

Abstract
Lung cancer, primarily non-small-cell lung cancer (NSCLC), is the leading cause of cancer deaths in the United States and worldwide. While early detection significantly improves five-year survival, there are no reliable diagnostic tools for early detection. Several exosomal microRNAs (miRs) are overexpressed in NSCLC, and have been suggested as potential biomarkers for early detection. The present paper develops a mathematical model for early stage of NSCLC with emphasis on the role of the three highest overexpressed miRs, namely miR-21, miR-205 and miR-155. Simulations of the model provide quantitative relationships between the tumor volume and the total mass of each of the above miRs in the tumor. Because of the positive correlation between these miRs in the tumor tissue and in the blood, the results of the paper may be viewed as a first step toward establishing a combination of miRs 21, 205, 155 and possibly other miRs as serum biomarkers for early detection of NSCLC.
Introduction
Lung cancer is the leading cause of cancer-related deaths in the United States and worldwide, and non-small cell lung cancer (NSCLC) constitutes 85% of lung cancer deaths [1, 2]. Five years survival rate for NSCLC is significantly higher for those diagnosed at early stage [3], but there are no reliable tools for early detection of lung cancer. Most lung cancers are first diagnosed on symptoms. Approximately 10% of patients present brain metastasis at the time of initial diagnosis and their mean survival is 4 months [4]. Hence, there is a need for novel noninvasive biomarkers for early lung cancer diagnosis [5].
Exosomes are nano-vesicles of size 30-100 nm in diameter, surrounded by a lipid bilayer, and containing fuctional proteins, mRNAs and microRNAs (miRs). Exosomes are released by various cells, including cancer cells [6]. A growing body of evidence suggests that exosomal miRs may be used as serum biomarkers for prognosis of malignant tumors [5, 7]. Furthermore, exosomal miRs inhibitors have been evaluatedas anti-tumor drugs in experimental and clinical work for several types of cancer, including lung cancer [8, 9].
In the present paper we develop a mathematical model that relates the role of the exosomal miRs in lung cancer tissue to cancer cells proliferation and invasion. Since there is a positive correlation between exosomal miRs in serum and tissue in lung cancer [10–12], the model may serve as a first step toward establishing miRs as reliable serum biomarkers for early detection.
A simple schematic of a cell proliferation in the context of cancer is shown in Fig 1. When epidermal growth factor (EGF) ligands to its receptor EGFR, it initiates activation of the Ras-Raf-MEK-ERK pathway [13, 14] and the PI3K-AKT pathway [15–17]. Both pathways lead to cell proliferation [18–21] through activation of mTOR [16, 20, 22]. EGF-EGFR is negatively regulated by ERK [14, 23].
Fig 1. Two pathways, Ras-Raf-MEK-ERK and PI3K-AKT lead to the cell proliferation.
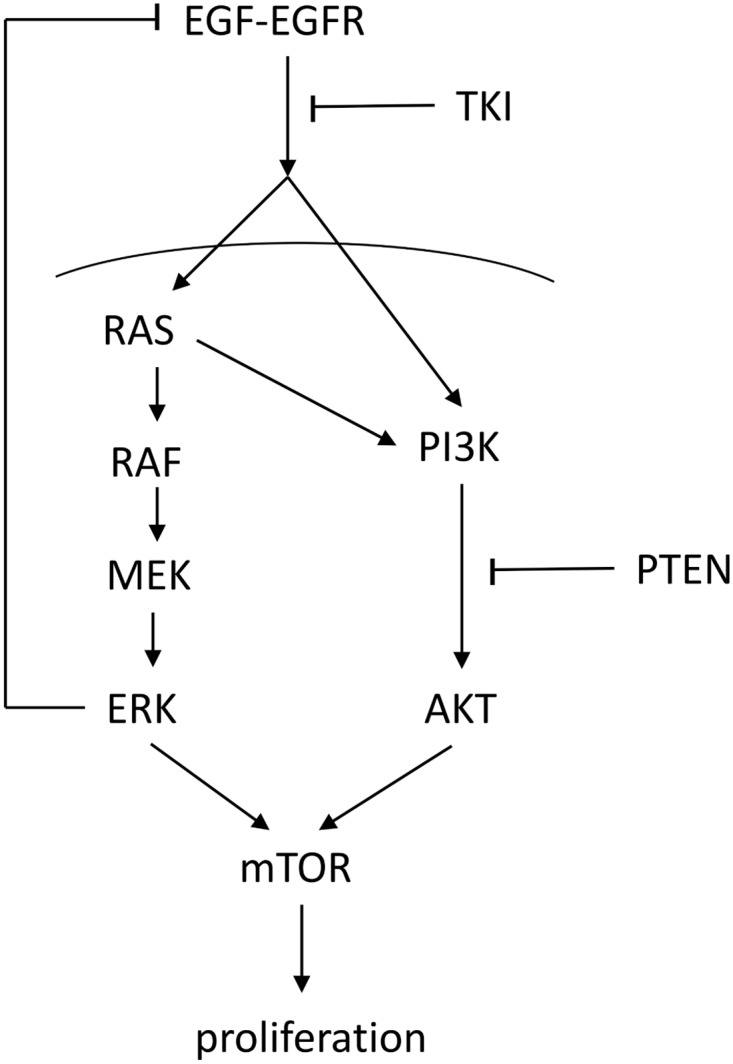
Sharp arrows indicate activation/enhancement and blocked arrows indicate inhibition.
EGFR tyrosine kinase inhibitor (TKI) modulates the activation of both RAS and PI3K, thereby inhibiting the activation of the downstream pathways of EGF-EGFR [13, 24, 25]. PTEN modulates the activation of AKT through converting PIP3, generated by PI3K, to PIP2 by dephosphorylation [15]. When DNA damage occurs, a signaling pathway activates Apaf-1 and caspase 9, forming an apoptosome, which leads to apoptosis through activation of caspase 3 [26, 27].
In NSCLC, the most expressed exosomal miRs are miR-21, miR-155 and miR-205 [10]. In Fig 2(a), we simplified the network of Fig 1 by using MAPK and AKT to represent the Ras-Raf-MEK-ERK and PI3K-AKT pathways. Fig 2(a) also shows the effect of overexpression of miR-21 and miR-205 on NSCLC proliferation. Recent studies established that miR-21 blocks TKI [8, 25], and thus promotes activation of the MAPK and AKT pathways. Also, miR-21 and miR-205 block PTEN [28, 29] and thus promote the activation of the AKT-mTOR pathway. Fig 2(b) shows the effect of overexpression of miR-155. MiR-155 blocks Apaf-1 expression [30] and thus also cellular apoptosis when DNA damage occurs. Hence overexpressions of miR-21 and miR-205 give rise to uncontrolled proliferation, while overexpression of miR-155 leads to reduced apoptosis. MiR-21 and miR-205 have also other targets; in particular it was suggested that miR-21 targets tumor suppressors involved in apopotsis, including Apaf-1, Pdcd4, RhoB and Faslg [31, 32]; hence overexpression of miR-21 reduces apoptosis. However, for simplicity, we focus in this paper on what seems to be the main targets of miR-21 and miR-205 in NSCLC, as shown in Fig 2.
Fig 2. Abbreviated version of Fig 1 depicting the roles of miR-21, miR-205 and miR-155.

MAPK represents the Ras-Raf-MEK-ERK signaling pathway and AKT represents the PI3K-AKT signaling pathway. (a) MiR-21 blocks TKI; miR-21 and miR-205 block PTEN. (b) MiR-155 blocks Apaf-1+caspase 9. Sharp arrows indicate activation/enhancement and blocked arrows indicate inhibition.
In this paper we consider growth and invasion of lung tumor associated with mutations in EGFR, MAPK and AKT, and its treatment by anti exosomal miRNAs (miR-21, miR-205 and miR-155). We use the mathematical model to determine the efficacy of these drugs under these mutations.
We consider two aspects of tumor progression: (i) Invasion, in which a tumor planar front progresses, in time, away from the main body of the tumor; (ii) Proliferation, in which a small spherical tumor grows in time. In order to focus on the role of the exosomal-miRs, we do not include in the model the immune responses and angiogenesis; thus the model represents an early stage of lung cancer.
Mathematical model
The mathematical model is based on the network shown in Fig 2. For simplicity we use just one variable, MAPK, to represent the Ras-Raf-MEK-ERK signaling pathway, and AKT to represent the PI3K-AKT signaling pathway. We also combine miR-205 with miR-21 in modeling their effect on blocking PTEN. Table 1 lists the variables used in the mathematical model in unit of g/cm3.
Table 1. List of variables in unit of g/cm3.
| Notation | Description |
|---|---|
| E | EGF-EGFR concentration |
| M | active MAPK concentration |
| P3 | PI3K concentration |
| A | active AKT concentration |
| T | TKI concentration |
| P | PTEN concentration |
| Ap | Apaf-1-caspase 9 apoptosome concnetration |
| EC | cancer-shed exosome concentration |
| m1 | exosomal miR-21 concentration (inlcuding miR-205) |
| cellular miR-21 concentration (inlcuding miR-205) | |
| m2 | exosomal miR-155 concentration |
| cellular miR-155 concentration | |
| C | cancer cells density |
| N | normal healthy cells density |
Equations for proteins As in [33], the dynamics of the proteins within cancer cells will appear in the form
where C0 is the steady state density of cancer cells.
Equation for EGF-EGFR (E). The equation for EGF-EGFR is given by
| (1) |
The coefficient λE is the production rate of the EGF-EGFR complex and the factor 1/(1 + M/KME) is the inhibition by ERK [14]; dE is the degradation rate of E.
Equation for MAPK (M). The MAPK pathway is activated by the EGF-EGFR [13, 14], a process resisted by TKI [8, 13]. Hence
| (2) |
where dM is the degradation rate of M.
Equation for AKT (A). The activation of the AKT pathway initiates with the activation of PI3K (P3) by EGF-EGFR directly and also through Ras (which is activated by EGF-EGFR) [15, 16, 19, 20]. In view of the TKI inhibition of EFG-EGFR [24, 25], the equation for P3 takes the form
where S is the concentration of Ras.
AKT is activated by PI3K which is negatively regulated by PTEN [15, 25], so that
We assume that the turnover of PI3K is very fast (the half-life of PI3K is very short [34]) and deduce from the steady state equation of P3 that
Substituting this into the equation for AKT, we obtain
Assuming also that the concentration of Ras is proportional to that of MAPK, i.e. S = μM, we obtain the following equation for AKT:
| (3) |
where λA = λAP λP3/dP3, KMA = KSP/μ.
Equation for TKI (T). The production of TKI is inhibited by miR-21 [8, 25]. Recalling that miR-21 is only a fraction of m1, we write the equation for TKI in the form
| (4) |
Equation for PTEN (P). The expression of PTEN is inhibited by both miR-21 and miR-205 [28, 29]. Hence
| (5) |
Equation for Apaf-1-caspase 9 apoptosome (Ap). The expression of Apaf-1 is down-regulated by miR-155 [30]. Hence
| (6) |
Equation for exosome (EC). Cancer cells shed exosomes at a rate λEcC. We assume that exosomes are degraded, releasing their miRs, when merging with cancer cells. Taking the rate of this degradation to be , the equation for the concentration of exosomes is given by
| (7) |
where the term DEcΔEC represents dispersion (or diffusion) of exosomes.
Equations for exosomal miR-21 (m1) and exosomal miR-155 (m2). MiR-21 and miR-155 are released from exosomes when exosomes merge with cancer cells. We take the exosomal production rate of miR-21 to be λm1EC ⋅ C/(KC + C), and obtain the equation
| (8) |
Similarly, the equation for miR-155 is given by
| (9) |
Equations for miR-21 () and miR-155 () in cancer cells. Since and lie in cancer cells, they diffuse with same coefficient as cancer cells. Hence, the equations for and are given by
| (10) |
and
| (11) |
respectively.
We will apply the mathematical model to consider two phases of tumor progression of lung cancer, invasion and proliferation.
Model for tumor invasion
Equation for cancer cell (C) in tumor invasion. The equation for cancer cell is the following:
| (12) |
Invasion of cancer cells is driven by competition for space and resources [35, 36]. At the early stage of tumor invasion resources are not limited, hence cells undergo migration in the direction of decreased gradient of cancer cells density. On the left-hand side of Eq (12) the term χ∇ ⋅ (C∇C) represents the directed migration of cancer cells in response to the competition for space; χ is the ‘directed migration coefficient’. We assume a logistic growth with rate which depends on both MAPK and AKT, since both pathways lead to cell replication; this accounts for the first term on the right-hand side of Eq (12). In addition to natural apoptosis, at rate dCC, damage to cancer cells (at rate proportional to dDC) leads to apoptosis by formation of the Apaf-1-caspase 9 apoptosome [26, 27]; this is accounted for by the second term on the right-hand side of Eq (12).
Boundary and initial conditions for tumor invasion model. We assume that a solid tumor lies in the the half plane x < 0, and model the progression of the tumor front in the direction of increasing x. We assume that the tumor front is planar, and that it moves in the interval 0 ≤ x ≤ 2 from x = 0 cm towards the end-point x = 2 cm. We impose the following boundary conditions for the cancer cells, exosomes and miRs:
| (13) |
Since initially the cancer is confined to x < 0, we take zero initial conditions:
| (14) |
Model for tumor proliferation
Equations for cancer cells (C) and normal healthy cells (N) in tumor proliferation. In the tumor invasion model, the directed migration coefficient χ represents the directed movement of the invading tumor cells. The precise range of the parameter χ is unknown. In order to visualize significant advance of the migrating tumor front we took χ to be the ‘relatively’ large. However, in the tumor proliferation model, proliferating cells grow faster than migrating cells [37–39], and the competition for space is primarily a competition with normal healthy cells (with density N) [40]. We therefore assume that the directed movement of cells is determined by the condition that the total cell density, C + N, is constant at each point in the tumor. The term −χ∇ ⋅ (C∇C) in Eq (12) is then neglected and replaced by the term , where represents the velocity of cells. The equation for cancer cells is given by
| (15) |
where the competition for space with the normal healthy cells is represented by the term εN in the logistic growth. We assume that most exosomes shed by cancer cells release their content when they make contact with nearby cancer cells, and therefore keep Eq (7) unchanged. The equation for normal healthy cells, N, is given by
| (16) |
The competition for space with cancer cells is represented by the term εC in the logistic growth term [40].
To simplify the computations, we assume that the tumor is spherical and denote its moving boundary (i.e. its radius) by r = R(t). We also assume that all the densities and concentrations are radially symmetric, that is, functions of (r, t), where 0 ≤ r ≤ R(t). In particular, , where is the unit radial vector.
Equation for u: We assume that the combined densities of healthy and cancer cells in the tumor is constant (θ), and take
| (17) |
We also assume that DN = DC. Adding Eqs (16) and (15), we obtain
so that
We assume that the free boundary r = R(t) moves with the velocity of cells:
| (18) |
Boundary and initial conditions for tumor proliferation model. We impose the boundary conditions:
| (19) |
We assume that the concentrations of proteins TKI, PTEN and Apaf-1 which inhibit tumor growth are ‘relatively’ high, i.e., above the steady state, so that they initially decrease as the tumor begins to increase. We also assume that the remaining proteins which are involved in promoting tumor growth, are initially below their steady state. One choice of initial conditions is given below:
| (20) |
Results
Results for tumor invasion
In simulating the invasion of cancer cells we use the model Eqs (1)–(12), with boundary Conditions (13) and initial Conditions (14), and with the parameters of Tables 2 and 3.
Table 2. Summary of parameter values.
| Notation | Description | Value used | References |
|---|---|---|---|
| DEc | diffusion coefficient of exosomes | 1.23 × 10−4 cm2day−1 | [82] & estimated |
| Dm1 | diffusion coefficient of miR-21 | 0.13028 cm2day−1 | [61, 83] & estimated |
| Dm2 | diffusion coefficient of miR-155 | 0.13028 cm2day−1 | [61, 83] & estimated |
| DC | diffusion coefficient of cancer cells | 8.64 × 10−7 cm2day−1 | [77] |
| DN | diffusion coefficient of normal healthy cells | 8.64 × 10−7 cm2day−1 | [77] |
| χ | directed migration coefficient | 3 × 10−4 − 3 × 10−2 cm5g−1day−1 | [33] & estimated |
| λE | production rate of EGF-EGFR | 1.1741 × 10−3 day−1 g/cm3 | [47] & estimated |
| λM | production rate of MAPK | 1.6499 × 10−2 day−1 | [47, 48] & estimated |
| λA | production rate of AKT | 2.9422 × 10−3 day−1 | [67, 68] & estimated |
| λMA | activation rate of AKT by MAPK (Ras) | 1/2 | [75] & estimated |
| λT | production rate of TKI | 8.4013 × 10−4 day−1 g/cm3 | estimated |
| λP | production rate of PTEN | 2.3352 × 10−4 day−1 g/cm3 | [47, 70] & estimated |
| λAp | production rate of Apaf-1 | 4.4095 × 10−3 day−1 g/cm3 | [73] & estimated |
| λEc | production rate of exosome by NSCLC cells | 9.81 × 10−9 day−1 | [58] & estimated |
| λm1 | production rate of miR-21 & miR-205 by EC | 0.8626 × 10−3 day−1 | [59, 74] & estimated |
| λm2 | production rate of miR-155 by EC | 0.4313 × 10−3 day−1 | [59, 74] & estimated |
| λC1 | growth rate of NSCLC cells due to MAPK | 0.6133 day−1 | [77] & estimated |
| λC2 | growth rate of NSCLC cells due to AKT | 0.3067 day−1 | [77] & estimated |
| λN | growth rate of normal healthy cells | 0.092 day−1 | [77] & estimated |
| ε | competition for space coefficient | 0.1 | [40] |
| dE | degradation rate of EGF-EGFR | 0.8318 day−1 | [47] & estimated |
| dM | degradation rate of MAPK | 0.6931 day−1 | [63–66] |
| dA | degradation rate of AKT | 0.6931 day−1 | [67, 68] |
| dT | degradation rate of TKI | 0.3466 day−1 | [69] & estimated |
| dP | degradation rate of PTEN | 22.1807 day−1 | [70, 71] |
| dAp | degradation rate of Apaf-1 | 2.7726 day−1 | [73] |
| dEc | degradation rate of exosome | 21.8 day−1 | fitted |
| dm1 | degradation rate of miR-21 and miR-205 | 0.5545 day−1 | [74] |
| dm2 | degradation rate of miR-155 | 0.5545 day−1 | [74] |
| dC | natural death rate of cancer cells | 0.023 day−1 | [77] & estimated |
| dD | death rate of cancer cells due to DNA damage | 0.414 day−1 | [77] & estimated |
| dN | natural death rate of normal healthy cells | 0.023 day−1 | [77] & estimated |
Table 3. Summary of parameter values (continued).
| Notation | Description | Value used | References |
|---|---|---|---|
| E0 | S.S. 1concentration of EGF-EGFR | 7.0573 × 10−4 g/cm3 | [47] |
| M0 | S.S. concentration of MAPK | 8.4 × 10−6 g/cm3 | [48, 50] |
| P30 | S.S. concentration of PI3K | 1.56 × 10−6 g/cm3 | [53, 54] |
| A0 | S.S. concentration of AKT | 9.362 × 10−7 g/cm3 | [53] |
| T0 | S.S. concentration of TKI | 8.5050 × 10−5 g/cm3 | estimated |
| P0 | S.S. concentration of PTEN | 1.88 × 10−7 g/cm3 | [47] |
| Ap0 | S.S. concentration of Apaf-1-caspase 9 | 2.84 × 10−5 g/cm3 | [57] |
| EC0 | S.S. concentration of C-shed exosome | 3.6 × 10−10 g/cm3 | [58] |
| m10 | S.S. concentration of miR-21 and miR-205 | 2.8 × 10−13 g/cm3 | [59, 60] |
| m20 | S.S. concentration of miR-155 | 1.4 × 10−13 g/cm3 | [10, 59] |
| C0 | S.S. density of cancer cell | 0.4 g/cm3 | [62] |
| N0 | S.S. density of normal healthy cells | 0.14 g/cm3 | [62] & estimated |
| CM | carrying capacity of NSCLC cells | 0.8 g/cm3 | [62] |
| KME | inhibition of EGF-EGFR by ERK | 3.936 × 10−5 g/cm3 | [53, 54] & estimated |
| KTM | inhibition of MAPK by TKI | 8.5050 × 10−5 g/cm3 | estimated |
| KTA | inhibition of AKT by TKI | 8.5050 × 10−5 g/cm3 | estimated |
| KPA | inhibition of AKT by PTEN | 1.88 × 10−7 g/cm3 | [47] |
| KmT | inhibition of TKI by miR-21 | 0.56 × 10−13 g/cm3 | [59, 60] & estimated |
| KmP | inhibition of PTEN by miR-21 and miR-205 | 0.56 × 10−13 g/cm3 | [59, 60] & estimated |
| Km2 | inhibition of Apaf-1 by miR-155 | 0.28 × 10−13 g/cm3 | [10, 59] & estimated |
| KMA | half-saturation of MAPK (Ras) in AKT activation | 8.4 × 10−6 g/cm3 | [48, 50] & estimated |
| KC | half-saturation of cancer cell on merging with exosome | 0.4 g/cm3 | [62] & estimated |
| KM | half-saturation of MAPK on cancer cell proliferation | 8.4 × 10−6 g/cm3 | [48, 50] & estimated |
| KA | half-saturation of AKT on cancer cell proliferation | 9.362 × 10−7 g/cm3 | [47] & estimated |
| KAp | half-saturation of Apaf-1 on NSCLC cell apoptosis | 2.84 × 10−5 g/cm3 | [57] & estimated |
1S.S. refers to steady state.
We explore how specific mutations affect the invasion of the tumor front, and how anti-miR drugs slow the invasion. We consider four cases: (i) the control case (with unspecified mutations), (ii) new mutation in EGFR, (iii) new mutation in MAPK, (iv) new mutation in AKT. In the control case all the parameters are taken to be the same as Tables 2 and 3, and χ is taken to be 3 × 10−2 cm5g−1day−1. In the case of mutations in EGFR, MAPK or AKT, χ is unchanged but the production rates λE, λM and λA are increased by some factor. The first row of Fig 3 shows the spatial profile of cancer cell density C(x, t) in the control case, and in the three cases of mutations (in EGFR, MAPK and AKT) at different time points t = 5, 15, 30, 60 days. We see that under each of the three mutations the tumor advanced at day 60 by at least 10% more than in the control case. We note however that although the sizes of the invasion under the three mutations are nearly the same, we accounted for the three mutations differently, increasing the production rates of EGFR by a factor 1.3, of MAPK by a factor 1.6 and of AKT by a factor 1.8. The ratios between these factors suggest that a mutation of EGFR increases tumor invasion more than a mutation of MAPK, and a mutation of MAPK increases tumor invasion more than a mutation of AKT. These suggestions, however, need to be verified experimentally.
Fig 3. Invasion of cancer cells with density C(x, t).

The parameters are as in Tables 2 and 3, and χ = 3 × 10−2 cm5g−1day−1. (a) The first row: The control case; a mutation in EGFR where λE is increased by 1.3-fold; a mutation in MAPK where λM is increased by 1.6-fold; a mutation in AKT where λE is increased by 1.8-fold. (b) The second row: Using anti-miR-21, where λm1 is reduced by a factor 2 compared to the first row. (c) The third row: Using both anti-miR-21 and anti-miR-155, where λm1 and λm2 are reduced by a factor 2 compared to the first row. The time is in unit of day, and x is in unit of cm.
The second row of Fig 3 shows the effect of anti-miR-21 drug in the control case and in the cases of EGFR, MAPK and AKT mutations. We note that anti-miR-21 reduces the rate of invasion by approximately 17%. When both anti-miR-21 and anti-miR-155 are combined, the reduction is by 40%, as seen in the third row of Fig 3.
Fig 4 simulates the dynamics of the total linear mass of the cancer cells in the control case and the cases of the three mutations when (i) no drug is applied, (ii) anti-miR-21 is applied, and (iii) both anti-miR-21 and anti-miR-155 are applied. We see that, by day 60: (i) each mutation increased the total cancer linear mass by approximately 25% compared to the control case; (ii) anti-miR-21 reduced the total tumor linear mass by approximately 40%; and (iii) in combination with anti-miR-155 the reduction was 65%, and the total linear tumor mass did not grow faster than in the untreated control case.
Fig 4. Total linear mass of cancer cells.

The mutations and anti-miRs are the same as in Fig 3, and all parameters are the same as in Fig 3. In the legend, anti-m1 indicates anti-miR-21, and anti-m1&m2 indicates the combination of anti-miR-21 and anti-miR-155. The time is in unit of day, and the mass is in unit of g.
Similar results can be obtained in the case of multiple mutations. Fig 5 illustrates the case of two mutations (the first row), with reduction in invasion front by approximately 40% at day 60 when the cancer is treated with both anti-miR-21 and anti-miR-155 drugs (the second row).
Fig 5. Invasion of cancer cells with density C(x, t).

The parameters are as in Tables 2 and 3, and χ = 3 × 10−2 cm5g−1day−1. (a) Mutations in both EGFR and MAPK, where λE and λM are increased by 1.15-fold and 1.3-fold, respectively; mutations in both EGFR and AKT, where λE and λA are increased by 1.15-fold and 1.4-fold, respectively; mutations in both MAPK and AKT, where λM and λA are increased by 1.3-fold and 1.4-fold, respectively. (b) Using both anti-miR-21 and anti-miR-155, where both λm1 and λm2 are reduced by a factor 2 compared to the panels in (a). The time is in unit of days, and x is in unit of cm.
Cancer invasion depends on the directed migration coefficient χ. In [33] the range of the parameter χ was taken to be (3 × 10−4, 3 × 10−2) cm5g−1day−1. In the simulations of Figs 3–5, we took the largest value χ = 3 × 10−2 cm5g−1day−1 in order to visualize the invasion of the tumor front over a relatively short period of time. It is reasonable to expect that both tumor invasion and total mass will decrease if χ is decreased. This is illustrated in Fig 6 in the case of a tumor with the same EGFR mutation as in Figs 3 and 4. We denote by Rχ the distance traveled by the tumor front by day 60, and by Mχ the total linear mass of the cancer cells by day 60. Fig 6 shows the growth of Rχ and Mχ (at day 60) as χ increases from 3 × 10−4 to 3 × 10−2 cm5g−1day−1: Rχ increases by a factor 8 and Mχ increases by a factor 11 approximately.
Fig 6. The growth of Rχ and Mχ.

(a) The distance that the invasion front traveled by day 60 (Rχ, in unit of cm). (b) The total linear mass of cancer cells at day 60 (Mχ, in unit of g). χ ranges from 3 × 10−4 to 3 × 10−2 cm5g−1day−1. All other parameter values are the same as in the EGFR-mutation case of Fig 3.
We next apply anti-miR-21 and anti-miR-155 drugs to the tumor (by reducing λm1 and λm2 to λm1/2 and λm2/2, as in Figs 3 and 4) and denote the corresponding Rχ and Mχ by and . We represent the efficacy of the anti-miRs drugs by and , that is, by the percentage of reduction in Rχ and Mχ. Fig 7 shows that the efficacy of the drug increases as the directed migration coefficient χ increases. The efficacy ϕR is approximately 33% when χ = 3 × 10−4g−1day−1, and it increases to 40% when χ = 3 × 10−4g−1day−1. The efficacy ϕM is 57% when χ = 3 × 10−4g−1day−1, and it increases to 68% when χ = 3 × 10−4g−1day−1.
Fig 7. The growth of ϕR and ϕM.

(a) The efficacy (ϕR) of anti-miR drugs in reducing the distance traveled by the tumor front at day 60. (b) The efficacy (ϕM) of anti-miR drugs in reducing the total linear mass of cancer cells at day 60. χ varies from 3 × 10−4 to 3 × 10−2 cm5g−1day−1. The anti-miR drugs reduce both λm1 and λm2 by a factor 2. All other parameter values are the same as Fig 6.
From Figs 6 and 7 we conclude that as χ increases, the tumor invasion and total mass increase, while at the same time the efficacy of anti-miRs drug also increases. The same results (not shown here) hold for other mutations as well as for the control case.
Fig 8 shows the relationship between the invasion distance in the control case to the total mass of miR-21 and the total mass of miR-155 after the first 60 days. Since the concentration of miRs in serum are positively correlated to their concentration in lung cancer tissue [10–12], Fig 8 suggests that miR-21 and miR-155 could potentially be used as serum biomarkers for NSCLC, in line with suggestions made in [5, 7].
Fig 8. Growth of invasion distance as a function of the total mass of m1 and total mass of m2.

(a) The invasion distance of cancer cells in the control case as a function of the total mass of miR-21 and miR-205. (b) The invasion distance of cancer cells as a function of the total mass of miR-155. All the parameter values are the same as the control case in Fig 3.
Results for tumor proliferation
The simulations of proliferation of cancer cells are based on the model Eqs (1)–(8), (16)–(18) with boundary Conditions (19) and initial Conditions (20). We increase both λC1 and λC2 by a factor 1.4 compared to the values in tumor invasion model in order to account for the fact that proliferating cells grow faster than migrating cells [37–39]. We also increase the steady state C0 from 0.4 g/cm3 to 0.46 g/cm3 to reflect the fact that invading cancer cells have sparser density than proliferating cells. We take the steady state density of healthy cells, N0, to be 0.14 g/cm3 so that Eq (17) holds. All the other parameter values are the same as in Tables 2 and 3. We take the initial tumor radius to be R(0) = 0.01 cm.
Fig 9 shows the average concentrations of all the variables over a period of 60 days. Most of the concentrations are either monotone increasing or monotone decreasing in time: the cell growth inhibitors TKI, PTEN and Apaf-1 are decreasing, while the cell growth promoters are increasing. The only exception is the average density of E. It is initially increasing since MAPK density is small. But MAPK continues to increase (as T keeps decreasing), and after a few days the inhibition by MAPK (or actually ERK, see Fig 1) forces E to decrease, and it does so until it reaches a steady state.
Fig 9. Average densities/concentrations of all the variables in the model.

λC1 and λC2 are increased by a factor 1.4. θ = 0.6 g/cm3, C0 = 0.46 g/cm3 and N0 = 0.14 g/cm3. All other parameter values are the same as in Tables 2 and 3.
We note that in estimating some of the parameters of the model equations we assumed steady-state of the various variables (cells, proteins, miRs). The steady state of the variables in Fig 9 agree approximately with those steady state values, and this establishes consistency of our assumed steady-state values. In particular, the average density of cancer cells stabilize at 0.4631 g/cm3, and the average density of normal healthy cells stabilize at 0.1337 g/cm3, while C + N remains approximately equal to 0.6 g/cm3 at the entire time.
Fig 10 shows the growth of the tumor radius and volume, and of the total mass of m1 and m2 for the first 60 days. From these profiles we can deduce relations between the total volume of the tumor and the total mass of m1 and of m2, at day 60. These relations are shown in Fig 11.
Fig 10. The growth of tumor radius R(t), tumor volume, total mass of m1 and total mass of m2 for the first 60 days.

λC1 and λC2 are increased by a factor 1.4. θ is taken to be 0.6 g/cm3. All other parameter values are the same as in Tables 2 and 3.
Fig 11. Volume of tumor as a function of the total mass of miR-21 and that of miR-155.

(a) The volume of tumor as a function of the total mass of miR-21 after the first 60 days. (b) The volume of tumor as a function of the total mass of miR-155 after the first 60 days. All the parameter values are the same as Fig 10.
Fig 11 may suggest that miR-21 and miR-155 could be used as biomarkers for determining the volume of NSCLC when this volume is still extremely small; however this suggestion is highly speculative at this time, since other miRs shed from both the cancer cells and immune cells are also circulating in the blood.
We note that the corresponding Fig 8 simulates a different situation, where the tumor is already well established in {x < 0} and its front begins to invade into {x > 0}.
Treatment
It is well known that cancer cells in NSCLC lose sensitivity to anti-tumor drugs, for example to paclitaxel, gefitinib and cisplatin, and that some anti-miRs can restore some of this sensitivity. We use our model to explore the effect of anti-miR combined with paclitaxel, gefitinib and cisplatin.
Paclitaxel drugs (PTX) block progression of mitosis by protecting microtubules against disassembly and preventing chromosomes from achieving metaphase spindle configuration [41]. Researches have observed that paclitaxel-treated cells have defects in mitotic spindle assembly, chromosome segregation and cell division [41]. Experiments in vivo by Yung et al. [42] show that anti-miR-21 reduces tumor volume in NSCLC, and the combination of paclitaxel and anti-miR-21 demonstrated greater ability to reduce cancer cell proliferation than either agent administered alone. The simulations in Fig 12(a) mimic this experiment; the effect of PTX is accounted for by reducing 1.4λCi to 1.3λCi (i = 1, 2), and the effect of anti-miR-21 is accounted for by reducing λm1 to λm1/2. We note however that in our model the cancer is at an earlier stage and its volume is much smaller compared to the volume of 0.8 cm3 in [42].
Fig 12. Tumor growth under different drugs. EGFR mutation is accounted for by increasing λE by factor 3.

(a) Paclitaxel inhibits the division of cells, where λCi are reduced from 1.4λCi to 1.3λCi (i = 1, 2); anti-miR-21 is accounted for by reducing λm1 to λm1/2. (b) Both anti-miR-21 and the gefitinib, a drug that inhibits the EGFR, reduce the growth of tumor; anti-miR-21 is accounted for by reducing λm1 to λm1/2, and EGFR-inhibitor gefitinib is accounted for by decreasing 3λE to 1.5λE. (c) Anti-miR-155 is accounted for by reducing λm2 to λm2/1.2 and cisplatin, a drug that induces cancer cell apoptosis, is accounted for by increasing dC and dD by factor 1.1.
Gefitinib is a drug used in the treatment of NSCLC. It blocks the production of EGF-EGFR and thus obstructs the MAPK and AKT pathways [43]. Tumor cells that are initially sensitive to gefitinib may eventually lose sensitivity due to the emergence of acquired resistance [44]. Alternative mechanisms are currently being explored aimed to overcome the development of gefitinib resistance in the patients of NSCLC [44, 45]. Recent studies [8, 25] show that miR-21 modulates gefitinib sensitivity. In particular, Shen et al. [8] demonstrated in vivo that reduction in miR-21 significantly restored gefitinib sensitivity by up-regulation of PTEN expression and the inactivation of AKT and MAPK pathways. We can use our model to represent the experimental results of Shen et al. [8]. We account for the effect of gefitinib by reducing 3λE, in the case of EGFR mutation, to 1.5λE, and the effect of anti-miR-21 by reducing λm1 to λm1/2. Fig 12(b) shows that anti-miR-21 alone reduces the growth of tumor volume, but in combination with gefitinib the reduction is significantly larger. This is in qualitative agreement with the results in [8], although here again our model considers an early stage of a tumor whereas, in [8], the tumor volume is already 0.5 cm3.
Cisplatin induces cancer cell apoptosis by inhibition of DNA synthesis and repair in cell cycle [46]. The efficacy of cisplatin is initially high, but in the majority of cancer patients it eventually drops due to cisplatin resistance. Many mechanisms of cisplatin resistance have been described, including changes in cellular uptake, drug efflux, increased detoxification of the drug, inhibition of apoptosis, and increased DNA repair [46]. Experiments by Zang et al. [30] show that down-regulation of miR-155 can enhance the sensitivity of lung cancer cells to cisplatin treatment through the induction of DNA damage and apoptosis via the restoration of the mitochondrial apoptotic pathway. The simulation in Fig 12(c) shows that anti-miR-155 alone reduces the tumor volume, but in combination with cisplatin the reduction is significantly higher.
Discussion and conclusion
Worldwide, lung cancer is the leading cause of cancer deaths, and approximately 85% of lung cancer cases are NSCLC [1, 2]. Five years survival rate for NSCLC is significantly higher for those diagnosed at early stage [3]. Unlike mammography for breast cancer or colonoscopy for colon cancer there are no reliable tools for early detection of lung cancer; most lung cancers are first diagnosed on symptoms. Hence, there is increased focus on identifying biomarkers for detection of NSCLC at early stage [5].
A growing body of evidence suggests that exosomal miRs may be used as serum biomarkers for prognosis of malignant tumors [5, 7]. In NSCLC the highest overexpressed miRs are miR-21, miR-205 and miR-155 [6, 10]. Since exosomal miRs concentration in the blood are positively correlated to their concentrations in tissue [10–12], it is important to understand how the concentrations of miRs 21, 205 and 155 in NSCLC tissue are related to the progression of the cancer, both in terms of tumor growth and tumor-front invasion.
In the present paper we developed a mathematical model that relates the role of the above exosomal miRs in tissue to cancer cells proliferation and invasion. MiRs 21 and 205 regulate cell proliferation through MAPK and PI3K-AKT pathways, while miR-155 regulates apoptosis through the Apaf-1-Caspase 9 complex. The mathematical model includes, separately, invasion and proliferation phases of NSCLC. In invasion, the ‘directed migration coefficient’ χ plays a critical role: the tumor front increases as χ increases. In order to visualize this monotonic relation, we used a ‘relatively’ large χ; in vivo this parameter may be much smaller.
In the model of tumor proliferation, what makes the tumor volume grow is the fact that the combined densities of cancer cells (C) and healthy cells (N) is limited, i.e, C + N = constant. Simulations of the proliferating model show qualitative agreement with experimental results in the treatment of NSCLC. In these experiments, miR-21 and miR-155 are used to re-establish sensitivity of cancer cells to specific chemotherapeutic drugs.
Simulations of the model of tumor proliferation establish quantitative relations between the total mass of over-expressed miRs (21, 205, 155) and tumor volume. Because of the positive correlation between miRs in cancer tissue and serum [10–12], the present model may be viewed as a first step toward establishing a combination of miRs 21, 205, 155 and possibly additional miRs as serum biomarkers for early detection of NSCLC. As more experimental and clinical data become available, the model could then be refined by estimating more precisely some of the parameters, by expanding the genetic network of Fig 2, and by precisely relating the concentrations of miRs in serum to miRs in lung tissue.
Parameter values
In the sequal we shall use the following conversion of units: 1Da = 1g/mol, so that
| (21) |
Also, 1 Molar = 1 mol/L = 10−3 mol/cm3. Hence 1nM = 10−12 mol/cm3, and
| (22) |
Steady state concentrations
In some estimating parameters, we use steady state equations; we denote the steady state concentrations of species X by X0.
EGF-EGFR Cancer cells express 2 − 3 × 106 EGFR proteins per cell [47]. We take the average to be 2.5 × 106 EGFR per cell, or fraction of a mole, per cell, where NA = 6.022 × 1023 is Avogadro’s number. The molecular weight of EGFR is 170kDa [47]. By Eq (21), the mass of EGFR in one cell is . Assuming that one cell has a volume of 10−9 cm3, we find that the concentration of EGFR is 7.0573 × 10−4 g/cm3. We assume that the concentration of EGF-EGFR is not limited by the availability of EGF, hence E0 = 7.0573 × 10−4 g/cm3.
MAPK The molar concentration of Ras is 0.4μM [48] and its molecular weight is 21kDa [49]. Hence, by Eq (22), the concentration of Ras is M01 = (0.4 × 103) × 21 × 10−9 g/cm3 = 8.4 × 10−6 g/cm3. The molar concentration of Raf is 0.013μM [48, 50], and its molecular weight is 72kDa [48]. Hence the concentration of Raf is M02 = (0.013 × 103) × 72 × 10−9 g/cm3 = 0.936 × 10−6 g/cm3. The molar concentration of MEK is 1.4μM [48, 49] and its molecular weight is 43kDa [51]. Hence the concentration of MEK is M03 = (1.4 × 103) × 43 × 10−9 g/cm3 = 6.02 × 10−5 g/cm3. The molar concentration of ERK is 0.96μM [48, 49] and its molecular weight is 41kDa [52]. Hence the concentration of MEK is M04 = (0.96 × 103) × 41 × 10−9 g/cm3 = 3.936 × 10−5 g/cm3. All the M0i are of the same order of magnitude. In estimating parameters, we shall use the steady state concentration M0 of the MAPK. We take it to be that of Ras, that is M0 = M01, since steady state of Ras leads to steady state in the MAPK pathway.
PI3K The molar concentration of PI3K is 8nM [53, 54], and its molecular weight is 195kDa [55]. Hence, by Eq (22), the concentration of PI3K is P30 = 8 × 195 × 10−9 g/cm3 = 1.56 × 10−6 g/cm3.
AKT The molar concentration of AKT is 15.1nM [47], and its molecular weight is 62kDa [47]. Hence, by Eq (22), the concentration of AKT is given by A0 = 15.1 × 62 × 10−9 g/cm3 = 9.362 × 10−7 g/cm3.
PTEN The molar concentration of PTEN is 4nM [56], and its molecular weight is 47kDa [56]. Hence, by Eq (22), the concentration of PTEN is P0 = 4 × 47 × 10−9 g/cm3 = 1.88 × 10−7 g/cm3.
TKI TKI inhibits the activation of PI3K, and PTEN inhibits the activation of AKT. We assume that these two inhibitions are proportional, that is T0/E0 = P0/P30. Hence T0 = P0E0/P30 = 8.5050 × 10−5 g/cm3.
Apaf-1 The molar concentration of Apaf-1 ranges from 0.1μM to 0.5μM [57]. We take it to be 0.2μM. The molecular weight of Apaf-1 is 142kDa [57]. Hence, by Eq (22), the concentration of Apaf-1 is (0.2 × 103) × 142 × 10−9 g/cm3 = 2.84 × 10−5 g/cm3. We assume that the concentration of Apaf-1-caspase-9 apoptosome is not limited by the availability of caspase 9, hence the concentration is Ap0 = 2.84 × 10−5 g/cm3.
Exosome In breast cancer, 106 cancer cells release 5 × 108 exosomes in 24 hours [58]. Assuming that the number of cancer cells in lung cancer is 4 × 108 per cm3, and taking the average diameter of exosomes to be 70nm, we estimate the mass density of EC by EC0 = 3.6 × 10−10 g/cm3.
MiR-21, miR-205 and miR-155 For simplicity, we assume steady state of Eqs (10) and (11), so that . We also assume that the cellular concentration of miR-21 is proportional to the exosomal concentration of miR-21, in the sense that , where m10 is the steady state of m1. Similarly, we assume , where m20 is the steady state of m2. In the simulations, we take γ = 10, but the simulations do not change qualitatively if we use different values of γ of the same order of magnitude.
The range of molar concentration of miR-21 in healthy individuals is 0.1-0.326 amol/μL [59, 60], and we take it to be 0.2 amol/μL, that is . The molecular weight of miR-21 is 7kDa [61]. Hence, by Eq (22), the cellular concentration of miR-21 is (0.2 × 10−3) × 7 × 10−9 g/cm3 = 1.4 × 10−12 g/cm3. The concentrations of miR-205 and miR-155 are approximately the same as concentration of miR-21 [10]. Since in our model we combine miR-21 and miR-205, we take , while . Hence, m20 = 1.4 × 10−13g/cm3 and m10 = 2.8 × 10−13 g/cm3.
NSCLC We take, for the invasion model, CM = 0.8 g/cm3 and C0 = 0.4 g/cm3 [62].
Parameter estimation
In the sequal, in expressions of enhancement of the form or inhibition of the form , the parameter K, the ‘half-saturation’ of X, will be taken to be the steady state of X. Thus KMA = KM = M0, KA = A0, KAp = Ap0 and KC = C0, and KME = M0, KTM = KTA = T0, KPA = P0. However, we make an exception in the case of miRs; we assume that the inhibition of protein expressions by miRs is more significant than inhibition by signaling proteins, and take KmT = m10/5, KmP = m10/5 and Km2 = m20/5.
For a species with concentration X and half-life t1/2, the dynamics of its degradation or death is given by
Parameter estimation for Eq (1): The half-life of EGFR ranges from 8 to 24 hours [47]. We take it to be 20 hours, i.e. t1/2 = 5/6 days, so that dE = ln2/t1/2 = 0.8318 days−1. From the steady state of Eq (1) with KME = M0, we deduce that λE = 2dEE0 = 1.1741 × 10−3 day−1 ⋅ g/cm3.
Parameter estimation for Eq (2): The half-life of KRAS is 12 hours [63]; the half-life of Raf is 30 hours [64]; the half-life of MEK is 8 hours [65], and the half-life of ERK is 24 hours [66]. We take the half-life of the combined pathway MAPK to be 24 hours, that is t1/2 = 1 day, then dM = 0.6931 day−1. From the the steady state of Eq (2) with KTM = T0, we find that λM = 2dMM0/E0 = 1.6499 × 10−2 day−1.
Parameter estimation for Eq (3): The half-life of AKT ranges from 12 to 36 hours [67, 68]. We take it to be 24 hours, that is, t1/2 = 1 day, so that dA = 0.6931 day−1. We assume that the activation of AKT pathway by Ras is weaker than the activation by EGF-EGFR, and take λMA = 1/2. From the steady state of Eq (3) with KTA = T0, KPA = P0 and KMA = M0, we find that λA = (16/5) ⋅ dAA0/E0 = 2.9422 × 10−3 day−1.
Parameter estimation for Eq (4): The half-life of the TKI drugs erlotinib, ASD9291 and sunitinib are 36, 50 and 40-60 hours [69]. We take the half-life of TKI to be 48 hours, that is, t1/2 = 2 days. Hence dT = 0.3466 day−1. From the steady state equation of Eq (4) with KmT = m10/5, we find that λT = 28.5dTT0 = 8.4013 × 10−4 day−1 ⋅ g/cm3.
Parameter estimation for Eq (5): The half-life of PTEN is 45 minutes [70, 71], that is t1/2 = 0.03125 days, so that dP = 22.1807 day−1. From the steady state of Eq (5) with KmP = m10/5, we find that λP = 56dPP0 = 2.3352 × 10−4 day−1 ⋅ g/cm−3.
Parameter estimation for Eq (6): The half-life of Apaf-1 is 1.81h [72]; the half-life of caspase-9 is 6.6h [73]. We assume that Apaf-1-caspase-9 apoptosome is as stable as caspase-9, and take the half-life of the apoptosome to be 6h, that is t1/2 = 0.25 days. Then dAp = 2.7726 day−1. From the steady state of Eq (6) with Km2 = m20/5, we get λAp = 56dApAp0 = 4.4095 × 10−3 day−1 ⋅ g/cm3.
Parameter estimation for Eq (7): The rate of breakdown of exosomes upon contact with cancer cells is unknown. We take this rate to be dEc = 21.8 day−1. From the steady state of Eq (7) with KC = C0, we get λEc = dEcEC0/(2C0) = 9.81 × 10−9 day−1.
Parameter estimation for Eq (8): The half-life of miRs is greater than 24 hours [74]; we take half-life of miR-21 to be 30 hours, i.e. t1/2 = 1.25 day. Hence dm1 = 0.5545 day−1. From the steady state of Eq (8) with KC = C0, we get λm1 = 2dm1m10/EC0 = 0.8626 × 10−3 day−1.
Parameter estimation for Eq (9): We take the half-life of miR-155 to be 30 hours [74], and then dm2 = 0.5545 day−1. From the steady state of Eq (9) with KC = C0, we obtain λm2 = 2dm2m20/EC0 = 0.4313 × 10−3 day−1.
Parameter estimation for Eq (12): The most common mutations in NSCLC occur in tumor suppressors TP53 and ALK, and in oncogenes PTEN, EGFR, KRAS, LKB1 and BRAF, and mutations seem to occur more frequently in MAPK (KRAS, BRAF) than in PI3K-AKT [21, 75, 76]. We accordingly assume that the proliferation rate of cancer cells through the MAPK pathway is higher than the proliferation rate through the AKT pathway, and take λC1 = 2λC2. We also assume that in steady state
and take λC = 0.46 day−1 [77]. Hence λC1 = (4/3)λC = 0.6133 day−1 and λC2 = (2/3)λC = 0.3067 day−1.
From the steady state equation of Eq (12) with KM = M0, KA = A0, KAp = Ap0 and C0 = CM/2, we get
We assume that apoptosis rate through intrinsic apoptosis pathway (Apaf-1/Caspace-9) is higher than apoptosis through the extrinsic signaling pathway [78], and take dD to be larger than dC, so that dD = 0.9λC and 2dC = 0.1λC. Hence dD = 0.414 and dC = 0.023.
Diffusion coefficients: We take DC = 8.64 × 10−7 cm2day−1 [77]. Diffusion of a sphere is inversely proportional to its diameter. We assume that the average diameter of exosome and cells are 70nm and 10μm respectively. Then we get . By [79], we have the relation , where DV and MV are the diffusion coefficient and molecular weight of vascular endothelial growth factor (VEGF), respectively, and DV = 8.64 × 10−2 [77], and MV = 24kDa [80]. The molecular weight of miR-21 is Mm1 = 7 kDa, hence Dm1 = 0.13028 cm2day−1, and similarly Dm2 = 0.13028 cm2day−1. The directed migration coefficient χ is taken to be in the range 3 × 10−4 − 3 × 10−2 cm5g−1day−1 [33]. In cell invasion, χ should be much larger than in cell growth, so we take χ = 2 × 10−2 cm5g−1day−1 for the model of tumor invasion.
Parameter estimation for Eq (16): Cancer cells can survive in hostile environment better than normal cells, so the apoptosis rate dN should be somewhat larger than dC; we take dN = 1.1dC = 0.0253 day−1 and λN = 0.8λC = 0.368 day−1. Since cancer cells replication is less susceptible to damage, dDN should be larger than dD; we take dDN = 1.1dD = 0.4554 day−1. We choose ε = 0.1, as in [40]. We assume that C0 in the proliferation phase to be somewhat larger than the average density 0.4 g/cm3 in the invasion phase; we take C0 = 0.46 g/cm3 and N0 = 0.14 g/cm3.
Sensitivity analysis
We performed sensitivity analysis on some of the production parameters of the system Eqs (1)–(9), (15)–(18); we also included the important parameter dEc which was only fitted. Following the method of [81] we performed Latin hypercube sampling and generated 1000 samples to calculate the partial rank correlation coefficients (PRCC) and the p-values with respect to the tumor radius at day 60. We have taken the range of each parameter from 1/2 to twice its value in Table 2. The results are shown in Fig 13.
Fig 13. Statistically significant PRCC values (p-value < 0.01) for R(t) at day 60.

We see that the production rates that increase proliferation through EGF-EGFR → MAPK and EGF-EGFR → AKT pathways, namely, (λE, λM) and (λE, λM, λMA) are positively correlated to tumor radius. On the other hand the production rates of cell-replication inhibitors, λT and λP, and the production rate of apoptosis-promotor apoptosome, λAp, are negatively correlated. Since miR-21 blocks the inhibitors T and P, so if λm1EC grows the tumor volume will increase. Hence λm1 is positively correlated and dEc is negatively correlated.
Computational method
We employ moving mesh method to numerically solve the free boundary problem for the tumor proliferation model. To illustrate this method, we take Eq (16) as example and rewrite it as the following form:
| (23) |
where F represents the term in the right hand side of Eq (16). Let and denote numerical approximations of i-th grid point and , respectively, where τ is the size of time-step. The discretization of Eq (23) is derived by the fully implicit finite difference scheme:
| (24) |
where , , , and . The mesh moves by , where is solved by the velocity equation.
Acknowledgments
This work is supported by the Mathematical Biosciences Institute and the National Science Foundation (Grant DMS 0931642), the National Natural Science Foundation of China (Grant No. 11501568), and the Fundamental Research Funds for the Central Universities and the Research Funds of Renmin University of China.
Data Availability
All relevant data are within the paper.
Funding Statement
AF is supported by the Mathematical Biosciences Institute and the National Science Foundation (Grant DMS 0931642). XL is supported by the National Natural Science Foundation of China (Grant No. 11501568), the Fundamental Research Funds for the Central Universities and the Research Funds of Renmin University of China, and the Mathematical Biosciences Institute and the National Science Foundation (Grant DMS 0931642).
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer Statistics 2015. CA CANCER J CLIN. 2015;65:5–29. 10.3322/caac.21254 [DOI] [PubMed] [Google Scholar]
- 2. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–86. 10.1002/ijc.29210 [DOI] [PubMed] [Google Scholar]
- 3. Henschke CI, McCauley DI, Yankelevitz DF, Naidich DP, McGuinness G, Miettinen OS, et al. Early Lung Cancer Action Project: overall design and findings from baseline screening. Lancet. 1999;354(9173):99–105. 10.1016/S0140-6736(99)06093-6 [DOI] [PubMed] [Google Scholar]
- 4. Ali A, Goffin JR, Arnold A, Ellis PM. Survival of patients with non-small-cell lung cancer after a diagnosis of brain metastases. Curr Oncol. 2013;20(4):300–6. 10.3747/co.20.1481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. An T, Qin S, Xu Y, Tang Y, Huang Y, Situ B, et al. Exosomes serve as tumour markers for personalized diagnostics owing to their important role in cancer metastasis. J Extracell Vesicles. 2015;4:27522 10.3402/jev.v4.27522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Frydrychowicz M, Kolecka-Bednarczyk A, Madejczyk M, Yasar S, Dworacki G. Exosomes-structure, biogenesis and biological role in non-small-cell lung cancer. Scand J Immunol. 2015;81(1):2–10. 10.1111/sji.12247 [DOI] [PubMed] [Google Scholar]
- 7. Properzi F, Logozzi M, Fais S. Exosomes: the future of biomarkers in medicine. Biomark Med. 2013;7(5):769–778. 10.2217/bmm.13.63 [DOI] [PubMed] [Google Scholar]
- 8. Shen H, Zhu F, Liu J, Xu T, Pei D, Wang R, et al. Alteration in Mir-21/PTEN expression modulates gefitinib resistance in non-small cell lung cancer. PLoS One. 2014;9(7):e103305 10.1371/journal.pone.0103305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lei L, Huang Y, Gong W. MiR-205 promotes the growth, metastasis and chemoresistance of NSCLC cells by targeting PTEN. Oncol Rep. 2013;30(6):2897–902. [DOI] [PubMed] [Google Scholar]
- 10. Rabinowits G, Gerçel-Taylor C, Day J, Taylor D, Kloecker G. Exosomal MicroRNA: a diagnostic marker for lung cancer. Clin Lung Cancer. 2009;10(1):42–46. 10.3816/CLC.2009.n.006 [DOI] [PubMed] [Google Scholar]
- 11. Zhao W, Zhao JJ, Hang L, Xu QF, Zhao YM, Shi XY, et al. Serum miR-21 level: a potential diagnostic and prognostic biomarker for non-small cell lung cancer. Int J Clin Exp Med. 2015;8(9):14759–63. [PMC free article] [PubMed] [Google Scholar]
- 12. Liu R, Liao J, Yang M, Shi Y, Peng Y, Wang Y, et al. Circulating miR-155 expression in plasma: a potential biomarker for early diagnosis of esophageal cancer in humans. J Toxicol Environ Health A. 2012;75(18):1154–62. 10.1080/15287394.2012.699856 [DOI] [PubMed] [Google Scholar]
- 13. Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. 10.1038/sj.onc.1210422 [DOI] [PubMed] [Google Scholar]
- 14. Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279–3290. 10.1038/sj.onc.1210421 [DOI] [PubMed] [Google Scholar]
- 15. Yip PY. Phosphatidylinositol 3-kinase-AKT-mammalian target of rapamycin (PI3K-Akt-mTOR) signaling pathway in non-small cell lung cancer. Transl Lung Cancer Res. 2015;4(2):165–176. 10.3978/j.issn.2218-6751.2015.01.04 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fumarola C, Bonelli MA, Petronini PG, Alfieri RR. Targeting PI3K/AKT/mTOR pathway in non small cell lung cancer. Biochemical Phamacology. 2014;90:197–207. 10.1016/j.bcp.2014.05.011 [DOI] [PubMed] [Google Scholar]
- 17. Papadimitrakopoulou V, Adjei A. The Akt/mTOR and mitogen-activated protein kinase pathways in lung cancer therapy. J Thoracic Oncology. 2006;1(7):749–751. 10.1016/S1556-0864(15)30399-3 [DOI] [PubMed] [Google Scholar]
- 18. Normanno N, De Luca A, Binanco C, Strizziand L, Mancino M, et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2–16. 10.1016/j.gene.2005.10.018 [DOI] [PubMed] [Google Scholar]
- 19. Marinov M, Fischer B, Arcaro A. Targeting mTOR signaling in lung cancer. Critical Review in Oncology/Hematology. 2007;63:172–182. 10.1016/j.critrevonc.2007.04.002 [DOI] [PubMed] [Google Scholar]
- 20. Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–430. 10.1038/nature04869 [DOI] [PubMed] [Google Scholar]
- 21. Siegelin MD, Borczuk AC. Epidermal growth factor receptor mutations in lung adenocarcinoma. Laboratory Investigation. 2014;94:129–137. 10.1038/labinvest.2013.147 [DOI] [PubMed] [Google Scholar]
- 22. Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011;36(6):320–328. 10.1016/j.tibs.2011.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kang HW, Crawford M, Fabbri M, Nuovo G, Garofalo M. A mathematical model for MicroRNA in lung cancer. PLoS ONE. 2013;8(1):e53663 10.1371/journal.pone.0053663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sgambato A, Casaluce F, Maione P, Rossi A, Rossi E, Napolitano A, et al. The role of EGFR tyrosine kinase inhibitors in the first-line treatment of advanced non small cell lung cancer patients harboring EGFR mutation. Curr Med Chem. 2012;19(20):3337–52. [DOI] [PubMed] [Google Scholar]
- 25. Li B, Ren S, Li X, Wang Y, Garfield D, Zhou S, et al. MiR-21 overexpression is associated with acquired ressistance of EGFR-TKI in non-small cell lung cancer. Lung cancer. 2014;83:146–153. 10.1016/j.lungcan.2013.11.003 [DOI] [PubMed] [Google Scholar]
- 26. Soengas MS, Alarcon RM, Yoshida H, Giaccia AJ, Hakem R. Apaf-1 and caspase-9 in p53-dependent apoptosis and tumor inhibition. Science. 1999;284:156–159. 10.1126/science.284.5411.156 [DOI] [PubMed] [Google Scholar]
- 27. Bratton SB, Salvesen GS. Regulation of the Apaf-1-caspase-9 apoptosome. J Cell Science. 2010;123:3209–314. 10.1242/jcs.073643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang JG, Wang JJ, Zhao F, Liu Q, Jiang K, Yang GH. MicroRNA-21(miR-21) represses tumor supressor PTEN and promotes growth and invasion in non-small cell lung cancer (NSCLC). Clin Chim Acta. 2010;411:846–852. 10.1016/j.cca.2010.02.074 [DOI] [PubMed] [Google Scholar]
- 29. Zhang G, Hou X, Li Y, Zhao M. MiR-205 inhibits cell apoptosis by targeting phosphatase and tensin homolog deleted on chromosome ten in endometrial cancer ishikawa cells. MBC Cancer. 2014;14:440 10.1186/1471-2407-14-440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zang YS, Zhong YS, Fang Z, Li B, An J. MiR-155 inhibits the sensitivity of lung cancer cells to cisplatin via negative regulation of Apaf-1 expression. Cancer Gene Therapy. 2012;19:773–778. 10.1038/cgt.2012.60 [DOI] [PubMed] [Google Scholar]
- 31. Hatley ME, Patrick DM, Garcia MR, Richardson JA, Bassel-Duby R, van Rooij E, et al. Modulation of K-Ras-dependent lung tumorigenesis by MicroRNA-21. Cancer Cell. 2010;18(3):282–93. 10.1016/j.ccr.2010.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang Y, Meng H, Peng Q, Yang X, Gan R, et al. Downregulation of microRNA-21 expression restrains non-small cell lung cancer cell proliferation and migration through upregulation of programmed cell death 4. Cancer Gene Ther. 2015;22(1):23–9. 10.1038/cgt.2014.66 [DOI] [PubMed] [Google Scholar]
- 33. Kim Y, Roh S, Lawler S, Friedman A. MiR451 and AMPK mutual antagonism in glioma cell migration and proliferation: a mathematical model. PLoS One. 2011;6(12):e28293 10.1371/journal.pone.0028293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Salamon R, Backer J. PIP3: Tool of Choice for the Class I PI 3-kinases. Bioessays. 2013;35(7):602–11. 10.1002/bies.201200176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Quaranta V, Rejniak KA, Gerlee P, Anderson AR. Invasion emerges from cancer cell adaptation to competitive microenvironments: quantitative predictions from multiscale mathematical models. Semin Cancer Biol. 2008;18(5):338–48. 10.1016/j.semcancer.2008.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kam Y, Rejniak KA, Anderson AR. Cellular modeling of cancer invasion: integration of in silico and in vitro approaches. J Cell Physiol. 2012;227(2):431–8. 10.1002/jcp.22766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. aand Qian Xie CFG, Su YL, Koeman J, Khoo SK, Gustafson M, et al. Proliferation and invasion: Plasticity in tumor cells. PNAS. 2005;102(30):10528–10533. 10.1073/pnas.0504367102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Matus DQ, Lohmer LL, Kelley LC, Schindler AJ, Kohrman AQ, et al. Invasive cell fate requires G1 cell-cycle arrest and Histone Deacetylase-mediated changes in gene expression. Developmental Cell. 2015;35(2):162–174. 10.1016/j.devcel.2015.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gil-Henn H, Patsialou A, Wang Y, Warren MS, Condeelis JS, Koleske AJ. Arg/Abl2 promotes invasion and attenuates proliferation of breast cancer in vivo. Oncogene. 2013;32:2622–2630. 10.1038/onc.2012.284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jain HV, Clinton SK, Bhinder A, Friedman A. Mathematical modeling of prostate cancer progression in response to androgen ablation therapy. Proc Natl Acad Sci USA. 2011;108(49):19701–6. 10.1073/pnas.1115750108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Weaver B. How Taxol/paclitaxel kills cancer cells. Mol Biol Cell. 2014;25(18):2677–81. 10.1091/mbc.E14-04-0916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yung BC, Li J, Zhang M, Cheng X, Li H, Yung EM, et al. Lipid Nanoparticles Composed of Quaternary Amine-Tertiary Amine Cationic Lipid Combination (QTsome) for Therapeutic Delivery of AntimiR-21 for Lung Cancer. Mol Pharmaceutics. 2016;13(2):653–662. 10.1021/acs.molpharmaceut.5b00878 [DOI] [PubMed] [Google Scholar]
- 43. Tamura K, Fukuoka M. Gefitinib in non-small cell lung cancer. Expert Opin Pharmacother. 2005;6(6):985–93. 10.1517/14656566.6.6.985 [DOI] [PubMed] [Google Scholar]
- 44. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7(3):169–81. 10.1038/nrc2088 [DOI] [PubMed] [Google Scholar]
- 45. Jo U, Park KH, Whang YM, Sung JS, Won NH, Park JK, et al. EGFR endocytosis is a novel therapeutic target in lung cancer with wild-type EGFR. Oncotarget. 2014;5(5):1265–78. 10.18632/oncotarget.1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Florea AM, Busselberg D. Cisplatin as an anti-tumor Drug: Cellular mechanisms of activity, drug resistance and induced side effects. Cancers. 2011;3(1):1351–1371. 10.3390/cancers3011351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sorkin A, Duex JE. Quantitative analysis of endocytosis and turnover of epidermal growth factor (EGF) and EGF receptor. Curr Protoc Cell Biol. 2010;15(14). 10.1002/0471143030.cb1514s46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fujioka A, Terai K, Itoh RE, Aoki K, Nakamura T, et al. Dynamics of the Ras/ERK MAPK cascade as monitored by fluorescent probes. J Biol Chem. 2006;281(13):8917–8926. 10.1074/jbc.M509344200 [DOI] [PubMed] [Google Scholar]
- 49. Roskoski RJ. ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacological Research. 2012;66:105–143. 10.1016/j.phrs.2012.04.005 [DOI] [PubMed] [Google Scholar]
- 50. Kaur H, Park CS, Lewis JM, Haugh JM. Quantitative model of Ras-phosphoinositide 3-kinase signalling cross-talk based in co-operative molecular assembly. Biochem J. 2006;393:235–243. 10.1042/BJ20051022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Callans L, Naama H, Khandelwal M, Plotkin R, Jardines L. Raf-1 Protein Expression in Human Breast Cancer Cells. Annals of Surgical Oncology. 1995;2(1):38–42. 10.1007/BF02303700 [DOI] [PubMed] [Google Scholar]
- 52. Akinleye A, Furqan M, Mukhi N, Ravella P, Liu D. MEK and the inhibitors: from bench to bedside. J Hematol Oncol. 2013;6:27 10.1186/1756-8722-6-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Legewie S, Herzel H, Westerhoff H, Bluthgen N. Recurrent design patterns in the feedback regulation of the mammalian signalling network. Mol Syst Biol. 2008;17:190 10.1038/msb.2008.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Leander R, Dai S, Schlesinger LS, Friedman A. A mathematical model of CR3/TLR2 crosstalk in the context of francisella tularensis infection. PLOS Compt Biol. 2012;8(11):e1002757 10.1371/journal.pcbi.1002757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cantrell DA. Phosphoinositide 3-kinase signalling pathways. J Cell Sci. 2001;114(8):1439–45. [DOI] [PubMed] [Google Scholar]
- 56. Meyer R, D’Alessandro LA, Kar S, Kramer B, She B, Kaschek D, et al. Heterogeneous kinetics of AKT signaling in individual cells are accounted for by variable protein concentration. Front Physiol. 2012;3:1–14. 10.3389/fphys.2012.00451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Reubold T, Wohlgemuth S, Eschenburg S. A new model for the transition of APAF-1 from inactive monomer to caspase-activating apoptosome. J Biol Chem. 2009;284(47):32717–24. 10.1074/jbc.M109.014027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Riches A, Campbell E, Borger E, Powis S. Regulation of exosome release from mammary epithelial and breast cancer cells—a new regulatory pathway. Eur J Cancer. 2014;50(5):1025–34. 10.1016/j.ejca.2013.12.019 [DOI] [PubMed] [Google Scholar]
- 59. Komatsu S, Ichikawa D, Takeshita H, Tsujiura M, Morimura R, Nagata H, et al. Circulating microRNAs in plasma of patients with oesophageal squamous cell carcinoma. Br J Cancer. 2011;105(1):104–111. 10.1038/bjc.2011.198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Komatsu S, Ichikawa D, Tsujiura M, Konishi H, Takeshita H, Nagata H, et al. Prognostic impact of circulating miR-21 in the plasma of patients with gastric carcinoma. Anticancer Res. 2013;33(1):271–6. [PubMed] [Google Scholar]
- 61. Bader AG, Brown D, Stoudemire J, Lammers P. Developing therapeutic microRNAs for cancer. Gene Therapy. 2011;18:1121–1126. 10.1038/gt.2011.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Szomolay B, Eubank T, Roberts RD, Marsh CB, Friedman A. Modeling the inhibition of breast cancer growth by GM-CSF. J Theor Biol. 2012;303:141–151. 10.1016/j.jtbi.2012.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shukla S, Allam US, Ahsan A, Chen G, Krishnamurthy PM, Marsh K, et al. KRAS protein stability is regulated through SMURF2: UBCH5 complex-mediated β-TrCP1 degradation. Neoplasia. 2014;16:115–128. 10.1593/neo.14184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schumacher C, Cioffi CL, Sharif H, Haston W, Monia BP, Wennogle L, et al. Exposure of human vascular smooth muscle cells to Raf-1 antisense oligodeoxynucleotides: cellular responses and pharmacodynamic implications. Mol Pharmacol. 1998;53(1):97–104. [DOI] [PubMed] [Google Scholar]
- 65. Wang PY, Rao JN, Zou T, Liu L, Xiao L, Yu TX, et al. Posttranscriptional regulation of MEK-1 by polyamines through the RNA-binding protein HuR modulating intestinal epithelial apoptosis. Biochem J. 2010;426(3):293–306. 10.1042/BJ20091459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Satoh M, Parent AD, Zhang JH. Inhibitory Effect With Antisense Mitogen-Activated Protein Kinase Oligodeoxynucleotide Against Cerebral Vasospasm in Rats. Stroke. 2002;33:775–781. 10.1161/hs0302.103734 [DOI] [PubMed] [Google Scholar]
- 67. Lee JH, Shin SH, Kang S, Lee YS, Bae S. A novel activation-induced suicidal degradation mechanism for Akt by selenium. Int J Mol Med. 2008;21(1):91–7. [PubMed] [Google Scholar]
- 68. Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. Akt Forms an Intracellular Complex with Heat Shock Protein 90 (Hsp90) and Cdc37 and Is Destabilized by Inhibitors of Hsp90 Function. J Biol Chem. 2002;277(42):39858–66. 10.1074/jbc.M206322200 [DOI] [PubMed] [Google Scholar]
- 69. Peters S, Zimmermann S, Adjei AA. Oral epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of non-small cell lung cancer: Comparative pharmacokinetics and drug-drug interactions. Cancer Treatment Reviews. 2014;40:917–926. 10.1016/j.ctrv.2014.06.010 [DOI] [PubMed] [Google Scholar]
- 70. Faratian D, Goltsov A, Lebedeva G, Sorokin A, Moodie S, Mullen P, et al. Systems biology reveals new strategies for personalizing cancer medicine and confirms the role of PTEN in resistance to trastuzumab. Cancer Res. 2009;69(16):6713–20. 10.1158/0008-5472.CAN-09-0777 [DOI] [PubMed] [Google Scholar]
- 71. Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol. 2000;20(14):5010–8. 10.1128/MCB.20.14.5010-5018.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Vreeland AC, Yu S, Levi L, de Barros Rossetto D, Noya N. Transcript Stabilization by the RNA-Binding Protein HuR Is Regulated by Cellular Retinoic Acid-Binding Protein 2. Molecular and Cellular Biology. 2014;34(12):2135–2146. 10.1128/MCB.00281-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. von Roretz C, Lian XJ, Macri AM, Punjani N, Clair E, Drouin O, et al. Apoptotic-induced cleavage shifts HuR from being a promoter of survival to an activator of caspase-mediated apoptosis. Cell Death Differ. 2013;20:154–168. 10.1038/cdd.2012.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McDonald D. Understanding miRNA turnover: a study of miRNA lalf-life. Broad Insitute. 2010;.
- 75. Dearden S, Stevens J, Wu YL, Blowers D. Mutation incidence and coincidence in non small-cell lung cancer: meta-analyses by ethnicity and histology (mutMap). Annals of Oncology. 2013;24:2371–2376. 10.1093/annonc/mdt205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lung Cancer Mutation Panel (EGFR, KRAS, ALK). Quest Diagnosis; Available from: http://www.questdiagnostics.com/testcenter/TestDetail.action?ntc=91216
- 77. Liao KL, Bai XF, Friedman A. Mathematical modeling of interleukin-27 induction of anti-tumor T cells response. PLoS ONE. 2014;9(3):e91844 10.1371/journal.pone.0091844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mcllwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2013;5(4):a008656 10.1101/cshperspect.a008656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Young ME. Estimation of diffusion coefficients of proteins. Biotechnology and Bioengineering. 1980;XXII:947–955. 10.1002/bit.260220504 [DOI] [Google Scholar]
- 80. Shui YB, Wang X, Hu JS, Wang SP, Garcia CM, Potts JD, et al. Vascular endothelial growth factor expression and signaling in the lens. Invest Ophthalmol Vis Sci. 2003;44(9):3911–9. 10.1167/iovs.02-1226 [DOI] [PubMed] [Google Scholar]
- 81. Marino S, Hogue IB, Ray CJ, Kirschner DE. A methodology for performing global uncertainty and sensitivity analysis in systems biology. J Theor Biol. 2008;254(1):178–96. 10.1016/j.jtbi.2008.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hao W, Crouser ED, Friedman A. Mathematical model of sarcoidosis. Proc Natl Acad Sci USA. 2014;111(45):16065–70. 10.1073/pnas.1417789111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hao W, Friedman A. The LDL-HDL Profile Determines the Risk of Atherosclerosis: A Mathematical Model. PLoS ONE. 2014;9(3):e90497 10.1371/journal.pone.0090497 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.