy D., Bourassa S., Langelier M.F., Riccio A.A., Pascal J.M., Moon K.M., Foster L.J., Ning Z., et al. Quantitative site-specific ADP-ribosylation profiling of DNA-dependent PARPs. DNA Repair (Amst.) 2015;38:68–79. doi: 10.1016/j.dnarep.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 19.Mortusewicz O., Ame J., Schreiber V., Leonhardt H. Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res. 2007;35:7665–7675. doi: 10.1093/nar/gkm933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murai J., Huang S.Y., Das B.B., Renaud A., Zhang Y., Doroshow J.H., Ji J., Takeda S., Pommier Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72:5588–5599. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zahradka P., Ebisuzaki K. A shuttle mechanism for DNA-protein interactions. Eur. J. Biochem. 1982;127:579–585. [PubMed] [Google Scholar]
- 22.Dawicki-McKenna J.M., Langelier M.F., DeNizio J.E., Riccio A.A., Cao C.D., Karch K.R., McCauley M., Steffen J.D., Black B.E., Pascal J.M. PARP-1 activation requires local unfolding of an autoinhibitory domain. Mol. Cell. 2015;60:755–768. doi: 10.1016/j.molcel.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clark N.J., Kramer M., Muthurajan U.M., Luger K. Alternative modes of binding of poly(ADP-ribose) polymerase 1 to free DNA and nucleosomes. J. Biol. Chem. 2012;287:32430–32439. doi: 10.1074/jbc.M112.397067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eustermann S., Videler H., Yang J.C., Cole P.T., Gruszka D., Veprintsev D., Neuhaus D. The DNA-binding domain of human PARP-1 interacts with DNA single-strand breaks as a monomer through its second zinc finger. J. Mol. Biol. 2011;407:149–170. doi: 10.1016/j.jmb.2011.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mendoza-Alvarez H., Alvarez-Gonzalez R. Poly(ADP-ribose) polymerase is a catalytic dimer and the automodification reaction is intermolecular. J. Biol. Chem. 1993;268:22575–22580. [PubMed] [Google Scholar]
- 26.Pion E., Ullmann G.M., Ame J.C., Gerard D., de Murcia G., Bombarda E. DNA-induced dimerization of poly(ADP-ribose) polymerase-1 triggers its activation. Biochemistry. 2005;44:14670–14681. doi: 10.1021/bi050755o. [DOI] [PubMed] [Google Scholar]
- 27.Ali A.A., Timinszky G., Arribas-Bosacoma R., Kozlowski M., Hassa P.O., Hassler M., Ladurner A.G., Pearl L.H., Oliver A.W. The zinc-finger domains of PARP1 cooperate to recognize DNA strand breaks. Nat. Struct. Mol. Biol. 2012;19:685–692. doi: 10.1038/nsmb.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Langelier M.F., Planck J.L., Servent K.M., Pascal J.M. Purification of human PARP-1 and PARP-1 domains from escherichia coli for structural and biochemical analysis. Methods Mol. Biol. 2011;780:209–226. doi: 10.1007/978-1-61779-270-0_13. [DOI] [PubMed] [Google Scholar]
- 29.Wang Z.Q., Auer B., Stingl L., Berghammer H., Haidacher D., Schweiger M., Wagner E.F. Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev. 1995;9:509–520. doi: 10.1101/gad.9.5.509. [DOI] [PubMed] [Google Scholar]
- 30.Prendergast F.G., Meyer M., Carlson G.L., Iida S., Potter J.D. Synthesis, spectral properties, and use of 6-acryloyl-2-dimethylaminonaphthalene (acrylodan). A thiol-selective, polarity-sensitive fluorescent probe. J. Biol. Chem. 1983;258:7541–7544. [PubMed] [Google Scholar]
- 31.Chen I., Dorr B.M., Liu D.R. A general strategy for the evolution of bond-forming enzymes using yeast display. Proc. Natl. Acad. Sci. U.S.A. 2011;108:11399–11404. doi: 10.1073/pnas.1101046108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hibbs R.E., Talley T.T., Taylor P. Acrylodan-conjugated cysteine side chains reveal conformational state and ligand site locations of the acetylcholine-binding protein. J. Biol. Chem. 2004;279:28483–28491. doi: 10.1074/jbc.M403713200. [DOI] [PubMed] [Google Scholar]
- 33.Albertazzi L., Arosio D., Marchetti L., Ricci F., Beltram F. Quantitative FRET analysis with the EGFP-mCherry fluorescent protein pair. Photochem. Photobiol. 2009;85:287–297. doi: 10.1111/j.1751-1097.2008.00435.x. [DOI] [PubMed] [Google Scholar]
- 34.Sapsford K.E., Berti L., Medintz I.L. Materials for fluorescence resonance energy transfer analysis: Beyond traditional donor-acceptor combinations. Angew. Chem. Int. Ed Engl. 2006;45:4562–4589. doi: 10.1002/anie.200503873. [DOI] [PubMed] [Google Scholar]
- 35.Haince J.F., McDonald D., Rodrigue A., Dery U., Masson J.Y., Hendzel M.J., Poirier G.G. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008;283:1197–1208. doi: 10.1074/jbc.M706734200. [DOI] [PubMed] [Google Scholar]
- 36.Ikejima M., Noguchi S., Yamashita R., Ogura T., Sugimura T., Gill D.M., Miwa M. The zinc fingers of human poly(ADP-ribose) polymerase are differentially required for the recognition of DNA breaks and nicks and the consequent enzyme activation. other structures recognize intact DNA. J. Biol. Chem. 1990;265:21907–21913. [PubMed] [Google Scholar]
- 37.Steffen J.D., Tholey R.M., Langelier M.F., Planck J.L., Schiewer M.J., Lal S., Bildzukewicz N.A., Yeo C.J., Knudsen K.E., Brody J.R., et al. Targeting PARP-1 allosteric regulation offers therapeutic potential against cancer. Cancer Res. 2014;74:31–37. doi: 10.1158/0008-5472.CAN-13-1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Langelier M.F., Servent K.M., Rogers E.E., Pascal J.M. A third zinc-binding domain of human poly(ADP-ribose) polymerase-1 coordinates DNA-dependent enzyme activation. J. Biol. Chem. 2008;283:4105–4114. doi: 10.1074/jbc.M708558200. [DOI] [PubMed] [Google Scholar]
- 39.Tao Z., Gao P., Hoffman D.W., Liu H. Domain C of human poly(ADP-ribose) polymerase-1 is important for enzyme activity and contains a novel zinc-ribbon motif. Biochemistry. 2008;47:5804–5813. doi: 10.1021/bi800018a. [DOI] [PubMed] [Google Scholar]
- 40.Gibbs-Seymour I., Fontana P., Rack J.G., Ahel I. HPF1/C4orf27 is a PARP-1-interacting protein that regulates PARP-1 ADP-ribosylation activity. Mol. Cell. 2016;62:432–442. doi: 10.1016/j.molcel.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Slade D., Dunstan M.S., Barkauskaite E., Weston R., Lafite P., Dixon N., Ahel M., Leys D., Ahel I. The structure and catalytic mechanism of a poly(ADP-ribose) glycohydrolase. Nature. 2011;477:616–620. doi: 10.1038/nature10404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sharifi R., Morra R., Appel C.D., Tallis M., Chioza B., Jankevicius G., Simpson M.A., Matic I., Ozkan E., Golia B., et al. Deficiency of terminal ADP-ribose protein glycohydrolase TARG1/C6orf130 in neurodegenerative disease. EMBO J. 2013;32:1225–1237. doi: 10.1038/emboj.2013.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.