

Abstract
Eight novel 2-(2, 6-dioxopiperidin-3-yl)phthalimidine EM-12 dithiocarbamates 9 and 10, N-substituted 3-(phthalimidin-2-yl)-2, 6-dioxopiperidines 11–14 and 3-substituted 2, 6-dioxopiperidines 16 and 18 were synthesized as tumor necrosis factor-α (TNF-β) synthesis inhibitors. Synthesis involved utilization of a novel condensation approach, a one-pot reaction involving addition, iminium rearrangement and elimination, to generate the phthalimidine ring required for the creation of compounds 9–14. Agents were, thereafter, quantitatively assessed for their ability to suppress the synthesis on TNF-β in a lipopolysaccharide (LPS)-challenged mouse macrophage-like cellular screen, utilizing cultured RAW 264.7 cells. Whereas compounds 9, 14 and 16 exhibited potent TNF-α lowering activity, reducing TNF-α by up to 48% at 30 μM, compounds 12, 17 and 18 presented moderate TNF-α inhibitory action. The TNF-α lowering properties of these analogues proved more potent than that of revlimid (3) and thalidomide (1). In particular, N-dithiophthalimidomethyl-3-(phthalimidin-2-yl)-2, 6-dioxopiperidine 14 not only possessed the greatest potency of the analogues to reduce TNF-α synthesis, but achieved this with minor cellular toxicity at 30 μM. The pharmacological focus of the presented compounds is towards the development of well-tolerated agents to ameliorate the neuroinflammation that is commonly associated with neurodegenerative disorders, epitomized by Alzheimer’s disease and Parkinson’s disease.
Keywords: thalidomide, revlimid, N-substituted EM-12, dithiocarbamates, 3-substituted 2, 6-dioxopiperidines, iminium rearrangement, neurodegenerative diseases, TNF-α inhibition
1. Introduction
Inflammation of the central nervous system, neuroinflammation, is now recognized as a key feature of all neurodegenerative disorders that, with the progressively aging population, are becoming increasingly prominent.1 Alzheimer’s disease (AD) and Parkinson’s disease (PD) are prime examples of debilitating disorders that impact over 5 million and one million Americans, respectively. Neuroinflammation incorporates a wide spectrum of complex cellular responses that often contribute to the pathogenesis and can ultimately drive the progression of a neurological disorder.2a–c The normal adult brain possesses low or undetectable levels of peripheral inflammatory cell subtypes. In the mature brain a primary intrinsic immune-competent cell type is the microglial cell. In the normal, inactivated state microglial cells display a ramified morphology.2c However, during either chronic or acute neuroinflammation, microglia activation rapidly occurs that results in an alteration in morphology and a subsequent release of many inflammatory mediators.2a–c Key amongst these is the pro-inflammatory cytokine, tumor necrosis-alpha (TNF-α), a potent activator of the immune system that can induce immune cell activation leading to the rapid recruitment of neighboring resting microglia or astrocytes within the adjacent brain microenvironment.2b,c
Pharmacological interventions that are able to reset the fine and often lost balance between resting and activated glial cells could be of significant clinical benefit.2c TNF-α protein is a key candidate therapeutic target as - based on data from many clinical, cell biology and animal studies - abnormal regulation of this protein is strongly associated with an unregulated glial cell activity and resulting neuroinflammation.2b The identification of novel agents which can restore the normal function of activated CNS glial cells by means of reducing the pro-inflammatory effects of TNF-α protein within brain represent a viable mechanism of action for the management of clinical disease.2c
Thalidomide (1) (Figure 1) is effective in alleviating erythema nodosum leprosum associated with leprosy and aphthous ulcers in AIDS patients.3a–d Although the mechanism via which this drug ameliorates these inflammatory diseases remains to be fully elucidated, thalidomide is known to induce a wide range of immunomodulatory actions. Amongst this diversity of activities is a TNF-α lowering action, mediated by destabilizing TNF-α mRNA levels at the level of its 3′-untranslated region to reduce the rate of synthesis of TNF-α protein.4a,b As the close thalidomide analogue, lenalidomide (revlimid (3) (Figure 1)), likewise possesses TNF-α action,3c,d the backbone holds promise as a multi-template for development of biologically active compounds. In addition, as thalidomide (1) displays a moderate degree of lipophilicity (C log D value −0.83)6a that allows it access to the brain, analogues bearing equivalent physicochemical properties may provide promise in the treatment of neurodegenerative diseases exacerbated by neuroinflammation. Additionally, as the TNF-α activity of 1 is not particularly potent, requiring administration of high doses that are often both sedative and associated with adverse effects,5 the synthesis of novel and more effective TNF-α lowering agents is a worthy goal. In this regard, several N-substituted 3-(phthalimidin-2-yl)-2, 6-dioxopiperidines and 3-substituted 2, 6-dioxopiperidines are described that demonstrate promising anti-TNF-α potency.
Figure 1.
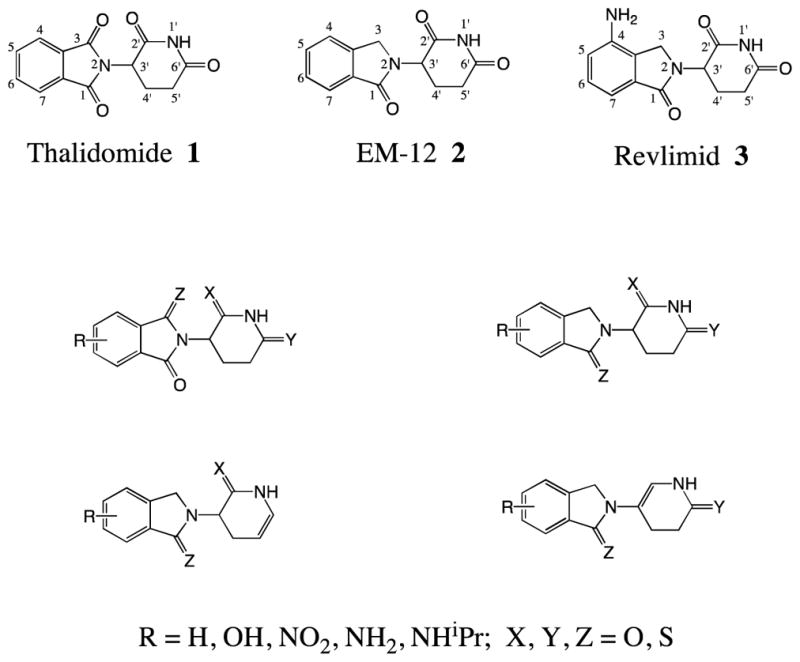
Known thalidomide, EM-12, revlimid and sulfur containing analogues possessing TNF-α action
2. Results and discussion
2.1. Chemistry
Prior studies have identified a number of thalidomide analogues with improved TNF-α lowering properties in models of cellular and CNS neuroinflammation. In this regard, compounds in Figure 1 have demonstrated encouraging results regarding anti-TNF-α potency associated with tolerable cellular toxicity.6a–d
N-substituted thalidomide derivatives have been reported with potent antitumor activities.7 Indeed, several N-alkylated thalidomide analogues have been screened in cell culture and then further evaluated in animals, in which a higher antitumor activity than that of thalidomide was observed.8a–f In general, when a sulfur atom was introduced into the thalidomide pharmacophore, a more potent antitumor activity was detected.9 Up to now the biological properties of N-dithiocarbamate substituted 3-(phthalimidin-2-yl)-2, 6-dioxopiperidines have not been reported. Based on our unpublished observations and a structural analysis of specific known compounds possessing TNF-α inhibitory activity, we generated two novel N-dithiocarbamate substituted 3-(phthalimidin-2-yl)-2, 6-dioxopiperidines, 9 and 10, as likely TNF-α inhibitors (Scheme 1).
Scheme 1.

Reagents and conditions: (i) Formaldehyde (37% solution in water), N2, reflux, 0.5 h; (ii) thionyl chloride, DMF, 0 °C, 1 h; (iii) trifluoroacetic acid, CH2Cl2, N2, rt, 22.5 h; (iv) phthaldialdehyde, THF, N2, rt, 71 h; (v) carbon disulfide, cyclohexylamine (for 9), piperidine (for 10), CH3CN, N2, rt, 42 to 46 h; (vi) glutarimide (for 11), dithioglutarimide (for 12), KOH, CH3CN, N2, rt, 16 to 18 h; (vii) phthalimide (for 13), dithiophthalimide (for 14), KOH, CH3CN, N2,rt, 18 h.
Compounds 11–14 were synthesized as shown in Scheme 1. The rationale underpinning the conception of these agents is based on green chemistry,10 whereby two potentially active agents may, under appropriate conditions, derive from a single compound; thereby providing a form of ‘medicinal economy’. Specifically, under physiological conditions, the use of a methylene linkage to connect two nitrogen atoms, as in 11–14, may permit the occurrence of simple metabolism to allow generation of the corresponding 2-(2, 6-dioxopiperidin-3-yl)phthalimidine EM-12 (2) (Figure 1), glutarimide, dithioglutarimide, phthalimide or dithiophthalimide. Importantly, these latter agents all possess a degree of TNF-α lowering potency.6a Previously, we and others have demonstrated that 2-(2, 6-dioxopiperidin-3-yl)phthalimidine EM-12 (2) generates TNF-α inhibitory activity with a low cellular toxicity6c,e and that dithiophthalimide is likewise active and well tolerated.6a As illustrated in Table 1, compound 14, containing such a methylene linkage that simultaneously connects the 2-(2, 6-dioxopiperidin-3-yl)phthalimidine EM-12 and dithiophthalimide moieties, proved to be the most potent TNF-α lowering candidate amongst the eleven analyzed compounds.
Table 1.
Inhibition of LPS-induced TNF-α production in RAW 264.7 cells, cell viability and calculated lipophilicity of assayed compounds 9–14, 16–20 are shown. The anti-TNF-α properties of the analogs are compared with those of revlimid 3
| Compd | TNF-α activity (30 μM) Control = 100% |
Cell viability (30 μM) Control = 100% |
C log D a | Fold to 1 anti-TNF-α activity b | ||
|---|---|---|---|---|---|---|
| % Control | P value | % Control | P value | |||
| 3 | 109 ± 10 | 0.4482 | 106 ± 3 | 0.1252 | − 1.31 | 1 |
| 9 | 55 ± 3 | < 0.001 | 103 ± 2 | 0.1555 | +1.39 | 45 |
| 10 | 96 ± 1 | 0.0734 | 112 ± 1 | < 0.0001 | +0.65 | 4 |
| 11 | 79 ± 3 | 0.0087 | 102 ± 2 | 0.2028 | − 1.66 | 21 |
| 12 | 63 ± 2 | 0.0184 | 94 ± 3 | 0.1488 | − 0.72 | 37 |
| 13 | 88 ± 2 | 0.0571 | 98 ± 2 | 0.4333 | − 0.48 | 12 |
| 14 | 52 ± 2 | 0.0009 | 82 ± 6 | 0.0427 | +0.27 | 48 |
| 16 | 55 ± 4 | 0.0092 | 109 ± 3 | 0.0762 | − 1.75 | 45 |
| 17 | 61 ± 5 | 0.0154 | 116 ± 2 | 0.0163 | − 1.98 | 39 |
| 18 | 67 ± 5 | 0.0043 | 84 ± 2 | 0.0018 | − 2.13 | 33 |
| 19 | 104 ± 3 | 0.4214 | 95 ± 5 | 0.5793 | − 1.93 | −4 |
| 20 | 96 ± 2 | 0.5184 | 96 ± 4 | 0.6212 | − 2.04 | 4 |
Presented values are mean ± S.E.M. of n = 3 measurements. A Students t-test was used to assess for statistically significant changes; P<0.05 was considered significant.
C log D – calculated log D values were determined at pH 7.0 (CompuDrug, Pallas).
Fold to thalidomide (1) anti-TNF-α activity was defined as (100 − TNF-α activity % Change of every compound)/(100 − TNF-α activity % Change contrasted with 1), TNF-α activity % control for thalidomide was equal to 99% of levels achieved in the presence of vehicle. The TNF-activity of compound 17 compares favorably with that of A10 in Ref. 6c.
For the syntheses of compounds 9–14, we adopted our own novel condensation approach to allow formation of the phthalimidine ring. This one-pot reaction involved addition, rearrangement and elimination. A potential mechanism underpinning this is shown in Figure 2. The process proved to be simple and the product yield was good (ranging from 52% to 89%), thereby providing a shortcut for synthesis of 2-(2, 6-dioxopiperidin-3-yl)phthalimidine EM-12 analogues. In chemistry more generally, such dehydration spanning a nitrogen atom to form a lactam is, likewise, interesting and has not previously been reported. By contrast, previously published synthetic routes to yield EM-12 are greater than a single step, as is evident from the work of Luzzio and colleagues6f where synthesis was achieved in 6 steps in a yield of 25%. Other reports describe the use of a radical inducing reagent or UV light. In our one pot reaction, however, synthesis is undertaken at room temperature and a yield up to 80% is achievable, even if undertaken in neutral or weakly basic reaction mediums.
Figure 2.

Possible Mechanism Forming Intermediate 8
Consequent to the introduction of hetero-atoms, such as sulfur and nitrogen, into thalidomide that provided significant TNF-α inhibitory activity, we designed compound 16, in which the 1, 3-carbonyl groups of thalidomide are replaced by sulfonyl groups, and compounds 17a and 18, in which the 1, 3–dioxo and 1-oxo of thalidomide were substituted by imino groups, respectively (Scheme 2, Figure 3).
Scheme 2.

Reagents and conditions: (i) 1,2-Benzenedisulfonyl dichloride, Et3N, THF, N2, reflux, 24 h; (ii) 1,3-diiminoisoindoline, Et3N, THF, N2, reflux, 98 h; (iii) 3-iminoisoindolinone, Et3N, THF, N2, reflux, 72 h; (iv) 2,3-pyridinedicarboxylic anhydride, AcOH, N2, reflux, 7.5 h; (v) 3,4-pyridinedicarboxylic anhydride, AcOH, N2, reflux, 6.5 h.
Figure 3.

Possible Mechanism Forming Compound 17
For the synthesis of 17a, as illustrated in Figure 3, the principal product is 17.6c This can be attributed to the presence of a 1, 3-H transfer, as depicted in Figure 3, under the reaction conditions indicated. Two primary amino groups may prove more advantageous to support the intramolecular elimination of ammonia. The condensation of reactants 3-iminoisoindolinone and 15 normally afforded product 18, which has been confirmed by two correlative peaks of 3′-H/1-C and 3;-H/3-C in the (1H-detected) heteronuclear multiple-bond correlation (HMBC) spectrum. Syntheses of compounds 19 and 20 proved straightforward using the methods defined in Scheme 2, and the yield of 20 proved to be high (93%).
2.2. TNF-α inhibitory activity
Inhibition of LPS-induced TNF-α production in RAW 264.7 cells, cell viability and computed lipophilicity (C log D value) of assessed compounds 9–14, 16–20 are shown in Table 1. The biological activities of the above analogues were compared to those of revlimid (3). In addition to thalidomide (1), 3 is a credible TNF-α inhibitor,11a,b and is both approved for and effective in the for treatment of multiple myeloma and specific myelodysplastic syndromes.12a–f Herein, compounds 9, 12, 14 and 16–18 possessed more potent TNF-α inhibitory activity than that of revlimid (3) as well as thalidomide (1) in our assay model, which has now been extensively characterized.6c Indeed, compounds 9, 14 and 16 not only showed the most potency as TNF-α inhibitors amongst all eleven assayed compounds (contrasting markedly with revlimid (3)) but appeared well tolerated, albeit 14 was associated with a mild decline in cell viability at 30 μM. Parenthetically, the TNF-α activity of compound 17, whose chemistry is reported for the first time herein, compares favorably to that reported by Tweedie et al.,6c (agent A10), demonstrating the consistency of the assay across time.
The C log D values of our analogues, interestingly, ranged from lipophilic (9: +1.39) to water-soluble (16: −1.75) (Table 1), suggesting that their potency as TNF-α inhibitors related more to their structural configuration rather than to a physicochemical characteristic, such as lipophilicity, that would be predicted to augment cellular uptake. Clearly, structural configuration together with physicochemical properties impact the ability of a compound to suitably orientate, dock and then appropriately interact with a required target, such as one regulating TNF-α protein synthesis, and are thereby fundamental to its TNF-α lowering effects. However, regulation of TNF-α synthesis by thalidomide (1) and analogues is not mediated via a classical receptor or enzyme-based interaction for which structure-activity relations are generally available but, instead, appears to involve complex post-transcriptional regulatory actions mediated at the level of the 3′-UTR of TNF-α mRNA.4a,b
In general, mRNAs are amenable to several forms of post-transcriptional regulation, which include pre-mRNA splicing and maturation (3′polyadenylation and 5′ capping), mRNA nuclear export to the cytoplasm, appropriate sub-cytoplasmic localization, stabilization and translation.13a–c Two major kinds of trans acting factors that recognize specific cis elements (RNA sequences) within the target mRNA are involved in the regulation of these steps, RNA-binding proteins (RBPs) and microRNAs. For TNF-α gene expression, the role of RBPs has been widely explored, whereas the part of microRNAs remains to be more fully characterized.13c,d For inflammatory cytokines like TNF-α, in particular, but also for IL-2, IL-6 and interferon-γ, synthesis is tightly regulated at the level of mRNA stability, thereby permitting rapid responses to external stimuli, as occurs for LPS. The presence of adenylate-uridylate-rich elements (AREs) within the 3′-UTR of TNF-α mRNA plays a primary role in post-transcriptional repression, targeting it for rapid degradation or inhibition of translation.13a,b p38 MAPK has been implicated as a major signaling cascade facilitating the stability of TNF-α via its 3′-UTR ARE cis elements, which have been shown to be mediated through interactions with RBPs.13a–d In particular, proteins such as HuR, whose translocation to the cytoplasm is potently induced by oxidative stress and other aberrant stimuli,13c have been associated with promoting transcript stabilization. Upon export to the cytoplasm, HuR binds and stabilizes ARE-containing transcripts and conveys them to translational machinery. Conversely, RNA-binding proteins such as tristetraprolin (TTP) and related proteins (e.g., butyrate response factor 1 and 2) have a major regulatory role in accelerating the degradation of bound mRNAs, albeit the precise mechanisms accounting for this degradation are incompletely understood but they include the action of several cellular structures known to breakdown labile mRNAs, the proteasome, exosome and RNA processing-body.13a–c
It widely accepted that LPS challenge of RAW 264.7 cells extends the half-life of TNF-α mRNA, allowing release of its translational repression. By contrast, administration of thalidomide (1) induces an increase in translational blockade and a shortening of the TNF-α mRNA half-life from 30 min to 17 min.4a,14a,b Although yet to be elucidated, interactions between thalidomide analogs and the binding of HUR as well as TTP related RBPs with 3′-UTR cis elements likely underpins alterations in the rate of TNF-α synthesis. In this regard, small chemical inhibitors that disrupt HuR:ARE binding have been described.14c,d As yet, insufficient compounds have been assessed to provide structure-activity relations, but such a target, or a strengthening of TTP:ARE binding, represents a potential means via which thalidomide analogs may effectively regulate TNF-α protein levels.
These same properties of thalidomide analogues may or may not be vital for pharmacological interactions with other targets, such as nitric oxide,15a,b known to be regulated by thalidomide (1) and are a focus of current studies. In the event that the inhibitory activity of designed compounds proves ideal, in part or in whole, it not only provides a basis to manipulate TNF-α levels to define its pharmacological role in health and disease, but also affords structural information to allow investigation of the target and its regulatory elements.
In this regard, these TNF-α lowering data are sufficiently promising to indicate that further study of the pharmacology and biology of these agents is warranted in models of neurodegenerative diseases where a component of the disease pathology is associated with neuroinflammation.
3. Conclusion
Novel 2-(2, 6-dioxopiperidin-3-yl)phthalimidine EM-12 dithiocarbamates 9 and 10, N-substituted 3-(phthalimidin-2-yl)-2, 6-dioxopiperidines 11–14 and 3-substituted 2, 6-dioxopiperidines 16 and 18 were designed, prepared and assessed for TNF-α lowering activity. Synthesis involved a novel condensation approach for the generation of analogues of 2-(2, 6-dioxopiperidin-3-yl)phthalimidine EM-12 (2). In addition to N-substituted 3-(phthalimidin-2-yl)-2, 6-dioxopiperidines 14 and 9, 3-(o-benzenedisulfonimid-2-yl)-2, 6-dioxopiperidine 16 proved to be a potent anti-TNF-α candidate agent. These novel compounds thereby provide potential promise as immunomodulatory drugs candidates whose biological and pharmacological actions warrant further assessment in animal models of inflammatory disorders. Particularly relevant would be a neuroinflammatory focus, as invariably occurs in neurodegenerative conditions, exemplified by Alzheimer’s disease and Parkinson’s disease.
4. Experimentals
4.1. Chemistry
Melting points (uncorrected) were measured with a Fisher-Johns apparatus. 1H NMR, and 13C NMR were recorded on a Bruker (Bellevica, MA) AC-300 spectrometer. MS (m/z) data were measured on an Agilent 5973 GC-MS (CI). Elemental analyses were performed by Atlantic Microlab, Inc. (Norcross, GA). All reactions involving non-aqueous solutions were performed under an inert atmosphere.
4.1.1. 1-Hydroxymethyl-3-(tert-butoxycarbonylamino)-2, 6-dioxopiperidine (5)
A mixture of 3-(tert-butoxycarbonylamino)-2, 6-dioxopiperidine 4 (7.10 g, 31.1 mmol) and formaldehyde (37% solution in water, 37.2 ml) was refluxed under an atmosphere of nitrogen for 0.5 h. After cooling, isolated precipitate was recrystallized with acetone to afford product 5 (5.85 g, 72.9 %) as white crystals: mp 216.5–217.5 °C; 1H NMR (DMSO-d6) δ 7.20 (d, J = 6.0 Hz, 1H, NH), 6.04 (t, J = 10.5 Hz, 1H, OH), 5.05–4.92 (m, 2H, CH2OH), 4.38–4.22 (m, 1H, C3-H), 2.89–2.59 (m, 2H), 1.99–1.83 (m, 2H) and 1.41 (s, 9H, CH3) ppm; 13C NMR (DMSO-d6) δ 172.1, 171.9, 155.8, 78.5, 62.7, 51.3, 31.6, 28.5 and 23.6 ppm; MS (CI/CH4), m/z 257 (M-1).
4.1.2. 1-Chloromethyl-3-(tert-butoxycarbonylamino)-2, 6-dioxopiperidine (6)
Thionyl chloride (7.35 g, 61.8 mmole) was dropwise added to a solution of compound 5 (5.83 g, 22.6 mmole) in DMF (17 mL) at 0 °C. The mixture was reacted for 1 h maintained at the same temperature. Thereafter, it was poured onto ice (60 g). Precipitate was isolated and was washed with ice water to pH 7 ca. The crude product was recrystallized with acetone to afford 6 (5.7 g, 90.5 %) as a white solid: mp 134.0–135.0 °C; 1H NMR (DMSO-d6) δ 7.31 (d, J = 6.1 Hz, 1H, NH), 5.49 (s, 2H, CH2Cl), 4.58–4.30 (m, 1H, C3-H), 3.08–2.67 (m, 2H), 2.10–1.87 (m, 2H) and 1.48 (s, 9H, CH3) ppm; 13C NMR (DMSO-d6) δ 171.5, 171.1, 155.8, 78.7, 51.3, 48.5, 31.4, 28.5 and 23.3 ppm; MS (CI/CH4), m/z 276 (M+).
4.1.3. 3-Amino-1-chloromethyl-2, 6-dioxopiperidine trifluoroacetate (7)
Trifluoroacetic acid (28.4 mL) was dropwise added into a solution of compound 6 (4.0 g, 14.5 mmol) in dichloromethane (274 mL) at room temperature. The mixture was reacted for 22.5 h under an atmosphere of nitrogen at the same temperature. Thereafter, it was concentrated and precipitated with ether. Isolated solid was dried overnight to afford product 7 (4.05 g, 96.2 %) as a purplish salt; 1H NMR (DMSO-d6) δ 8.80 (s, 3H, NH3+), 5.56 and 5.50 (AB system, J = 9.2 Hz, 2H, CH2Cl), 4.51–4.28 (m, 1H, C3-H), 3.01–2.71 (m, 2H) and 2.31–1.90 (m, 2H) ppm; 13C NMR (DMSO-d6) δ 170.2, 169.2, 159.0, 158.5, 49.9, 48.1, 30.6 and 21.3 ppm; MS (CI/CH4), m/z 288 (M-2).
4.1.4. 2-(1-Chloromethyl-2, 6-dioxopiperidin-3-yl)phthalimidine (8)
A mixture of phthaldialdehyde (2.74 g, 20.4 mmol) and compound 7 (5.94 g, 20.4 mmol) in THF (1.7 L) was stirred under an atmosphere of nitrogen at room temperature for 71 h. Thereafter, solvent was removed, and the crude product was purified with chromatography on silica gel (CH3CN/CH2Cl2 = 1/4) to afford product 8 (3.1 g, 51.7 %) as a white solid: mp 180.5–181.0 °C; 1H NMR (CDCl3) δ 7.94–7.43 (m, 4H, Ar-H), 5.59 and 5.51 (AB system, J = 8.1 Hz, 2H, CH2Cl), 5.30–5.19 (m, 1H, C3′-H), 4.50 and 4.36 (AB system, J = 16.5 Hz, 2H, C3-H), 3.15–2.85 (m, 2H) and 2.46–2.13 (m, 2H) ppm; 13C NMR (CDCl3) δ 169.4, 169.3, 168.6, 141.4, 132.1, 131.3, 128.3, 124.2, 122.9, 52.5, 47.2, 47.1, 31.9 and 22.3 ppm; MS (CI/CH4), m/z 294 (M+2). Anal. Calcd for C14H13ClN2O3: C, 57.44; H, 4.48; N, 9.57. Found: C, 57.37; H, 4.46; N, 9.17.
4.1.5. [3-(1-Oxo-1, 3-dihydro-1H-isoindol-2-yl)-2, 6-dioxopiperidin-1-yl]methyl cyclohexyldithiocarbamate (9)
A mixture of compound 8 (37.0 mg, 0.126 mmol), carbon disulfide (19.2 mg, 0.252 mmol) and cyclohexylamine (25.1 mg, 0.253 mmol) in acetonitrile (7 mL) was reacted for 42.5 h under an atmosphere of nitrogen at room temperature. After removing solvent, the residues were separated with chromatography on silica gel (MeOH/CH2Cl2 = 1/20) to afford product 9 (36.0 mg, 66.7%) as a yellow crystals: mp 128.0–129.0 °C; 1H NMR (CDCl3) δ 8.75 (s, 1H, NH), 7.95–7.38 (m, 4H, Ar-H), 5.31–5.01 (m, 3H, C3′-H and CH2S), 4.49–4.25 (m, 2H, C3-H) and 3.14–1.05 (m, 15H, C5,4-H and cyclohex-H) ppm; 13C NMR (CDCl3) δ 191.2, 171.3, 170.2, 169.3, 141.3, 132.1, 131.3, 128.3, 124.2, 122.9, 55.8, 52.6, 47.4, 43.3, 31.8, 31.4, 25.3, 24.7 and 22.3 ppm; MS (CI/CH4), m/z 432 (MH+). Anal. Calcd for C21H25N3O3S2: C, 58.44; H, 5.84; N, 9.74. Found: C, 58.05; H, 5.88; N, 9.47.
4.1.6. [3-(1-Oxo-1, 3-dihydro-1H-isoindol-2-yl)-2, 6-dioxopiperidin-1-yl]methyl piperidin-1-carbodithioate (10)
A mixture of compound 8 (33.0 mg, 0.113 mmol), carbon disulfide (17.2 mg, 0.226 mmol) and piperidine (19.2 mg, 0.225 mmol) in acetonitrile (6 mL) was reacted for 46.0 h under an atmosphere of nitrogen at room temperature. After removing solvent, the residues were separated with chromatography on silica gel (MeOH/CH2Cl2 = 1/15) to afford product 10 (33.5 mg, 71.0 %) as a white powder: mp 180.0–181.5 °C; 1H NMR (CDCl3) δ 7.90–7.41 (m, 4H, Ar-H), 5.79 and 5.68 (AB system, J = 14.5 Hz, 2H, CH2S), 5.29–5.18 (m, 1H, C3′-H), 4.49 and 4.30 (AB system, J = 16.5 Hz, 2H, C3-H), 4.24 (s, br, 2H, pip-C2,6-H), 3.81 (s, br, 2H, pip-C2,6-H), 3.09–2.81 (m, 2H), 2.41–2.11 (m, 2H) and 1.79–1.54 (m, 6H, pip-C3,4,5-H) ppm; 13C NMR (CDCl3) δ 192.7, 170.3, 169.2, 141.4, 131.9, 131.4, 128.1, 124.0, 122.8, 52.4, 51.6, 47.1, 46.4, 32.0, 25.8, 24.1 and 22.5 ppm; MS (CI/CH4), m/z 258 (M-159). Anal. Calcd for C20H23N3O3S2: C, 57.53; H, 5.55; N, 10.06. Found: C, 57.80; H, 5.38; N, 9.95.
4.1.7. N-(1, 3-dioxopiperidin-2-yl)methyl-3-(1-oxo-1, 3-dihydro-1H-isoindol-2-yl)-2, 6-dioxopiperidine (11)
A mixture of glutarimide (36.0 mg, 0.318 mmol) and potassium hydroxide (42 mg, 0.750 mmol) in acetonitrile (42 mL) was stirred for an hour at room temperature. Thereafter, compound 8 (93.0 mg, 0.318 mmol) was added to the reaction system and stirred for a further 17 h under an atmosphere of nitrogen at the same temperature. After removing solvent, the residues were separated with chromatography on silica gel (CH3CN/CH2Cl2 = 1/3) to afford product 11 (68.0 mg, 57.9 %) as a white solid: mp 216.5–217.0 °C; 1H NMR (CDCl3) δ 7.88 (d, J = 9.0 Hz, 1H, C7-H), 7.57 (d, J = 8.5 Hz, 1H, C4-H), 7.46 (t, J = 9.0 Hz, 2H, C5,6-H), 5.85 (s, 2H, CH2N2), 5.19–5.10 (m, 1H, C3′-H), 4.52 and 4.35 (AB system, J = 16.5 Hz, 2H, C3-H), 3.08–1.81 (m, 10H, C5′,4′-H and glu-H) ppm; 13C NMR (CDCl3) δ 171.9, 170.3, 169.4, 169.3, 141.6, 132.0, 131.7, 128.2, 124.1, 123.0, 52.8, 47.4, 45.0, 32.9, 32.2, 22.8 and 17.0 ppm; MS (CI/CH4), m/z 300 (M-69). Anal. Calcd for C19H19N3O5H2O: C, 58.91; H, 5.46; N, 10.85. Found: C, 59.17; H, 5.38; N, 10.61.
4.1.8. N-(1, 3-dithioxopiperidin-2-yl)methyl-3-(1-oxo-1, 3-dihydro-1H-isoindol-2-yl)-2, 6-dioxopiperidine (12)
A mixture of dithioglutarimide (46.2 mg, 0.318 mmol), potassium hydroxide (42 mg, 0.750 mmol) and compound 8 (93.0 mg, 0.318 mmol) in acetonitrile (42 mL) was stirred for 16 h under an atmosphere of nitrogen at room temperature. After removing solvent, the residues were separated with chromatography on silica gel (CH3CN/CH2Cl2 = 1/3) to afford product 12 (33.0 mg, 25.8 %) as a yellow solid: mp 218.0–220.0 °C; 1H NMR (CDCl3) δ 7.91–7.43 (m, 4H, Ar-H), 5.22–5.12 (m, 1H, C3′-H), 5.07 and 4.94 (AB system, J = 10.1 Hz, 2H, CH2N2), 4.50 and 4.39 (AB system, J = 16.5 Hz, 2H, C3-H), 3.09–1.89 (m, 10H, C5′,4′-H and thioglu-H) ppm; 13C NMR (CDCl3) δ 201.7, 170.9, 169.8, 169.5, 141.6, 132.2, 131.6, 128.4, 124.4, 123.1, 53.1, 48.1, 43.6, 37.6, 32.0, 22.6 and 21.2 ppm; MS (CI/CH4), m/z 404 (M+3). Anal. Calcd for C19H19N3O3S2H2O: C, 54.40; H, 5.04; N, 10.01. Found: 54.08; H, 4.42; N, 9.48.
4.1.9. N-Phthalimidomethyl-3-(1-oxo-1, 3-dihydro-1H-isoindol-2-yl)-2, 6-dioxopiperidine (13)
A mixture of phthalimide (46.8 mg, 0.318 mmol) and potassium hydroxide (42 mg, 0.750 mmol) in acetonitrile (42 mL) was stirred for an hour at room temperature. Thereafter, compound 8 (93.0 mg, 0.318 mmol) was added to the reaction system and stirred for another 17 h under an atmosphere of nitrogen at the same temperature. After removing solvent, the residues were separated with chromatography on silica gel (CH3CN/CH2Cl2 = 1/3) to afford product 13 (67.0 mg, 52.2 %) as a white solid: mp 209.5–210.5 °C; 1H NMR (CDCl3) δ 7.95–7.40 (m, 8H, Ar-H), 5.83 and 5.72 (AB system, J = 13.5 Hz, 2H, CH2N2), 5.39–5.23 (m, 1H, C3′-H), 4.54 and 4.34 (AB system, J = 16.6 Hz, 2H, C3-H), 3.10–2.82 (m, 2H) and 2.46–2.11 (m, 2H) ppm; 13C NMR (CDCl3) δ 170.3, 169.6, 169.3, 167.0, 141.7, 134.3, 132.0, 131.7, 131.6, 128.2, 124.2, 123.6, 123.0, 52.5, 47.1, 43.6, 32.1 and 22.7 ppm; MS (CI/CH4), m/z 390 (M-13). Anal. Calcd for C22H17N3O5: C, 65.50; H, 4.25; N, 10.42. Found: C, 65.32; H, 4.12; N, 10.21.
4.1.10. N-Dithiophthalimidomethyl-3-(1-oxo-1, 3-dihydro-1H-isoindol-2-yl)-2, 6-dioxopiperidine (14)
A mixture of dithiophthalimide (57.0 mg, 0.318 mmol) and potassium hydroxide (42 mg, 0.750 mmol) in acetonitrile (42 mL) was stirred for an hour at room temperature. Thereafter, compound 8 (93.0 mg, 0.318 mmol) was added to the reaction system and stirred for another 17 h under an atmosphere of nitrogen at the same temperature. After removing solvent, the residues were separated with chromatography on silica gel (CH3CN/CH2Cl2 = 1/3) to afford product 14 (17.0 mg, 12.3 %) as a yellow gum. 1H NMR (CDCl3) δ 7.98–7.39 (m, 8H, Ar-H), 6.47 (s, 2H, CH2N2), 5.31–5.19 (m, 1H, C3′-H), 4.49 and 4.32 (AB system, J = 16.3 Hz, 2H, C3-H), 3.09–2.79 (m, 2H) and 2.49–2.07 (m, 2H) ppm; 13C NMR (CDCl3) δ 197.0, 170.4, 169.3, 169.1, 141.5, 134.8, 133.5, 132.0, 131.6, 128.2, 124.2, 123.6, 123.0, 52.5, 49.1, 47.1, 32.2 and 22.7 ppm; MS (CI/CH4), m/z 383 (M-52). Anal. Calcd for C22H17N3O3S21/3 H2O: C, 59.87; H, 4.03; N, 9.52. Found: C, 59.91; H, 3.71; N, 9.17.
4.1.11. 3-(o-Benzenedisulfonimid-2-yl)-2, 6-dioxopiperidine (16)
A mixture of 1, 2-benzenedisulfonyl dichloride (1.00 g, 3.63 mmol), 3-amino-2, 6-dioxopiperidine trifluoroacetate 15 (0.88 g, 3.63 mmol) and triethylamine (1.10 g, 10.87 mmol) in anhydrous THF (100 mL) was first reacted for 1.5 h at room temperature under an atmosphere of nitrogen, and, thereafter, it was refluxed for 24 h under an atmosphere of nitrogen. Following cooling, a grey crude product was filtered. This was recrystallized from hot acetone to provide product 16 (0.77 g, 64.2 %) as a purplish powder: mp 253 °C (dec.); 1H NMR (DMSO-d6) δ 11.08 (s, 1H, NH), 8.40–8.01 (m, 4H, Ar-H), 5.51–5.40 (m, 1H, C3′-H), 2.96–2.51 (m, 2H) and 2.50–2.30 (m, 2H) ppm; 13C NMR (DMSO-d6) δ 172.5, 169.1, 137.4, 135.8, 122.3, 59.0, 31.5 and 24.7 ppm; MS (CI/CH4), m/z 331 (MH+). Anal. Calcd for C11H10N2O6S2: C, 39.99; H, 3.05; N, 8.48. Found: C, 40.57; H, 3.22; N, 8.21.
4.1.12. 3-(1, 3-Diimino-1, 3-dihydro-2H-isoindol-1N-yl)-2, 6-dioxopiperidine (17)
A mixture of 1, 3-diiminoisoindoline (0.50 g, 3.44 mmol), 3-amino-2, 6-dioxopiperidine trifluoroacetate 15 (0.83 g, 3.43 mmol) and triethylamine (0.95 g, 9.39 mmol) in THF (200 mL) was refluxed for 98 h under an atmosphere of nitrogen. After cooling, a grey crude product was filtered. This was recrystallized from hot acetone to afford product 17 (0.67 g, 76.1 %) as a gray powder: mp 246 °C (dec.); 1H NMR (DMSO-d6) δ 10.72 (s, 1H, CONH), 8.51 and 8.39 (sh, 2H, CCNH and C=NH), 7.90–7.50 (m, 4H, Ar-H), 5.01 (t, J = 5.5 Hz, 1H, C3′-H), 2.61 (m, 2-H) and 2.04 (m, 2-H) ppm; 13C NMR (DMSO-d6) δ 173.8, 173.5, 170.6, 168.5, 139.8, 136.1, 131.1, 130.3, 121.5, 121.0, 59.2, 30.3 and 26.3 ppm; MS (CI/CH4), m/z 257 (MH+). Anal. Calcd for C13H12N4O2: C, 60.93; H, 4.72; N, 21.86. Found: C, 60.89; H, 4.82; N, 21.58.
4.1.13. 3-(1-Oxo-3-imino-1, 3-dihydro-2H-isoindol-2-yl)-2, 6-dioxopiperidine (18)
A mixture of 3-iminoisoindolinone (0.50 g, 3.42 mmol), 3-amino-2, 6-dioxopiperidine trifluoroacetate 15 (0.83 g, 3.43 mmol) and triethylamine (0.94 g, 9.29 mmol) in THF (200 mL) was refluxed for 72 h under an atmosphere of nitrogen. After removing solvent, crude product was recrystallized from hot acetone to afford product 18 (0.38 g, 43.2 %) as a white powder: mp 267 °C (dec.); 1H NMR (DMSO-d6) δ 11.15, 10.90 (2s, 2H, 2NH), 7.88–7.68 (m, 4H, Ar-H), 4.46 (t, J = 7.6 Hz, 1H, C3′-H), 2.71–2.59 (m, 2H) and 2.22–2.08 (m, 2H) ppm; 13C NMR (DMSO-d6) δ 173.4, 172.0, 169.2, 152.5, 136.6, 133.9, 132.5, 131.5, 123.1, 122.3, 59.5, 30.4 and 26.1 ppm; MS (CI/CH4), m/z 257 (M+). Anal. Calcd for C13H11N3O3: C, 60.70; H, 4.31; N, 16.33. Found: C, 60.67; H, 4.38; N, 16.20.
4.1.14. 3-(4-Aza-1, 3-dioxo-1, 3-dihydro-2H-isoindol-2-yl)-2, 6-dioxopiperidine (19)
A mixture of 2, 3-pyridinedicarboxylic anhydride (1.0 g, 6.71 mmol) and 3-amino-2, 6-dioxopiperidine trifluoroacetate 15 (1.63 g, 6.73 mmol) in acetic acid (51 mL) was refluxed under an atmosphere of nitrogen for 7.5 h. After removing solvent, crude product was recrystallized from acetone to afford product 19 (0.18 g, 10.3 %) as white needle crystals: mp 266.0–267.5 °C; 1H NMR (CDCl3) δ 8.95 (s, br, 1H, C5-H), 8.13 (d, J = 9.3 Hz, 1H, C7-H), 8.00 (s, 1H, NH), 7.61 (s, br, 1H, C6-H), 5.11–4.91 (m, 1H, C3′-H), 2.99–2.65 (m, 3H) and 2.21–2.09 (m, 1H) ppm; 13C NMR (CDCl3) δ 170.2, 167.2, 165.2, 165.0, 155.7, 151.3, 131.6, 127.7, 127.2, 49.6, 31.3 and 22.5 ppm; MS (CI/CH4), m/z 259 (M+). Anal. Calcd for C12H9N3O4: C, 55.60; H, 3.50; N, 16.21. Found: C, 55.03; H, 3.62; N, 15.74.
4.1.15. 3-(5-Aza-1, 3-dioxo-1, 3-dihydro-2H-isoindol-2-yl)-2, 6-dioxopiperidine (20)
A mixture of 3, 4-pyridinedicarboxylic anhydride (1.0 g, 6.71 mmol) and 3-amino-2, 6-dioxopiperidine trifluoroacetate 15 (1.63 g, 6.73 mmol) in acetic acid (51 mL) was refluxed under an atmosphere of nitrogen for 6.5 h. After removing solvent, crude product was recrystallized from acetone to afford product 20 (1.62 g, 93.1 %) as white crystals: mp 234.0–235.5 °C; 1H NMR (DMSO-d6) δ 11.18 (s, 1H, NH), 9.29–9.10 (m, 2H, C4-H, C6-H), 7.99 (d, J = 7.8 Hz, 1H, C7-H), 5.31–5.15 (m, 1H, C3′-H), 3.05–2.39 (m, 3H) and 2.19–2.02 (m, 1H) ppm; 13C NMR (DMSO-d6) δ 173.0, 169.9, 166.8, 166.4, 156.7, 144.7, 139.1, 125.7, 117.5, 49.6, 31.2 and 22.1 ppm; MS (CI/CH4), m/z 259 (M+). Anal. Calcd for C12H9N3O4: C, 55.60; H, 3.50; N, 16.21. Found: C, 55.65; H, 3.56; N, 15.99.
4.2. Biological assay
RAW 264.7 cells were cultured and treated with thalidomide analogues and LPS as has been previously described.6c In a manner similar to its use herein in cell culture, LPS has been found to elevate TNF-α levels in animals – both systemically as well as within the brain, and to be associated with neurodegenerative disease onset.16a,b The CellTiter 96 Aqueous One Solution Cell Proliferation Assay (Promega, Madison, WI) is a commercially used assay that is widely used to quantitatively determine cell proliferation, and this assay was utilized in our studies in line with the manufacturer’s guidelines. Changes in cellular health status can be determined by use of indirect measures related to the formation of a colored tetrazolium dye product that can be measured spectrophotometrically at 490 nm. An increase in absorbance at 490 nm is indicative of an increase in cell numbers and thus cell proliferation; and a decrease in absorbance is indicative of cell death. Data are expressed as a percentage change in Optical Densities (O.D.) compared to the appropriate control values. TNF-α protein levels were measured in culture media by use of an ELISA specific for mouse TNF-α protein (BioLegend MAX™ Mouse TNF-α ELISA Kit, BioLegend, San Diego, CA) and are expressed as a percentage change from the appropriate control.
Acknowledgments
This work was supported in part by the Intramural Research Program of the National Institute on Aging, National Institutes of Health (NIH) and by NIH grants (AG18379 and AG18884) to D.K.L. The authors declare that they have no conflicts of interest regarding the contents of this manuscript, and are grateful to Dr. Amy Newman and colleagues of the Medicinal Chemistry Section, National Institute on Drug Abuse, NIH, for use of NMR equipment.
References and notes
- 1.Irma MS. Alzheimer’s & Dementia. 2009;5:234. doi: 10.1016/j.jalz.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 2.(a) Frank-Cannon TC, Alto LT, McAlpine FE, Tansey MG. Mol Neurodegen. 2009;16:47. doi: 10.1186/1750-1326-4-47. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Frankola KA, Greig NH, Luo W, Tweedie D. CNS Neurol Disord Drug Targets. 2011;10:391. doi: 10.2174/187152711794653751. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Reale M, Greig NH, Kamal MA. Mini Rev Med Chem. 2009;9:1229. doi: 10.2174/138955709789055199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Walker SL, Waters MF, Lockwood DN. Lepr Rev. 2007;78:197. [PubMed] [Google Scholar]; (b) Van Veen NH, Lockwood DN, van Brakel WH, Ramirez J, Jr, Richardus JH. Cochrane Database Syst Rev. 2009;3:CD006949. doi: 10.1002/14651858.CD006949.pub2. [DOI] [PubMed] [Google Scholar]; (c) Ladizinski B, Shannon EJ, Sanchez MR, Levis WR. J Drugs Dermatol. 2010;9:814. [PubMed] [Google Scholar]; (d) Sissung TM, Thordardottir S, Gardner ER, Figg WD. Anticancer Agents Med Chem. 2009;9:1058. doi: 10.2174/187152009789735017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Moreira AL, Sampaio EP, Zmuidzinas A, Frindt P, Smith KA, Kaplan G. J Exp Med. 1993;177:1675. doi: 10.1084/jem.177.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sampaio EP, Sarno EN, Gallily R, Cohn ZA, Kaplan G. J Exp Med. 1991;173:699. doi: 10.1084/jem.173.3.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pillemer SR, Leakan RA, Sankar V, Manny J, Baum BJ. Arthritis Care & Research. 2004;51:505. doi: 10.1002/art.20416. [DOI] [PubMed] [Google Scholar]
- 6.(a) Zhu X, Giordano T, Yu QS, Holloway HW, Perry TA, Lahiri DK, Brossi A, Greig NH. J Med Chem. 2003;46:5222. doi: 10.1021/jm030152f. [DOI] [PubMed] [Google Scholar]; (b) Luo W, Yu QS, Tweedie D, Deschamps j, Parrish D, Holloway HW, Li Y, Brossi A, Greig NH. SYNTHESIS. 2008;21:3415. [Google Scholar]; (c) Tweedie D, Luo W, Short RG, Brossi A, Holloway HW, Li Y, Yu QS, Greig NH. J Neuroscience Methods. 2009;183:182. doi: 10.1016/j.jneumeth.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Baratz R, Tweedie D, Rubovitch V, Luo W, Hoffer BJ, Greig NH, Pick CG. J Neurochem. 2011 doi: 10.1111/j.1471-4159.2011.07377.x. In review. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Kaplan G, Sampaio EP. 5,385,901. US Patent. 1995; Chem Abstr. 1992;117:226313a. [Google Scholar]; (f) Luzzio FA, Mayorov AV, Ng SSW, Kruger EA, Figg WD. J Med Chem. 2003;46:3793. doi: 10.1021/jm020079d. [DOI] [PubMed] [Google Scholar]
- 7.Zahran MA-H, Salem TA-R, Samaka RM, Agwa HS, Awad AR. Bioorg & Med Chem. 2008;16:9708. doi: 10.1016/j.bmc.2008.09.071. [DOI] [PubMed] [Google Scholar]
- 8.(a) Caladas ED, Hosana CM, Miranda MCC, Souzana L, Lima JF. J Agric Food Chem. 2001;49:4521. doi: 10.1021/jf010124a. [DOI] [PubMed] [Google Scholar]; (b) Erian AW, Sherif SM. Tetrahedron. 1999;55:7957. [Google Scholar]; (c) Wood TF, Gardner JH. J Am Chem Soc. 1941;63:2741. [Google Scholar]; (d) Bowden K, Chana RS. J Chem Soc, Perkin Trans 2. 1990:2163. [Google Scholar]; (e) Beji M, Sbihi H, Baklouti A, Cambon AJ. Fluorine Chem. 1999;99:17. [Google Scholar]; (f) Goal A, Mazur SJ, Fattah RJ, Hartmam TL, Turpin JA, Huang M, Rice WG, Appella E, Inman JK. Bioorg Med Chem Lett. 2002;12:767. doi: 10.1016/s0960-894x(02)00007-0. [DOI] [PubMed] [Google Scholar]
- 9.Zhang H. CODEN: PIXXD2 WO 2006105697 A1 20061012 CAN 145: 397376 AN 2006: 1065842. China PCT Int Appl. 2006
- 10.Anastas PT, Warner JC. Green Chemistry: Theory and Practice. Oxford University Press; New York: 1998. p. 30. [Google Scholar]
- 11.(a) Chaulet C, Croix C, Alagille D, Normand S, Delwail A, Favot L, Lecron JC, Viaud-Massuard MC. Bioorg Med Chem Lett. 2011;21:1019. doi: 10.1016/j.bmcl.2010.12.031. [DOI] [PubMed] [Google Scholar]; (b) Gordon JN, Prothero JD, Thornton CA, Pickard KM, Di Sabatino A, Goggin PM, Pender SL, Macdonald TT. J Crohns Colitis. 2009;3:175. doi: 10.1016/j.crohns.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 12.(a) Muller GW, Sirling DI, Chen R. 5,635,517. US Patent. 1997; (b) Muller GW, Chen RS, Huang SY, Corral LG, Wong LM, Patterson RT, Chen Y, Kaplan G, Stirling D. Bioorg & Med Chem Lett. 1999;9:1625. doi: 10.1016/s0960-894x(99)00250-4. [DOI] [PubMed] [Google Scholar]; (c) Corral LG, Haslett PA, Muller GW, Chen R, Wong LM, Ocampo CJ, Patterson RT, Stirling DI, Kaplan G. J Immunol. 1999;163:380. [PubMed] [Google Scholar]; (d) Kiaei M, Petri S, Kipiani K, Gardian G, Choi DK, Chen J, Calingasan NY, Schafer P, Muller GW, Stewart C, Hensley K, Beal MF. J Neuroscience. 2006;26:2467. doi: 10.1523/JNEUROSCI.5253-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Muller GW, Chen R, Saindane MT, Ge C. 20060052609. US Patent Application. 2006; (f) Armoiry X, Aulagner G, Facon T. J Clin Pharmacy Therapeutics. 2008;33:219. doi: 10.1111/j.1365-2710.2008.00920.x. [DOI] [PubMed] [Google Scholar]
- 13.(a) Khera TK, Dick AD, Nicholson LB. Prog Retin Eye Res. 2010;29:610. doi: 10.1016/j.preteyeres.2010.08.003. [DOI] [PubMed] [Google Scholar]; (b) Patil CS, Liu M, Zhao W, Coatney DD, Li F, VanTubergen EA, D’Silva NJ, Kirkwood KL. Mol Ther. 2008;16:1657. doi: 10.1038/mt.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Abdelmohsen K, Kuwano Y, Kim HH, Gorospe M. Biol Chem. 2008;389:243. doi: 10.1515/BC.2008.022. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Stamou P, Kontoyiannis DL. Curr Dir Autoimmun. 2010;11:61. doi: 10.1159/000289197. [DOI] [PubMed] [Google Scholar]
- 14.(a) Han JH, Beutler B, Huez G. Biochim Biophys Acta. 1991;1090:22. doi: 10.1016/0167-4781(91)90032-h. [DOI] [PubMed] [Google Scholar]; (b) Seko Y, Cole S, Kasprzak W, Shapiro BA, Ragheb JA. Autoimmun Rev. 2006;5:299. doi: 10.1016/j.autrev.2005.10.013. [DOI] [PubMed] [Google Scholar]; (c) Chae M-J, Sung HY, Kim E-H, Lee M, KwaK H, Chae CH, Kim S, Park W-Y. Exp Mol Med. 2009;41:824. doi: 10.3858/emm.2009.41.11.088. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Cheneval D, Kastelic C, Fuerst P, Parker CN. J Biomol Screen. 2010;15:609. doi: 10.1177/1087057110365897. [DOI] [PubMed] [Google Scholar]
- 15.(a) Park E, Levis WR, Greig NH, Jung E, Schuller-Levis G. J Drugs Dermatol. 2010;9:330. [PMC free article] [PubMed] [Google Scholar]; (b) Naureckiene S, Edris W, Ajit SK, Katz AH, Sreekumar K, Rogers KE, Kennedy JD, Jones PG. J Pharmacol Toxicol Methods. 2007;55:303. doi: 10.1016/j.vascn.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 16.(a) Banks WA, Robinson SM. Brain Behav Immun. 2010;24:102. doi: 10.1016/j.bbi.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jaeger LB, Dohgu S, Sultana R, Lynch JL, Owen JB, Erickson MA, Shah GN, Price TO, Fleegal-Demotta MA, Butterfiled DA, Banks WA. Brain Behav Immun. 2009;23:507. doi: 10.1016/j.bbi.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]