

Abstract
Transplantation of human pluripotent stem cells (hPSC) differentiated into insulin-producing β cells is a regenerative medicine approach being investigated for diabetes cell replacement therapy. This report presents a multifaceted transplantation strategy that combines differentiation into stem cell-derived β (SC-β) cells with 3D printing. By modulating the parameters of a low-cost 3D printer, we created a macroporous device composed of polylactic acid (PLA) that houses SC-β cell clusters within a degradable fibrin gel. Using finite element modeling of cellular oxygen diffusion-consumption and an in vitro culture system that allows for culture of devices at physiological oxygen levels, we identified cluster sizes that avoid severe hypoxia within 3D-printed devices and developed a microwell-based technique for resizing clusters within this range. Upon transplantation into mice, SC-β cell-embedded 3D-printed devices function for 12 weeks, are retrievable, and maintain structural integrity. Here, we demonstrate a novel 3D-printing approach that advances the use of differentiated hPSC for regenerative medicine applications and serves as a platform for future transplantation strategies.
1. Introduction
Diabetes mellitus is a disease that affects hundreds of millions of people worldwide [1] and is characterized by loss of blood glucose control. This typically occurs through either autoimmune-mediated destruction of insulin-producing β cells found in islets of Langerhans within the pancreas or insulin resistance in peripheral tissue that leads to β cell failure. Common treatments for diabetes include insulin injections or drugs that either increase insulin sensitivity or increase insulin secretion from remaining β cells, but complications due to imprecise glucose control persist and are costly [2].
Replacement of insulin-producing β cells is a promising approach for controlling diabetes in patients. Transplantation of islets from cadaveric donors that contain β cells have been performed with patients via intrahepatic infusion and demonstrated improved blood glucose control over several years [3–6]. Most recently, differentiation of hPSC has been used to generate SC-β cells in vitro from both human embryonic stem cells (hESC) [7] and Type 1 diabetic patient-derived human induced pluripotent stem cells (hiPSC) [8]. These cells can be produced in almost unlimited quantities by suspension culture in spinner flasks, overcoming limitations in β cell supply from cadaveric islets, and have markedly similar characteristics compared to primary β cells, including gene expression and the ability to respond to glucose by secreting insulin both in vitro and in vivo. Importantly, transplanted SC-β cells control blood glucose in mouse models of diabetes [7–9].
Transplantation of SC-β cells would benefit from a device that is retrievable and macroporous because of the large number of cells necessary to treat a diabetic patient [3, 10]. There are currently no FDA-approved treatments using hPSC, and the safety of any such hPSC-based product needs to be assured, which can be achieved with removal of the transplanted cells. Transplantation of β cells benefits from the ability of the β cell to survive and function when transplanted in non-pancreatic locations. Most current clinical approaches with cadaveric islets rely on infusion into the liver, rendering them irretrievable [3–6]. Other transplantation sites used in research, such as the kidney capsule [7, 8, 11] or fat pad [12], are not viable for clinical transplantation. Large spaces, such as subcutaneous [13, 14], intraperitoneal [9, 15], or in the omentum [16], can potentially hold a sufficiently large cell-embedded device to convey a positive clinical outcome while also allowing for cell retrieval. Furthermore, much of the prior research has been focused on cellular encapsulation, which prevents vascularization of the transplanted graft, that causes cellular hypoxia, as oxygen is only delivered to the cells through diffusion, leading to either necrosis or greatly reduced function of transplanted islets [17, 18]. A macroporous device would allow vascularization of the graft, improving survival and function and reducing delays in glucose sensing, and can be loaded with fibrin, which is biocompatible and degradable, to further promote β cell survival and function along with host integration and vascularization [19–23].
3D printing affords us the ability to rapidly prototype several device designs. Recently, the cost of consumer-grade 3D printers has lowered to the point that they are affordable for most research laboratories. Devices with precise three-dimensional spatial configurations can be manufactured from low cost, biocompatible, and very slowly degrading materials, such as PLA [24]. PLA has been utilized in medical applications such as drug delivery systems and biofabrication due to its biocompatibility and high retention of structural integrity. It has been extensively studied in the past and is FDA approved for various bioengineering applications. In addition, the low viscosity of PLA allows for it to be compatible with a broader range of 3D printers, including those that have limited extruder nozzle pressure.
Here we present multifaceted strategy to produce a low-cost 3D-printed device for the subcutaneous transplantation of SC-β cells. We fabricated the macroporous device with PLA using 3D printing and distributed SC-β cells throughout the pores, secured in place with a fibrin gel. We determined the cluster size range that would prevent severe transient hypoxia with a finite element model of oxygen diffusion-consumption, developed a microwell-based approach for resizing SC-β cell clusters to be within this range, and demonstrated the ability of this approach to prevent hypoxia-induced death with in vitro culture under physiological oxygen. Upon transplantation into mice, SC-β cell-embedded 3D-printed devices are functional, secreting insulin in response to glucose for 12 weeks. Devices maintained structural integrity during this time and were retrievable from mice. This 3D-printing approach is a novel platform for development of a cell replacement therapy for diabetes.
2. Methods
2.1. Modeling and 3D printing of the device
The computer-aided drafting (CAD) design of the scaffold was generated by utilizing the Autodesk Inventor software. The CAD design was then converted to a G-code format using the KISSlicer softare. The following specifications of the 3D printer were selected for the generation of the device: 0.1 mm layer thickness, 0.2 mm extrusion width, 210 °C – 220 °C extruder temperature, 60 °C printer bed temperature, 10 mm/s loop speed, 37.5 mm/s solid speed, 52.5 mm/s sparse speed, and the addition of a prime pillar. 1.75mm natural PLA (Makergear; 1.75mm PLA-spooled, natural) was used as the 3D printing filament and the single-extruder 3D printer Makergear M2 (Makergear; M2 3D Printer - Assembled) was utilized to manufacture the device.
2.2. Stem cell culture and differentiation of SC-β cells
The Harvard University Embryonic Stem cell 8 line (HUES8) was utilized as the undifferentiated human stem cell line for this experiment [7]. The HUES8 cells were cultured using mTeSR1 (StemCell Technologies; 05850) in suspension culture inside 500mL spinner flasks (Corning; 89089-814) on a 9-Position stir plate (Chemglass) at 70 rpm. Each passage occurred every three days with fresh media change with mTeSR1 48 hr after the split. During the passage, the HUES8 clusters were single-cell dispersed using 6 mL PBS (Fisher; MT21040CV) and 6 mL Accutase (Innovative Cell Technologies; AT-104-500) for 6 min and were seeded at 6x105 cells/ml in mTeSR1 with 10 mM Y27632 (Abam; ab120129)
After 72 hr of culture in mTESR1, the following steps were followed to differentiate the HUES8 clusters into SC-β cell clusters [8]. Media preparation for the differentiation protocol is as follows. S1 media was prepared with MCDB131 (Cellgro; 15-100-CV) as the base media with 8 mM D-(+)-Glucose (Sigma; G7528), 2.46 g/L NaHCO3, (Sigma S3817), 2% FAF-BSA (Proliant; 68700), 1:50,000 ITS-X (Invitrogen; 51500056), 2 mM Glutamax (Invitrogen; 35050-079), 0.25 mM Vitamin C (Sigma;A4544), and 1% Pen/Strep (Corning; 30-002-Cl). S2 media was prepared with MCDB131,8 mM D-(+)-Glucose, 1.23 g/L NaHCO3, 2% FAF-BSA, 1:50,000 ITS-X, 2 mM Glutamax, 0.25 mM Vitamin C, and 1% Pen/Strep. S3 media was prepared with MCDB131,8 mM D-(+)-Glucose, 1.23 g/L NaHCO3, 2% FAF-BSA, 1:200 ITS-X, 2 mM Glutamax, 0.25 mM Vitamin C, and 1% Pen/Strep. S5 media was prepared with MCDB131,20 mM D-(+)-Glucose, 1.754 g/L NaHCO3, 2% FAF-BSA, 1:200 ITS-X, 2 mM Glutamax, 0.25 mM Vitamin C, 1% Pen/Strep, and 10 μg/ml Heparin (Sigma; H3149). S6 media was prepared with CMRL 1066 Supplemented (CMRLS) (Mediatech 99-603-CV) with 10% FBS (HyClone; 16777), and 1% Pen/Strep. All prepared media were sterile filtered using top-bottom 0.22 μm filtration units.
For days 1 and 2 of the differentiation, S1 media with 100 ng/ml Activin A (R&D Systems; 338-AC) and 3 PM Chir99021 (only on day 1) (Abcam; ab120890) were added. On days 4 and 5, S2 media with 50 ng/ml KGF (Peprotech; AF-100-19) was added. On day 7, S3 media with 10 PM Y27632, 50 ng/ml KGF, 0.25 μM Sant1 (Sigma; S4572), 2 μM Retinoic acid (RA) (Sigma; R2625), 200 nM LDN193189 (Sigma; SML0559), and 500 nM PdBU (EMD Millipore; 524390) was added. On days 8, 9, and 11, S3 media with 10 PM Y27632, 5 ng/ml Activin A, 50 ng/ml KGF, 0.25 μM Sant1, and 100 nM RA was added. On days 13, 14, 16, and 18, S5 media with 0.25 μM Sant1, 100 nM RA, 1 μM XXI (EMD Millipore; 565790), 10 μM Alk5i II (Enzo Life Sciences; ALX-270-445), 1 μM L-3,3’,5-Triiodothyronine (T3) (EMD Millipore;64245), 20 ng/ml Betacellulin (R&D Systems; 261CE050) were added. For days 20-35, S6 media with 10PM Alk5i II and 1mM T3 was fed every other day.
2.3. Immunohistochemistry
Differentiated SC-β cell clusters were collected and fixed with 4% PFA (Electron Microscopy Sciences; 15714) at 4°C overnight and embedded in histogel (Thermoscientific; HG-4000-012) for histological sectioning. The paraffin embedded sectioned slides were washed with Histoclear (Electron Microscopy Sciences; 64111-04) and rehydrated. Antigen retrieval on the slides were performed by using 0.05 M EDTA (Ambion; AM9262) and put into pressure cooker (Aptum Biologics; 2100 Retriever) for 2 hr. The slides were incubated in blocking solution (0.1% Triton-X (Acros Organics; 327371000) and 5% donkey serum (Jackson Immunoresearch; 017-000-121) in PBS) for 30 min at RT. Primary antibodies were added and incubated at 4°C overnight. Mouse anti-NKX6-1 (University of Iowa, Developmental Hybridoma Bank; F55A12-supernatant) with 1:100 dilution, rat anti-insulin (pro-)/C-peptide (Developmental Studies Hybridoma Bank; GN-ID4) with 1:300 dilution, and goat anti-human PDX-1/IPF1 (R&D Systems; AF2419) with 1:300 dilution were used as primary antibodies for this assay. Secondary antibodies conjugated to Alexa Fluor 488 and 594 were used to detect the primary antibodies. The slides were then washed with blocking solution and incubated with secondary antibodies for 2 hr at RT. For collection of images, the slides were treated with Vectashield (Vector; H-1200), covered with cover slides, and sealed with nail polish. Corresponding images were taken with the Leica DMI4000 fluorescence microscope. TUNEL assay (Roche; 12156792910) was performed on select slides as part of the immunohistochemistry process, with quantitative analysis achieved via manual cell counting.
2.4. Flow cytometry
The SC-β cell clusters were dispersed into single-cell suspension by using 0.05% Trypsin-EDTA (Gibco, ThermoFisher; 25300-054) at 37 °C for 10–20 min. The single cells were centrifuged for 3 min at 300 rcf, followed by aspiration of the supernatant, and fixed in 4% PFA at 4 °C for 30 min. The cells were washed once with PBS and incubated in blocking solution for 1 hr at 4 °C. Primary antibodies in blocking solution were added to the cells and incubated overnight at 4 °C. Mouse anti-NKX6-1 with 1:100 dilution and rat anti-insulin (pro-)/C-peptide with 1:300 dilution were used as primary antibodies for this assay. After incubation, the cells were washed twice with blocking solution and incubated with secondary antibodies for 2 hr at 4 °C. Secondary antibodies conjugated to Alexa Fluor 488 and 594 were used. Cells were then washed 3 times and resuspended in 500 Pl-700 Pl sorting buffer (0.5% BSA (Sigma; A8412) in PBS). The resuspended cells were filtered through a 40 Pm nylon mesh into flow cytometery tubes (BD Falcon; 352235) for analysis using the LSR-II flow cytometer (BD Biosciences). Quantification of the results was achieved with the FlowJo software.
2.5. Encapsulating SC-β cells inside the device
To encapsulate the SC-β cells inside the device, the 3D-printed devices were sprayed with 70% ethanol and dried inside a biosafety cabinet overnight. All five sides of the scaffold, except for the top side were wrapped with sealing film (Parafilm, MidSci; HS234526A) before the SC- β cell loading process. Fibrinogen from human plasma (EMD Millipore; 341576) was reconstituted in CMRLS without FBS and Pen/Strep at RT to make up 10 mg/ml fibrinogen solutions. The aliquots were stored in the −80 °C. Thrombin (Sigma) was reconstituted in PBS to make 50 IU/ml solution and was stored in −20 °C.
The fibrinogen solution and thrombin solutions were thawed and equilibrated to RT before the cell-loading process. SC-β cell clusters (5x106 cells) mixed with 120 μl–180 μl of 10 mg/ml fibrinogen were manually loaded into the device with a P1000 tip. Immediately following, 20 μl–60 μl of 50 IU/ml thrombin was added to the fibrinogen- SC-β cell cluster mixture inside the device. The cell-loaded device was incubated at RT for 3 min and moved to 37 °C incubator for additional 10 min of incubation. The sealing film was then removed from the device using sterile forceps and the SC-β cell cluster loaded device was kept in S6 media until transplantation.
2.6. Finite element modeling of clusters within hydrogel slab
Steady-state oxygen concentration profiles were calculated using finite element modeling in COMSOL Multiphysics [25] solving the species conservation equation
| (1) |
where D is the oxygen diffusivity coefficient (2.78x10−5 and 1.53x10−5 cm2/s for hydrogel and cells, respectively [25]), α is the solubility coefficient (1.27x10−9 and 1.02x10−9 mol/cm3/mmHg for hydrogel and cells, respectively [25]), pO2 is the local partial pressure of oxygen, V is cellular oxygen consumption rate, and vcell is the cellular volume (1817 μm3/cell determined by manual counting). D and α for the hydrogel was assumed to be the same as culture medium. V was assumed to follow Michaelis-Menten kinetics [26–28],
| (2) |
where Vmax is maximal oxygen consumption (27.6 amol/cell/s, determine with a Seahorse XF24(3) analyzer), pO2,cell is the partial pressure of oxygen the cell is exposed to, and Km is the Michaelis-Menten constant (0.44 mmHg [29]). The hydrogel slab was set at 5x5x1 mm, with the pO2 of the outer surface set at 40 mmHg. The inner cellular volume fraction within the slab was set and kept constant at 0.0956 to approximate that inside of the 3D-printed device while changing the number and size of clusters. The mesh was generated by setting the sequence type as physics-controlled mesh and element size as finer. The calculation with 100 μm diameter clusters had 231838 elements with an average quality of 0.6552, 123 μm diameter clusters had 123344 elements with an average quality of 0.6511, 171 μm diameter clusters had 41078 elements with an average quality of 0.6562, and 247 μm diameter clusters had 24656 elements with an average quality of 0.6579.
2.7. Resizing clusters
Two types of Aggrewells, Aggrewell 400EX and Aggrewell 400 (StemCell Technologies; 27840, 27845), were utilized in the resizing process. 2 ml of the Rinsing solution (StemCell Technologies; 07010) was added to each well of the Aggrewell 400EX plate, and the plate was centrifuged at 2000 rcf for 5 min to remove any present bubbles. The rinsing solution was aspirated, and the plate was washed once with CMRLS and kept at RT. 2 ml of CMRLS was added to each well of the Aggrewell 400 plate, and the plate was centrifuged at 2000 rcf for 5 min and kept at RT until further usage.
The SC-β cell clusters between 25 days and 34 days in the SC-β cell differentiation protocol were utilized in the resizing process. The selected SC-β cell clusters were washed once with PBS and treated with TryplE Express dispersion medium (Gibco, ThermoFisher; 12604-013) for 15 to 30 min. The cells were kept in the waterbath at 37 °C during the dispersion process. The cell count analysis was performed by the Vi-Cell automatic cell counter (Beckman Coulter; 731050) using the trypan blue dye exclusion method. The dispersed single cells in TryplE Express were spun down at 300 rcf for 3 min and were washed once with CMRLS. The cells were then reconstituted in CMRLS + 10 μM Alk5i II + 1 μM T3. 14.1x106 cells and 3.6x106 cells were seeded onto each well of the Aggrewell 400EX and Aggrewell 400, respectively. The cell-loaded Aggrewell plates were then centrifuged at 300 rcf for 3 min and were incubated in the 37 °C incubator for 48 hr.
2.8 Quantitative reverse transcription PCR
Gene expression was analyzed for both the resized (small) clusters and the large clusters 28 days into the differentiation protocol. The small clusters were collected for RNA extraction 48 hr after the resizing procedure. Total RNA was extracted using the RNeasy Mini Kit (Qiagen; 74014) including the optional step of on-column DNase Digestion using the RNase-Free DNase set (Qiagen; 79254). The extracted RNA was reverse-transcribed using the High Capacity Reverse Transcription Kit (Applied Biosystems; 4368814). Amplification of the cDNA via StepOne Plus Real-Time PCR (Applied Biosystems) was performed using PowerUP SYBR Green Master Mix (Applied Biosystems; A25742) and the following custom primers from Invitrogen were used (gene, right primer sequence, and left primer sequence): Insulin (INS), 5’-CAATGCCACGCTTCTGC-3’, 5’-TTCTACACACCCAAGACCCG-3’; NKX6-1, 5’-CCGAGTCCTGCTTCTTCTTG-3’, 5’-ATTCGTTGGGGATGACAGAG-3’; PDX1, 5’-CGTCCGCTTGTTCTCCTC-3’, 5’-CCTTTCCCATGGATGAAGTC-3’; Chromogranin A (CHGA), 5’-TGACCTCAACGATGCATTTC-3’, 5’-CTGTCCTGGCTCTTCTGCTC-3’; TBP (housekeeping gene), 5’-GCCATAAGGCATCATTGGAC-3’, 5’-AACAACAGCCTGCCACCTTA-3’. Data were analyzed using the CT method and normalized to the large clusters.
2.9 In vitro glucose-stimulated insulin secretion
In vitro glucose stimulated insulin secretion was used to measure the in vitro function of large and small clusters. Krebs buffer (Krb) was formulated as follows: 128 mM NaCl, 5 mM KCl, 2.7 mM CaCl2, 1.2 mM MgCl2, 1 mM Na2HPO4, 1.2 mM KH2PO4, 5 mM NaHCO3, 10 mM HEPES (Gibco, ThermoFisher; 15630-080), 0.1% BSA in Milli-Q filtered water. Krb was pre-warmed to 37 °C before the experiment. Large and small clusters 25–35 days in the differentiation protocol were collected in 1.7 ml microcentrifuge tubes (GeneMate; C-3262-1), washed twice with 1 mL of krb per tube, and underwent preincubation with 1 ml of 2 mM glucose in krb for 1 hr in a 37 °C incubator. The tube lids were open during all incubations to allow for air exchange. Following the preincubation, supernatant was removed, and the clusters were challenged with 1 mL of 2 mM glucose (low glucose) in krb. After 1 hr, 200 μl of supernatant was collected (low glucose sample). Remaining supernatant was removed, and the clusters were challenged with 1 ml of 20 mM glucose (high glucose) in krb for 1 hr in the incubator. At the end of the challenge, 200 μL of supernatant was collected (high glucose sample). The collected low glucose and the high glucose samples were processed for human insulin quantification using the Human Insulin ELISA kit (ALPCO; 80-INSH8-E01.1) and measured by an absorbance microplate reader (BioTek) at 450 nm. The insulin quantification was normalized via viable cell count using the Vi-Cell by single-cell dispersing the clusters with 0.05% Trypsin-EDTA.
2.10. In vitro hypoxia experiment
The bottom of 50-mL tubes (Corning; 352070) was removed, a thin layer of adhesive applied to the edges (NuSil; MED-1137), and a sheet of 0.005-in thick silicone rubber (Speciality Manufacturing; non-reinforced vulcanized gloss/gloss) attached to the opening [25]. After allowing the adhesive to set for 24 hr, the tubes were incubated with 70% ethanol for 24 hr, the ethanol solution removed, and then dried for 24 hr underneath a UV lamp in a biological safety cabinet. To evaluate SC-β cell clusters, 5x106 cells worth of clusters were plated onto silicone rubber as free floating clusters, embedded into 3D-printed devices, or embedded as resized clusters in 3D-printed devices cultured in CMRLS + 10 μM Alk5i II + 1 μM T3, then placed in an humidified HERACELL V105 160i incubator (Thermo Fisher) set for 5.3% oxygen, 5% carbon dioxide, and the balance nitrogen. After 48 hr, cells were removed and assessed using the Live/Dead Viability/Cytotoxicity Kit (Molecular Probes; L3224) and by fixation for histological examination.
2.11. Transplantation
All in vivo experiments were performed in accordance with the guidelines of the relevant committee. Immunocompromised Fox Chase SCID Beige mice aged 10–12 weeks were obtained from Charles River and transplanted subcutaneously into the left flank with regular (large) SC-β cell cluster-loaded devices as well as resized (small) SC-β cell cluster-loaded devices.
2.12. In vivo glucose-stimulated insulin secretion
At 2 wk and 12 wk time points, the mice underwent in-vivo glucose stimulated insulin secretion for quantitative measurement of human insulin level. The mice were fasted for 16 hr overnight before being injected intraperitoneal (IP) with 2 g D-(+)-glucose/1 kg body weight. At 0 and 30 minutes post injection, blood glucose was measured and blood was collected from the mice through facial vein puncture using a lancet (Feather 2017-01). Serum was separated from the collected blood sample using Microvettes (Sarstedt; 16.443.100) for quantification of serum human insulin level. The quantification was processed using the Human Insulin ELISA kit and measured by an absorbance microplate reader at 450 nm.
2.13. Retrieval of the 3D printed device
The 3D printed device was removed from the host and incubated in 0.1% Triton-X solution at RT on a rotator (MidSci) for 1 wk. After 1 wk, the device was removed from the solution, and the remaining tissue inside the device was manually removed through the usage of syringe needles and pipets.
2.14. Statistical Analysis
Determination of statistical significance was made using unpaired and paired t-tests, as indicated, with p<0.05 considered significant. Data is presented as mean ± standard error in the mean.
3. Results
3.1. Establishing 3D printer approach and device manufacture
We modulated the parameters of a MakerGear M2 3D printer to produce the small transplantation devices used in this study. These devices were designed using Autodesk Inventor and printed vertically utilizing the fused deposition modeling technique, an additive manufacturing technology (Figure 1a). The printer utilized PLA as the feedstock material by increasing the nozzle temperature of the extruder nozzle to the melting temperature of PLA. The extruded PLA is then printed layer-by-layer, gradually increasing in height over the printing period. We chose to fabricate devices from PLA due to its biocompatibility [24] and compatibility with the MakerGear M2 in contrast with other printer-compatible materials, such as acrylonitrile butadiene styrene (ABS), which is not biocompatible and thus not suitable for working with SC-β cells. A range of parameters including layer thickness, extrusion speeds, and temperatures were explored to produce consistent devices while minimizing the z-axis resolution (Table 1). PLA with extruder temperatures lower than 200 °C did not produce consistent extrusion width, resulting in an uneven surface level during the first few layers of the device, which disrupted the printer from continuing with the subsequent layers. The precise control of the bed temperature was found to be critical for initiating the 3D printing process. Setting the bed temperature to the glass transition temperature (Tg) of PLA at 60 °C allowed for the extruded layers to adhere quicker to the glass bed of the 3D printer, providing a solid foundation. Due to the intrinsic melting point of PLA between 180 °C – 210 °C, we found the inclusion of a prime pillar feature (orange structure in Figure 1b) in the 3D printing process to be necessary to increase the solidifying time for each layer of PLA, thereby maintaining the structural integrity of the device while minimizing stringing of unwanted PLA material (Figure 1b–d). Additionally, other 3D printing parameters including layer thickness, nozzle extrusion width, and the prime pillar feature were explored. Increasing layer thickness from 0.1 mm to 0.2 mm caused unwanted stringing and non-uniform pore shape (Figure S1a) but decreased the average pore size by almost half (Figure S1b). The exclusion of the prime pillar resulted in buildup of PLA residue on the extruder tip, affecting the precision of the 3D printer to create pores that are evenly sized and distributed throughout the device (Figure S1c). Each 3D-printed device had a pore size of 971±12 Pm in width and 1128±8 Pm in length and weighed approximately 0.275 g. On a cost-per-weight basis, the material cost of manufacturing one device is approximately $0.01, enabling economic production of many devices.
Figure 1.

3D printing approach for device fabrication. (a) Image of CAD design of device generated using Autodesk Inventor showing device dimensions. (b) Image of design converted to G-code format using KISSlicer showing device (grey) and prime pillar feature (orange). (c) Image of MakerGear M2 3D printing device and prime pillar. (d) Image of final 3D-printed device.
Table 1.
3D printer parameters used in study.
| 3D-printing specification | Range |
|---|---|
| Layer Thickness | 0.1 mm – 0.3 mm |
| Extrusion Width | 0.1 mm – 0.5 mm |
| Support | None/Prime pillar |
| Extruder Temperature PLA | 160 °C – 220 °C |
| Bed Temperature | 25 °C – 70 °C |
| Loop Speed | 5 mm/s - 15 mm/s |
| Solid Speed | 37.5 mm/s - 60 mm/s |
3.2. Production and loading of SC-β cells into 3D-printed device
In order to generate SC-β cells to load into 3D-printed devices for transplantation, we differentiated the HUES8 hESC line using our previously published directed differentiation protocol that utilizes a specific temporal combination of growth factors and small molecules to induce a β cell-like phenotype (Figure 2a) [7, 8]. Cells are cultured in suspension culture within spinner flasks in cellular aggregates consisting of >103 cells each to produce cells that co-express C-peptide, which is encoded by the insulin gene, with NKX6-1 and PDX1, transcription factors found in β cells (Figure 2b–c). Approximately 50% of cells express both C-peptide and NKX6-1, quantified with flow cytometric assessment of dispersed and immunostained cells (Figure 2d).
Figure 2.

Generation of SC-β cells from hESC. (a) Overview schematic of differentiation process to generate SC-β cells [7, 8]. (b) Histological section of SC-β cell clusters stained for C-peptide (green) and PDX1 (red) and with 4,6-diamidino-2-phenylindole (DAPI; blue). (c) Histological section of SC-β cell clusters stained for C-peptide (green) and NKX6-1 (red) and with DAPI (blue). (d) Flow cytometry dot plot of dispersed and fixed cells stained for C-peptide and NKX6-1. Scale bar=50 μm. Act A, activin A; Alk5i, Alk5 receptor inhibitor II; CHIR, CHIR9901; KGF, keratinocyte growth factor; LDN, LDN193189; PdbU, phorbol 12,13-dibutyrate; RA, retinoic acid; T3, triiodothyronine; Y, Y27632; AU, arbitrary units.
In order to load and secure SC-β cell clusters within the 3D-printed devices (Figure 3a), SC-β cell clusters (5x106 cells per mouse) were suspended in a fibrinogen solution and inserted into the device sealed with a wrapping that was disinfected with ethanol and UV light. Thrombin solution was immediately added directly into the cellular suspension within the device and manually shaken to maintain the cluster distribution. The gel cross-linked rapidly, being able to remain inside the device without the wrapping after 1–5 min (Figure 3b). Maintaining the three-dimensional spatial distribution of clusters likely helps maximum cell viability by providing more uniform oxygenation inside the device.
Figure 3.

Loading of SC-β cell clusters into 3D-printed device. (a) Schematic overview of cell loading process. (b) Image of fibrin-embedded clusters captured within 3D-printed device. Scale bar=500 μm.
3.3. Finite element modeling of clusters within hydrogel
Although our device design includes encapsulation of SC-β cell clusters within a fibrin gel, in part to promote vascularization, we recognized that there would be a transient period before host vascularization into the graft that the cells would be subjected to lower pO2. This was of particular concern in our scalable suspension stem cell culture system, as the cells are grown in large clusters that could be diffusion limiting for oxygen, and even low, non-zero, values of pO2 are known to reduce function in β cells [30]. In order to evaluate this assumption, we first performed finite element modeling of oxygen consumption- diffusion of cells within a hydrogel slab packaged into cluster sizes ranging from 100–247 μm diameter, keeping total cellular volume constant (Figure 4). This model assumed that the pO2 surrounding the hydrogel slab was 40 mmHg to approximate that found in the surrounding microvasculature. Large clusters sized 247 μm had severely low values of pO2 within the clusters, with many of the cells being exposed to virtually no oxygen. Smaller cluster sizes (100–171 μm) had much shallower gradients in pO2, with no cells approaching 0 mmHg. From this model, we established a range of cluster sizes that avoid severe hypoxia.
Figure 4.

Finite element modeling of oxygen concentration within cluster-embedded hydrogel slab. Cluster sizes ranging from 100-247 μm in diameter were evaluated, maintaining the total cellular volume within a 1-mm thick slab.
3.4. Establishment of SC-β cell cluster resizing approach
To avoid transient hypoxia within our 3D-printed device, we developed an approach to resize SC-β cell clusters to smaller sizes that are within the 100–171 μm diameter range identified in our finite element model (Figure 4). We took large SC-β cell clusters (Figure 5a), dispersed them into a single-cell suspension, and seeded 3x103 cells per 400 μm microwell (Figure 5b). After 48 hr, the cells reformed a small cluster (Figure 5c–d), with the average diameter of 143±5 μm (Figure 5e). The expression of the β cell genes insulin (INS), NKX6-1, PDX1, and chromogranin A (CHGA) was maintained (Figure 5f), and the clusters were able to increase insulin secretion in response to high glucose (Figure 5g).
Figure 5.

Resizing of SC-β cell clusters. (a) Clusters before dispersion. (b) Clusters dispersed into a single-cell suspension and seeded into microwells. (c) Reformed clusters after 24 hr incubation in microwells. (d) Reformed clusters recovered from microwells. (e) Measurement of average cluster diameter before (large) and after (small) resizing. n=40. Scale bar=400 μm. *p<0.01 (two-sided unpaired t-test). (f) Gene expression of resized small clusters 28 days into the differentiation protocol relative to large clusters (n=4). (g) Ratio of insulin secretion at high glucose over at low glucose for large and small SC-β cell clusters (n=3).
3.5. In vitro validation of device design
In order to further validate the findings of our mathematical model, we developed an in vitro culture system to test devices under defined values of pO2 comprised of a highly oxygen permeable silicone rubber surface on top of which devices or cell clusters can be cultured (Figure 6). The oxygen permeability of the silicone rubber results in a minimal oxygen concentration gradient through the material, allowing for pO2 in the incubator to match that of the bottom surface of the device being tested [25]. With this system, we evaluated survival of SC-β cell clusters not in a device (Figure 7a), within the device (Figure 7b), or resized into smaller clusters within the device (Figure 7c) after 48 hr of culture at 40 mmHg pO2. After this acute culture, we qualitatively assessed the cells using a viability dye that stains membrane-permeable cells, indicative of a dead cell, and observed substantial cell death for large clusters cultured within the device but not for the other two culture conditions (Figure 7d–f). We note that the dyes do not fully penetrate the clusters and that the observed membrane-permeable (red) cells have mostly detached from the cluster. To further evaluate the SC-β cell clusters, we fixed, sectioned, and treated clusters with a TUNEL assay to detect DNA fragmentation due to death and observed TUNEL+ cells within the central core of large clusters that were cultured within the device (Figure 7g). When quantified, there were more TUNEL+ cells in large clusters within the device compared to large clusters not in a device or small clusters within the device (Figure 7h). This in vitro assessment validates the prediction of our mathematical model that smaller clusters avoid severe hypoxia within our 3D-printed device.
Figure 6.
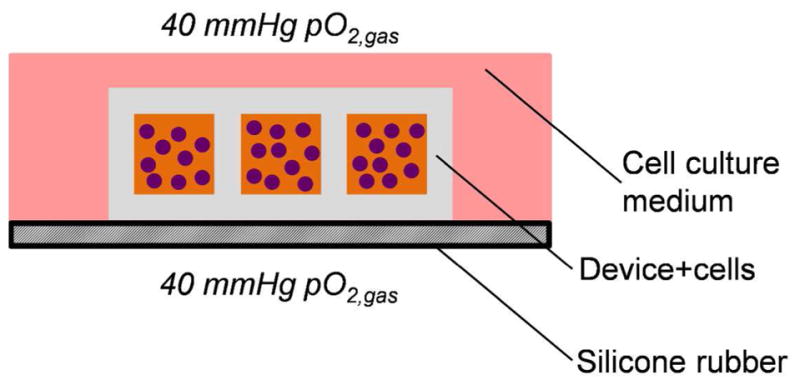
Schematic overview of in vitro culture system for testing devices at physiological oxygen.
Figure 7.

Resized cluster loaded into 3D-printed device are resistant to hypoxia-induced death. (a–c) Schematics illustrating test set up for (a) free floating large clusters, (b) large clusters loaded into device, and (c) small clusters loaded into device. (d–f) Representative images of clusters stained with a fluorescent viability dye after 48 hr of culture at 40 mmHg pO2,gas taken with the same exposure. The top images are composites of both the live (green) and dead (red) stains, middle images just dead stain, and bottom images just green stain. Panel (d) was cultured as indicated in panel (a), (e) as in (b), and (f) as in (c). Scale bar=200 μm (g) Histological section of SC-β cell clusters cultured as in (b) stained with a TUNEL assay (red) and with DAPI (blue). Scale bar=50 μm (h) Quantitation of TUNEL+ cells from histological sections. *p<0.01 (two-sided unpaired t-test).
3.6. Transplantation of 3D-printed device loaded with SC-β cells
We validated our 3D-printing approach by transplanting SC-β cell-loaded 3D-printed device subcutaneously into immunocompromised mice. We compared two groups of animals that were transplanted with either large or resized small clusters. After 2 wk in vivo, we evaluated the functional status of the graft with a glucose-stimulated insulin secretion assay. After fasting the mice overnight, serum was collected before and 30 min after an injection of glucose (Figure 8a). Mice that received devices with large clusters had detectable human insulin in the serum but did not respond to the glucose injection, which is consistent with an unhealthy graft. Mice that received devices with small clusters also had detectable human insulin in the serum. In addition, the glucose injection caused a 2.5±0.4 times increase in human insulin, indicating that the transplanted grafts were functional and glucose-responsive. This in vivo functional phenotype was observed up to 12 wk. After this 12 wk observation period, 3D-printed devices were still retrievable from the mice (Figure 8b). Devices maintained structural integrity and could be easily handled without risk of deformation. Some devices were placed in a decellularization solution to remove infilled mouse tissue and allow better visual inspection of the device (Figure 8c). No visible defects were noted. Removal of cells for histological processing from the retrieved device without destroying the tissue was difficult, but we observed the presence of SC-β cell clusters in the few intact pieces of tissue that we managed to remove (Figure 8d). This data demonstrates that our SC-β cell-embedded 3D-printed devices combined with cell cluster resizing to avoid hypoxia are functional when transplanted into mice and can be retrieved.
Figure 8.

3D-printed devices loaded with SC-β cells (5x106 cells per mouse) are functional after transplantation into mice. (a) In vivo glucose stimulated insulin secretion assays of mice 2 wk (n=6 for large clusters and n=7 for small clusters) and 12 wk (n=3 for small clusters) after transplantation. Mice were fasted overnight and serum collected before (0 min) and 30 min after an intraperitoneal injection of glucose. Human insulin was quantified from this serum using ELISA. (b) Image demonstrating handling of recovered 3D-printed device. (c) Image showing decellularized recovered 3D printed device. (d) Histological section of tissue recovered from device stained for C-peptide (green) and with 4,6-diamidino-2-phenylindole (DAPI; blue). We note that there are some red blood cells that autofluoresce green around the cellular cluster. Scale bar=25 μm. *p<0.01 (two-sided unpaired t-test when comparing large vs. smaller clusters, two-sided paried t-test when comparing 0 vs. 30 min).
4. Discussion
Here, we combined stem cell technology with 3D printing to develop a novel retrievable macroporous device for diabetes cell replacement therapy. 3D printing allowed us to rapidly prototype designs and fabricate devices with a defined three-dimensional structure (Figure 1). This was accomplished with a low cost, consumer-grade 3D printer by modulating printing parameters and inclusion of a cooling step after each extruded layer that allowed for fabrication of finer features, making this approach generally accessible to most research laboratories for investigation. Significant progress in the 3D printing market has made several low cost 3D printers available that could potentially fabricate devices described in this report. Encapsulation of cells within microcapsules for transplantation typically consisting of alginate has received significant attention over the last few decades [31] but suffer from not being retrievable. We demonstrated that our device is retrievable for at least 12 weeks after transplantation (Figure 8), and since PLA has a long degradation time, mechanical strength and retrievability can potentially be maintained for years. Furthermore, encapsulation prevents vascularization of cellular grafts, causing hypoxia and subsequently death or reduced function [17]. We have used finite element modeling of oxygen diffusion-consumption (Figure 4) in combination with cluster resizing (Figure 5) and a degradable fibrin gel (Figure 3) to reduce hypoxia and promote vascularization.
We exclusively used hPSC-derived cells in this study, which gives our approach several distinct advantages. SC-β cells can be generated at a large scale in spinner flasks (>108 cells per flask) [7, 8], helping to overcome the shortage of cadaveric islets available for diabetes cell replacement therapy. These cells could potentially be genetically engineered to avoid immune destruction [32]. SC-β cells can be derived from patient hiPSC [7] to avoid allogeneic rejection. In situations of autoimmunity, which occurs in Type 1 diabetes, patient SC-β cells combined with an immunotherapy to selectively suppress autoimmunity [33–35] is possible. Improvements in methodologies for generating SC-β cells in vitro will enable a consistent cell source that can be tailored to maximize transplantation outcomes, such as resizing of clusters to reduce hypoxia (Figure 5). Furthermore, while not tested in this study, cadaveric islets could be substituted for SC-β cells and used with our 3D printing approach.
A major advantage of 3D printing is the ability to augment device designs with additional functionality that is not feasible with alternative approaches, such as solvent casting and particulate leaching with polydimethylsiloxane, which has been used successfully as a retrievable device for cadaveric islets [16]. 3D bioprinting has been previously used to plot islets within composite hydrogels [36], and while in vivo utility of this approach has not yet been demonstrated, such an approach could be combined with our approach that uses a thermoplastic to create a device that is functional and retrievable while also providing encapsulation of cells to protect against immune attack. Immune tolerance of the cells and materials is a major challenge in the field [37], and the study presented here was not designed to address this problem. Further functionality could be added to our approach to help tackle this problem, such as anti-fibrotic or immune tolerating materials [9, 38]. Alternative PLA formulations that are Food and Drug Administration approved or formulated to withstand sterilization methods could be used. PLA could be substituted with polycaprolactone, and alternative degradable hydrogels, such as collagen, could substitute for fibrin. To minimize or eliminate time between transplantation and vascularization, materials engineered to release of pro-angiogenic compounds [39] or pre-vascularization of tissues could be used [40].
5. Conclusion
We have developed a macroporous retrievable 3D-printed device for the subcutaneous transplantation of SC-β cells. The device was fabricated with biocompatible PLA using a low cost, consumer-grade 3D printing. SC-β cell clusters were loaded into the pores suspended in a degradable fibrin gel, which secured clusters distributed throughout the device. Using finite element modeling, we calculated that cluster sizes smaller than 171 μm in diameter would avoid severe transient hypoxia that would occur after transplantation but before vascularization. Using an in vitro test setup that leveraged the high oxygen permeability of silicone rubber, we validated that resized clusters embedded within devices at physiological oxygen survive, consistent with the predictions of our model. Upon transplantation into mice, 3D-printed devices seeded with resized clusters are functional, secreting insulin in response to a glucose injection. Transplanted devices were functional for up to 12 wk, during which devices maintained their structural integrity and were retrievable. In the future, this approach will serve as a platform technology for more advanced 3D printing approaches for diabetes cell replacement therapy.
Supplementary Material
Acknowledgments
We thank and acknowledge the following people and organizations for assistance: Dominic (Nick) Thompson and Savannah Est of the Washington University Institute for Minimally Invasive Surgery for useful discussion; Patricia Widder for useful discussions and use of the MakerGear M2; Professor Donald Elbert for useful discussions; the Department of Pathology & Immunology Histology and Flow Cytometry Cores for technical assistance. This study was supported by the DRC at Washington University (NIH Grant No. 5 P30 DK020579). J.R.M. is an inventor on patent applications covering some content found in this manuscript.
References
- 1.Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract. 2010;87:4–14. doi: 10.1016/j.diabres.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 2.Zhang P, Zhang X, Brown J, Vistisen D, Sicree R, Shaw J, Nichols G. Global healthcare expenditure on diabetes for 2010 and 2030. Diabetes Res Clin Pract. 2010;87:293–301. doi: 10.1016/j.diabres.2010.01.026. [DOI] [PubMed] [Google Scholar]
- 3.Shapiro AM, Ricordi C, Hering BJ, Auchincloss H, Lindblad R, Robertson RP, Secchi A, Brendel MD, Berney T, Brennan DC, Cagliero E, Alejandro R, Ryan EA, DiMercurio B, Morel P, Polonsky KS, Reems JA, Bretzel RG, Bertuzzi F, Froud T, Kandaswamy R, Sutherland DE, Eisenbarth G, Segal M, Preiksaitis J, Korbutt GS, Barton FB, Viviano L, Seyfert-Margolis V, Bluestone J, Lakey JR. International trial of the Edmonton protocol for islet transplantation. N Engl J Med. 2006;355:1318–30. doi: 10.1056/NEJMoa061267. [DOI] [PubMed] [Google Scholar]
- 4.Shapiro AM, Lakey JR, Ryan EA, Korbutt GS, Toth E, Warnock GL, Kneteman NM, Rajotte RV. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med. 2000;343:230–8. doi: 10.1056/NEJM200007273430401. [DOI] [PubMed] [Google Scholar]
- 5.Qi M, Kinzer K, Danielson KK, Martellotto J, Barbaro B, Wang Y, Bui JT, Gaba RC, Knuttinen G, Garcia-Roca R, Tzvetanov I, Heitman A, Davis M, McGarrigle JJ, Benedetti E, Oberholzer J. Five-year follow-up of patients with type 1 diabetes transplanted with allogeneic islets: the UIC experience. Acta Diabetol. 2014;51:833–43. doi: 10.1007/s00592-014-0627-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bellin MD, Barton FB, Heitman A, Harmon JV, Kandaswamy R, Balamurugan AN, Sutherland DE, Alejandro R, Hering BJ. Potent induction immunotherapy promotes long-term insulin independence after islet transplantation in type 1 diabetes. Am J Transplant. 2012;12:1576–83. doi: 10.1111/j.1600-6143.2011.03977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pagliuca FW, Millman JR, Gurtler M, Segel M, Van Dervort A, Ryu JH, Peterson QP, Greiner D, Melton DA. Generation of functional human pancreatic beta cells in vitro. Cell. 2014;159:428–39. doi: 10.1016/j.cell.2014.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Millman JR, Xie C, Van Dervort A, Gurtler M, Pagliuca FW, Melton DA. Generation of stem cell-derived beta-cells from patients with type 1 diabetes. Nat Commun. 2016;7:11463. doi: 10.1038/ncomms11463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vegas AJ, Veiseh O, Gurtler M, Millman JR, Pagliuca FW, Bader AR, Doloff JC, Li J, Chen M, Olejnik K, Tam HH, Jhunjhunwala S, Langan E, Aresta-Dasilva S, Gandham S, McGarrigle JJ, Bochenek MA, Hollister-Lock J, Oberholzer J, Greiner DL, Weir GC, Melton DA, Langer R, Anderson DG. Long-term glycemic control using polymer-encapsulated human stem cell-derived beta cells in immune-competent mice. Nat Med. 2016;22:306–11. doi: 10.1038/nm.4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCall M, Shapiro AM. Update on islet transplantation. Cold Spring Harb Perspect Med. 2012;2:a007823. doi: 10.1101/cshperspect.a007823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hesse UJ, Sutherland DE, Gores PF, Sitges-Serra A, Najarian JS. Comparison of splenic and renal subcapsular islet autografting in dogs. Transplantation. 1986;41:271–4. doi: 10.1097/00007890-198602000-00028. [DOI] [PubMed] [Google Scholar]
- 12.Kroon E, Martinson L, Kadoya K, Bang A, Kelly O, Eliazer S, Young H, Richardson M, Smart N, Cunningham J, Agulnick A, D'Amour K, Carpenter M, Baetge E. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nature biotechnology. 2008;26:443–95. doi: 10.1038/nbt1393. [DOI] [PubMed] [Google Scholar]
- 13.Lacy PE, Hegre OD, Gerasimidi-Vazeou A, Gentile FT, Dionne KE. Maintenance of normoglycemia in diabetic mice by subcutaneous xenografts of encapsulated islets. Science (New York, NY. 1991;254:1782–4. doi: 10.1126/science.1763328. [DOI] [PubMed] [Google Scholar]
- 14.Motte E, Szepessy E, Suenens K, Stange G, Bomans M, Jacobs-Tulleneers-Thevissen D, Ling Z, Kroon E, Pipeleers D Beta Cell Therapy Consortium E-F. Composition and function of macroencapsulated human embryonic stem cell-derived implants: comparison with clinical human islet cell grafts. Am J Physiol Endocrinol Metab. 2014;307:E838–46. doi: 10.1152/ajpendo.00219.2014. [DOI] [PubMed] [Google Scholar]
- 15.Veiseh O, Doloff JC, Ma M, Vegas AJ, Tam HH, Bader AR, Li J, Langan E, Wyckoff J, Loo WS, Jhunjhunwala S, Chiu A, Siebert S, Tang K, Hollister-Lock J, Aresta-Dasilva S, Bochenek M, Mendoza-Elias J, Wang Y, Qi M, Lavin DM, Chen M, Dholakia N, Thakrar R, Lacik I, Weir GC, Oberholzer J, Greiner DL, Langer R, Anderson DG. Size- and shape-dependent foreign body immune response to materials implanted in rodents and non-human primates. Nature materials. 2015;14:643–51. doi: 10.1038/nmat4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pedraza E, Brady AC, Fraker CA, Molano RD, Sukert S, Berman DM, Kenyon NS, Pileggi A, Ricordi C, Stabler CL. Macroporous three-dimensional PDMS scaffolds for extrahepatic islet transplantation. Cell Transplant. 2013;22:1123–35. doi: 10.3727/096368912X657440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Colton CK. Oxygen supply to encapsulated therapeutic cells. Adv Drug Deliv Rev. 2014;67–68:93–110. doi: 10.1016/j.addr.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 18.Avgoustiniatos ES, Colton CK. Effect of external oxygen mass transfer resistances on viability of immunoisolated tissue. Ann N Y Acad Sci. 1997;831:145–67. doi: 10.1111/j.1749-6632.1997.tb52192.x. [DOI] [PubMed] [Google Scholar]
- 19.Beattie GM, Montgomery AM, Lopez AD, Hao E, Perez B, Just ML, Lakey JR, Hart ME, Hayek A. A novel approach to increase human islet cell mass while preserving beta-cell function. Diabetes. 2002;51:3435–9. doi: 10.2337/diabetes.51.12.3435. [DOI] [PubMed] [Google Scholar]
- 20.Riopel M, Li J, Trinder M, Fellows GF, Wang R. Fibrin supports human fetal islet-epithelial cell differentiation via p70(s6k) and promotes vascular formation during transplantation. Lab Invest. 2015;95:925–36. doi: 10.1038/labinvest.2015.74. [DOI] [PubMed] [Google Scholar]
- 21.Mason MN, Mahoney MJ. A novel composite construct increases the vascularization potential of PEG hydrogels through the incorporation of large fibrin ribbons. J Biomed Mater Res A. 2010;95:283–93. doi: 10.1002/jbm.a.32825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim JS, Lim JH, Nam HY, Lim HJ, Shin JS, Shin JY, Ryu JH, Kim K, Kwon IC, Jin SM, Kim HR, Kim SJ, Park CG. In situ application of hydrogel-type fibrin-islet composite optimized for rapid glycemic control by subcutaneous xenogeneic porcine islet transplantation. J Control Release. 2012;162:382–90. doi: 10.1016/j.jconrel.2012.07.018. [DOI] [PubMed] [Google Scholar]
- 23.Lim JY, Min BH, Kim BG, Han HJ, Kim SJ, Kim CW, Han SS, Shin JS. A fibrin gel carrier system for islet transplantation into kidney subcapsule. Acta Diabetol. 2009;46:243–8. doi: 10.1007/s00592-008-0073-4. [DOI] [PubMed] [Google Scholar]
- 24.Serra T, Mateos-Timoneda MA, Planell JA, Navarro M. 3D printed PLA-based scaffolds: a versatile tool in regenerative medicine. Organogenesis. 2013;9:239–44. doi: 10.4161/org.26048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Powers DE, Millman JR, Bonner-Weir S, Rappel MJ, Colton CK. Accurate control of oxygen level in cells during culture on silicone rubber membranes with application to stem cell differentiation. Biotechnology progress. 2010;26:805–18. doi: 10.1002/btpr.359. [DOI] [PubMed] [Google Scholar]
- 26.Wilson DF, Rumsey WL, Green TJ, Vanderkooi JM. The oxygen dependence of mitochondrial oxidative phosphorylation measured by a new optical method for measuring oxygen concentration. The Journal of biological chemistry. 1988;263:2712–8. [PubMed] [Google Scholar]
- 27.Dionne KE, Colton CK, Yarmush ML. Effect of oxygen on isolated pancreatic tissue. ASAIO Trans. 1989;35:739–41. doi: 10.1097/00002480-198907000-00185. [DOI] [PubMed] [Google Scholar]
- 28.Buchwald P. FEM-based oxygen consumption and cell viability models for avascular pancreatic islets. Theor Biol Med Model. 2009;6:5. doi: 10.1186/1742-4682-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilson DF, Erecinska M, Drown C, Silver IA. The oxygen dependence of cellular energy metabolism. Arch Biochem Biophys. 1979;195:485–93. doi: 10.1016/0003-9861(79)90375-8. [DOI] [PubMed] [Google Scholar]
- 30.Dionne KE, Colton CK, Yarmush ML. Effect of hypoxia on insulin secretion by isolated rat and canine islets of Langerhans. Diabetes. 1993;42:12–21. doi: 10.2337/diab.42.1.12. [DOI] [PubMed] [Google Scholar]
- 31.Lim F, Sun AM. Microencapsulated islets as bioartificial endocrine pancreas. Science (New York, NY. 1980;210:908–10. doi: 10.1126/science.6776628. [DOI] [PubMed] [Google Scholar]
- 32.Rong Z, Wang M, Hu Z, Stradner M, Zhu S, Kong H, Yi H, Goldrath A, Yang YG, Xu Y, Fu X. An effective approach to prevent immune rejection of human ESC-derived allografts. Cell stem cell. 2014;14:121–30. doi: 10.1016/j.stem.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, Herold KC, Lares A, Lee MR, Li K, Liu W, Long SA, Masiello LM, Nguyen V, Putnam AL, Rieck M, Sayre PH, Tang Q. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med. 2015;7:315ra189. doi: 10.1126/scitranslmed.aad4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grimm AJ, Kontos S, Diaceri G, Quaglia-Thermes X, Hubbell JA. Memory of tolerance and induction of regulatory T cells by erythrocyte-targeted antigens. Sci Rep. 2015;5:15907. doi: 10.1038/srep15907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Serr I, Furst RW, Achenbach P, Scherm MG, Gokmen F, Haupt F, Sedlmeier EM, Knopff A, Shultz L, Willis RA, Ziegler AG, Daniel C. Type 1 diabetes vaccine candidates promote human Foxp3(+)Treg induction in humanized mice. Nat Commun. 2016;7:10991. doi: 10.1038/ncomms10991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marchioli G, van Gurp L, van Krieken PP, Stamatialis D, Engelse M, van Blitterswijk CA, Karperien MB, de Koning E, Alblas J, Moroni L, van Apeldoorn AA. Fabrication of three-dimensional bioplotted hydrogel scaffolds for islets of Langerhans transplantation. Biofabrication. 2015;7:025009. doi: 10.1088/1758-5090/7/2/025009. [DOI] [PubMed] [Google Scholar]
- 37.Veiseh O, Tang BC, Whitehead KA, Anderson DG, Langer R. Managing diabetes with nanomedicine: challenges and opportunities. Nat Rev Drug Discov. 2015;14:45–57. doi: 10.1038/nrd4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vegas AJ, Veiseh O, Doloff JC, Ma M, Tam HH, Bratlie K, Li J, Bader AR, Langan E, Olejnik K, Fenton P, Kang JW, Hollister-Locke J, Bochenek MA, Chiu A, Siebert S, Tang K, Jhunjhunwala S, Aresta-Dasilva S, Dholakia N, Thakrar R, Vietti T, Chen M, Cohen J, Siniakowicz K, Qi M, McGarrigle J, Lyle S, Harlan DM, Greiner DL, Oberholzer J, Weir GC, Langer R, Anderson DG. Combinatorial hydrogel library enables identification of materials that mitigate the foreign body response in primates. Nature biotechnology. 2016;34:345–52. doi: 10.1038/nbt.3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferreira LS, Gerecht S, Fuller J, Shieh HF, Vunjak-Novakovic G, Langer R. Bioactive hydrogel scaffolds for controllable vascular differentiation of human embryonic stem cells. Biomaterials. 2007;28:2706–17. doi: 10.1016/j.biomaterials.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kolesky DB, Homan KA, Skylar-Scott MA, Lewis JA. Three-dimensional bioprinting of thick vascularized tissues. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:3179–84. doi: 10.1073/pnas.1521342113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.