

Abstract
Previously, we have shown that N,N-dimethylacetamide (DMA) prevents inflammation-induced preterm birth in a murine model, inhibits lipopolysaccharide (LPS)-induced increases in placental proinflammatory cytokines and upregulates the antiinflammatory cytokine interleukin-10 (IL-10). However, DMA’s mechanism of action remains to be elucidated. In the current study, we investigate how DMA produces its antiinflammatory effect. Using in vitro and ex vivo models, we show that DMA suppresses secretion of proinflammatory cytokines in lipopolysaccharide (LPS)-induced RAW 264.7 cells, TNFα-challenged JEG-3 cells and LPS-stimulated human placental explants. DMA significantly attenuated secretion of TNFα, IL-6, IL-10 and granulocyte macrophage colony stimulating factor (GM-CSF) from LPS-stimulated RAW 264.7 cells; IL-6 secretion from TNFα-stimulated JEG-3 cells; and TNFα, IL-6, IL-10, GM-CSF and Interleukin-8 (IL-8) from LPS-stimulated human placental explants. We further investigated whether DMA’s effect on cytokine expression involves the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and mitogen-activated protein kinase (MAPK) pathways. DMA (10 mM) significantly inhibited degradation of nuclear factor of kappa light polypeptide gene enhancer in B cell inhibitor α (IκBα) in LPS-stimulated RAW 264.7 cells, but there was no significant change in the expression of phosphorylated or native forms of downstream proteins in the MAPK pathway. In addition, DMA significantly attenuated luciferase activity in cells co-transfected with NF-κB-Luc reporter plasmid, but not with AP-1-Luc or CEBP-Luc reporters. Overall, our findings suggest that the antiinflammatory activity of DMA is mediated by inhibition of the NF-κB pathway via decreased IκBα degradation.
INTRODUCTION
Inflammation has now been implicated in a broad spectrum of disorders, ranging from cardiovascular to neurodegenerative disease (1,2). Infection and its resultant inflammation have been identified as the most common cause of spontaneous preterm birth (PTB) (3–5), which accounts for the majority of perinatal morbidity and mortality (4,6). Ascending infection and chorioamnionitis trigger the innate immune system, resulting in an inflammatory response that initiates the process of parturition (3–7). This paradigm has been the basis of recently developed in vivo models of inflammation-driven preterm birth, where preterm labor is induced by injecting lipopolysaccharide (LPS) into the uterine horn (8,9). While the incidence of PTB has increased in the United States over the last few decades, the availability of pharmacotherapy effective in preventing it remains elusive (5).
Toll-like receptors (TLRs) comprise an important subset of the pattern recognition receptors that trigger inflammatory signaling pathways controlled by nuclear factor kappa B (NF-κB) (10–15). Activation of TLRs results in a cascade of intracellular steps leading to activation of the inhibitory kappa B kinase (IKK) complex (16,17). This ultimately results in phosphorylation of IκBα, which is subsequently ubiquitinated and then degraded by the proteasome. Degradation of the inhibitory molecule IκBα releases the NF-κB dimer and allows its translocation to the nucleus (13). Nuclear translocation of NF-κB triggers the production of proinflammatory cytokines such as tumor necrosis factor (TNF)α and interleukin (IL)-1, which regulate the innate immune response and inflammation (13–15,18–21). Cytokines such as IL-1β, IL-6 and TNFα in turn increase the transcription of genes that mediate tissue remodeling, such as matrix metalloproteinases (MMPs) (22), and also increase synthesis of prostaglandins and other inflammatory mediators (23–27). In inflammatory cells, activation of NF-κB leads to the induction of inducible nitric oxide synthase (iNOS), which in turn leads to production of the powerful mediator nitric oxide (28). This well-orchestrated mechanism ultimately results in myometrial contraction, cervical ripening and rupture of membranes, leading to PTB (19,29–31). Therefore, NF-κB plays a central role in inflammation-associated PTB and represents one potential target for pharmacotherapeutic approaches aimed at controlling preterm labor and delivery (32–34).
Previously in our lab, we have shown that N, N-dimethylacetamide, a commonly available organic solvent, at a dose of 1.6 g/kg, significantly delayed PTB and rescued pups from being delivered prematurely in timed-pregnant C57BL/6 mice administered LPS (35). This study also examined the effect of DMA on expression of pro- and antiinflammatory cytokines and chemokines in mouse placental and uterine tissues. DMA was shown to downregulate proinflammatory cytokines and upregulate antiinflammatory cytokines in these tissues, suggestive of antiinflammatory action. These previous data led to the current work, in which we (1) attempt to elucidate the antiinflammatory mechanism of DMA in vitro and (2) further establish DMA’s antiinflammatory potential in the treatment of PTB by investigating its effect on inflamed human placental explants. In addition, the present work also investigates the effect of DMA on the canonical NF-κB and MAPK inflammatory pathways.
MATERIALS AND METHODS
Cell Culture
RAW 264.7 (American Type Culture Collection) and HEK-293 cells overexpressing TLR4, MD2 and CD14 genes (American Type Culture Collection, previously transfected by VP) were maintained in complete growth medium (CGM), using Dulbecco’s modified Eagle medium (DMEM) (Cellgro), while RPMI 1640 (Cellgro) was used for JEG-3 cells (American Type Culture Collection). Both media were supplemented with 10% fetal bovine serum (Atlanta Biologicals) and 1% penicillin-streptomycin (Cellgro). The serum-free media used to starve JEG-3 cells prior to experiments was composed of RPMI 1640 with 0.1% bovine serum albumin (BSA) and 1% penicillin-streptomycin. All cell lines were maintained at 37°C and 5% CO2 and grown to 80–90% confluence before subculturing or being used in experiments.
Cell Viability Assay
To determine the effect of the experimental conditions on cell viability, 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) (Alfa Aesar) assays were performed. RAW 264.7, HEK-293 and JEG-3 cells were seeded at 20,000, 25,000 and 30,000 cells/well, respectively, in 96-well plates. Following overnight incubation, cells were incubated with DMA (0.1, 1, and 10 mM) (Acros Organics), NF-κB inhibitor BAY 11-7082 (5 μM) (Enzo Lab Sciences), mitogen-activated protein kinase (MAPK) inhibitor SB202190 (25 μM, inhibiting virtually all MAPK activity) (Adipogen) or CGM for two hours. To mimic experimental conditions, RAW 264.7 and HEK-293 cells were incubated with LPS (1μg mL−1) (Sigma) from Escherichia coli 026:B6 and JEG-3 cells were incubated with recombinant human TNFα (20 ng mL−1) (R&D Systems). Twenty- four hours post-incubation, MTT was added to a final concentration of 0.5% and incubated further for two hours at 37°C and 5% CO2. The formed purple crystals were dissolved using dimethyl sulfoxide (DMSO) (BDH). The absorbance of the resulting purple solution at 570 nm was measured using an Opsys MR microplate reader (Dynex Technologies). At least three independent experiments were performed in quadruplicate wells.
Nitric Oxide Assay
Nitrite levels in cell culture supernatant were determined by Griess assay (Promega). Nitrite (NO−2) is a stable metabolite of NO in biological systems (36). Samples were prepared by seeding 1.5 × 106 RAW 264.7 cells in 25 cm2 flasks. After overnight incubation, cells were incubated for 2 h with DMA (0.1, 1, and 10 mM), BAY 11-7082 (5 μM, equal to its IC50 (37) or CGM. Cells were then incubated with LPS (1 μg mL−1) for 24 h. Three independent experiments were performed in duplicate. The Griess assay was performed following the manufacturer’s protocol. Nitrite concentrations in the samples were derived by linear regression from a standard curve generated with known concentrations of sodium nitrite as described by the manufacturer.
In Vitro Assay of Secreted Cytokines and Chemokines
RAW 264.7 and JEG-3 cells were seeded at 1.5 × 106 and 2 × 106 cells per 25 cm2 flask, respectively. RAW 264.7 cells were incubated overnight prior to incubation with DMA. JEG-3 cells were starved after the overnight incubation, 12–16 h prior to incubation with DMA, by replacing the CGM with serum-free media (SFM) (DMEM supplemented with 0.1% BSA and 1% penicillin-streptomycin). The media was then removed and the cells were washed with phosphate buffered saline (PBS). The cells were incubated for 2 h with DMA (0.1, 1 and 10 mM), BAY 11-7082 (5 μM) or SB202190 (25 μM, which inhibits >90% of MAPK activity) (37). No DMA was added to the media for control RAW 264.7 and JEG-3 cells. RAW 264.7 cells were incubated with LPS (1 μg mL−1) for 24 h. JEG-3 cells were stimulated with recombinant human TNFα (20 ng mL−1). Supernatants were collected and centrifuged at 1,000 × g for 10 min. The concentrations of TNFα, IL-6, IL-8, IL-10 and GM-CSF were determined using enzyme linked immunosorbent assay (ELISA) kits (eBioscience) following the manufacturer’s protocol. Three independent experiments were performed in duplicate, and analyses of each experimental sample were run in duplicate. The concentration of each cytokine was interpolated using a second-order polynomial (quadratic) equation generated from a standard curve in GraphPad Prism 6, as instructed by the manufacturer.
Ex Vivo Assay of Secreted Cytokines and Chemokines
Singleton placentae were obtained from uncomplicated term elective Cesarean deliveries after uneventful pregnancies (38–41 wks gestation, n = 7) and were collected in coordination with the New York Cord Blood Bank at the Albert Einstein College of Medicine, Bronx, NY. The protocol was approved and considered exempt by both Albert Einstein College of Medicine and St. John’s University’s Institutional Review Boards. Patients with chorioamnionitis, hypertension or preeclampsia, diabetes mellitus, fetal anomalies or Apgar scores <7 were excluded. Sections were taken from the maternal side of the placenta and washed thoroughly with cold sterile PBS. Blood vessels, blood clots, connective tissue and membranes were removed. The sections were finely minced, pooled in cold DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin and centrifuged at 1,000 × g for 10 min. Duplicate aliquots of 200 mg (wet weight) were incubated for two hours with DMA (0.1, 1 and 10 mM), BAY 11-7082 (5 μM), SB202190 (25 μM) or CGM for the control group. The explants were incubated with or without LPS (1 μg mL−1) for 20 h at 37°C and 5% CO2. The media was collected and ELISA was performed in duplicate wells for each sample to determine the concentrations of TNFα, IL-6, IL-8, IL-10 and GM-CSF, following the manufacturer’s protocol. The concentration of each cytokine was interpolated using a second-order polynomial (quadratic) equation generated from a standard curve in GraphPad Prism 6, as instructed by the manufacturer.
Tissue Explant Viability Assay
As the placental explants comprised a heterogeneous population of cells not suited for the MTT assay, a Pierce lactic dehydrogenase (LDH) assay kit (Thermo Fisher) was used to determine placental explant viability, following the manufacturer’s protocol. Briefly, 50 μL of supernatant were added into a 96-well plate in duplicate for each sample. An equal volume of Reaction Mixture was added and the reaction was allowed to proceed for 30 min at room temperature protected from light. Stop Solution was added, and the absorbance was read at 490 nm using a Opsys MRTM microplate reader.
Immunoblot Analysis
To determine the effect of DMA on various proteins, such as iNOS, nuclear factor of kappa light polypeptide gene enhancer in B cell inhibitor α (IκBα) and native and phosphorylated forms of p38, extracellular signal-regulated kinase (ERK) and c-jun N-terminal kinase (JNK), RAW 264.7 cells were seeded at 1.5 × 106 cells in 25 cm2 flasks and were allowed to grow for 48 h at 37°C and 5% CO2. The cells were incubated for 2 h with DMA (0.1, 1 and 10 mM), BAY 11-7082 (10 μM) or CGM. In addition, three more singleton placentae were obtained from uncomplicated term elective Cesarean deliveries after uneventful pregnancies and processed as described above. For iNOS expression, cells were incubated with LPS (0.1 μg mL−1) for six hours. To determine IκBα and the native and phosphorylated forms of p38, ERK and JNK, cells and explants were stimulated with LPS (1 μg mL−1) for 15 min. Whole-cell and explants lysates were prepared using a modified protocol as described by Abcam. Briefly, cells and explants were lysed in radio immunoprecipitation assay (RIPA) lysis buffer (G Biosciences) freshly supplemented before use with 1 mM of phenylmethylsulfonylfluoride and ethylenediaminetetraacetic acid-free protease inhibitor cocktail set III (Calbiochem) by repeated vortexing on ice for 30 min. Samples were then centrifuged at 14,000 × g for 20 min at 4°C and supernatants were collected as lysates. Protein concentrations of the samples were determined using bicinchoninic acid (BCA) protein assay (Thermo Fisher). Equal amounts of total protein were resolved in 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto PVDF membranes (Bio-Rad). The membranes were blocked for two hours at room temperature using 5% skim milk in TBST [20 mM Tris (pH 7.6), 150 mM NaCl, 0.1% Tween 20] and incubated at 4°C overnight with a 1:1,000 dilution of primary antibody for iNOS (Santa Cruz Biotechnology), β-actin, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), p38, p-p38, ERK 1/2, p-ERK1/2, JNK, p-JNK (Cell Signaling Technologies) or IκBα (Santa Cruz). The membranes were washed with TBST and then incubated at room temperature for one hour with a 1:1,000 dilution of horseradish peroxidase-linked secondary antibody (GE Healthcare) in 5% skim milk. Protein expression was detected using Pierce enhanced chemiluminescence (Thermo Fisher) and developed with clear blue X-ray films (Thermo Fisher) and analyzed using ImageJ software (National Institutes of Health). Three independent experiments were performed in duplicate.
Amplification, Collection and Purification of Plasmids
The NF-κB-Luc, AP-1-Luc, C/EBP-Luc and Renilla-Luc (Promega) reporter plasmids were amplified using competent E. coli (gift from Dr. Xingguo Cheng) transformed by the heat shock method. After growing the transformed bacteria, Endofree maxipreps (Qiagen) were used to collect and purify the amplified plasmids following the manufacturer’s protocol. The collected plasmids were dissolved using endotoxin-free Buffer TE, and the yield was determined by reading the absorbance at 260 nm using a BioSpectrometer kinetic (Eppendorf).
Transfection and Luciferase Assay
HEK 293 cells overexpressing TLR4, MD2 and CD14 genes were co-transfected with 100 ng of NF-κB-Luc, AP-1-Luc or C/EBP-Luc reporter plasmids and 10 ng of Renilla-Luc control plasmid using Lipofectamine 3000 reagent (Invitrogen) following the manufacturer’s protocol. Subsequently, the cells were incubated for two hours with DMA (0.1, 1 and 10 mM), BAY 11-7082 (10 μM) or CGM. Cells were incubated for an additional 24 h with or without LPS (1μg mL−1), and luciferase assay was performed as per the manufacturer’s instructions using a Synergy H1 microplate reader (Biotek) to measure luminescence. The ratio of firefly/renilla readings for each sample was taken. The effect of the treatment relative to the LPS-only treated group is expressed in fold luciferase activity, determined by dividing the ratio of the firefly/renilla readings in each experimental sample by the average ratio of the firefly/renilla readings in the LPS control group, as shown below. Three independent experiments were performed in triplicate.
Luciferase activity (fold) = ratio of firefly/renilla luciferase reading (each sample) / average of the ratio of firefly/renilla reading (LPS Control)
Statistical Analysis
Cell and tissue explant viability, luciferase assays, nitric oxide assays, cytokine levels and protein assays were analyzed using one-way analysis of variance (ANOVA). Post hoc analysis was performed using Dunnett’s test. Kaplan Meier curves for mouse pup survival were compared using the log rank test. The a priori p value was set at p <0.05. The statistical analysis was done using GraphPad Prism Version 6.
All supplementary materials are available online at www.molmed.org.
RESULTS
Cell Viability by MTT Assay
The highest concentration of DMA causing <10% cell death was determined to be 10 mM for RAW 264.7, JEG-3 cells and HEK-293/TLR4 cells (Supplementary Figure S1). Any change in cell viability at this concentration of DMA was not statistically significant. At 20 mM, viability was significantly decreased to approximately 75%, 80% and 85% in RAW 264.7, JEG-3 and HEK 293/TLR4 cells, respectively. One hundred mM DMA significantly decreased the viability of all cell lines. Thus, based on the aforementioned results, 10 mM DMA was the highest concentration used in our experiments.
DMA Regulates NO−2 Secretion and iNOS Expression in RAW 264.7 Cell
Nitrite levels were determined in supernatants 24 h after LPS incubation, as they were found to peak at this point in the time profile analysis conducted prior to the experiments (Supplementary Figure S2). As previously reported (35), LPS (1 μg mL−1) significantly increased NO−2 levels (≈65 μM) compared with the control (p <0.0001; Figure 1A). The NF-κB inhibitor BAY 11-7082 (5 μM) significantly reduced NO−2 levels to 20 μM (p <0.0001) compared with the control. DMA, at 10 mM, significantly reduced NO−2 levels by 15 μM (p <0.0001) compared with the control.
Figure 1.

DMA inhibits NO−2 secretion and iNOS expression. (A) NO−2 secretion was determined by Griess assay in LPS-stimulated RAW 264.7 cells after incubation with DMA and BAY 11-7082. (B) NO−2 secretion in LPS-stimulated RAW 264.7 cells after incubation with MMA. (C) Time profile of iNOS expression in LPS-stimulated RAW 264.7 cells. Data shown are means ± SEM of at least three independent experiments done in duplicate. * P ≤ 0.05; **** P≤ 0.0001. (D) Immunoblot analysis of iNOS expression after incubation with DMA and BAY 11-7082.
Monomethylacetamide (MMA) is the primary metabolite of DMA in vivo (39). We then determined whether MMA contributes to the antiinflammatory effect of DMA; ie, whether DMA might be a prodrug that needs to be converted to a more active form. MMA, at 10 mM, did not significantly alter LPS-induced NO−2 secretion in RAW264.7 cells. (Figure 1B). No detectable levels of NO−2 were found in either JEG-3 cells or human placental explants (data not shown). As expected, iNOS was significantly increased six hours after incubation with LPS (0.1 μg mL−1) as compared with controls (p <0.0001) (Figure 1D). Incubating cells with BAY 11-7082 (5 μM) significantly decreased LPS-induced iNOS expression (p < 0.0001) (Figure 2C). Similarly, DMA at 10 mM significantly reduced iNOS expression (p <0.05) after incubation with LPS (Figure 1C).
Figure 2.

DMA inhibits cytokine and chemokine secretion in LPS-stimulated RAW 264.7 cells. ELISA was performed to determine (A) TNFα, (B) IL-6, (C) GM-CSF and (D) IL-10 levels in RAW 264.7 cell culture supernatant after incubation with DMA and BAY 11-7082 in the presence of LPS after 24 h. Data shown are means ± SEM of at least three independent experiments done in duplicate. * P ≤ 0.05; ** P ≤ 0.01; *** P ≤ 0.001; **** P ≤ 0.0001.
DMA Inhibits Secretion of Cytokines and Chemokines in RAW 264.7 and JEG-3 Cells
LPS (1 μg mL−1) significantly stimulated the production of TNFα, IL-6, GM-CSF and IL-10 in RAW 264.7 cells (Figures 2A–D), but not in JEG-3 cells (data not shown). Increasing the concentration of LPS to 10 μg mL−1 did not affect levels of IL-6 in JEG-3 cell supernatants (Supplementary Figure S3, Panel A). However, TNFα significantly increased IL-6 secretion in JEG-3 cells when compared with the untreated control at various time points (Supplementary Figure S3, Panel B). Hence, experiments with JEG-3 cells were performed using TNFα as stimulant. The NF-κB inhibitor BAY 11-7082 was found to significantly reduce all cytokines and chemokines tested as compared with the LPS or TNFα controls (Figures 2 and 3).
Figure 3.
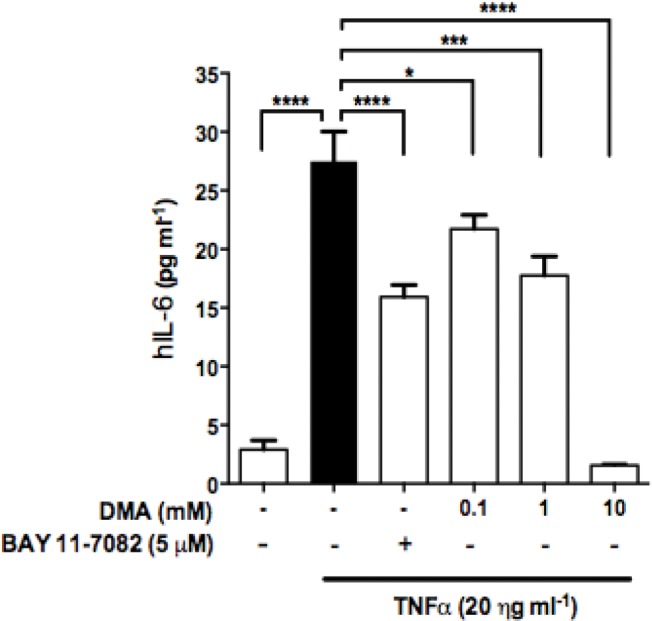
DMA inhibits IL-6 secretion in JEG-3 cells. ELISA was performed to determine IL-6 levels in JEG-3 cell supernatants after incubation with DMA and BAY 11-7082 in TNFα-stimulated JEG-3 cells. Data shown are means ± SEM of at least three independent experiments done in duplicate. * P ≤ 0.05; *** P ≤ 0.001; **** P ≤ 0.0001.
The incubation of RAW 264.7 cells with LPS produced a significant increase in TNFα levels (p <0.0001) (Figure 2A). DMA (0.1–10 mM) decreased the LPS-induced increase in TNFα levels in a concentration-dependent manner, with 10 mM of DMA producing a 90% decrease in secreted TNFα compared with the control (p <0.001) (Figure 2A). LPS also significantly increased IL-6 production in RAW 264.7 cells (100 ng mL−1; p <0.0001) (Figure 2B). DMA similarly reduced IL-6 secretion in a concentration-dependent manner, with the 10 mM concentration of DMA again significantly decreasing IL-6 levels compared with the control (p <0.05) (Figure 2B) . Incubation of RAW 264.7 cells with LPS significantly increased secretion of GM-CSF as well (p <0.01) (Figure 2C). Once again, DMA suppressed the LPS-induced increase in GM-CSF secretion in a concentration-dependent fashion, with 10 mM DMA causing a significant decrease in GM-CSF levels compared with the control (p <0.05) (Figure 2C). Lastly, reduction in LPS-induced IL-10 secretion was observed at all concentrations of DMA; however, only at 10 mM was statistical significance achieved compared with the LPS control (p <0.05) (Figure 2D).
The same cytokines were evaluated in JEG-3 cells stimulated with TNFα (20 ng mL−1). However, only IL-6 levels were increased in comparison to the untreated groups. DMA, in a concentration-dependent manner, reduced secretion of IL-6 (Figure 3). All the concentrations of DMA tested resulted in a statistically significant reduction in IL-6 production as compared with the TNFα control (p <0.05 at 0.1 mM, p <0.001 at 1 mM, p <0.0001 at 10 mM). At 10 mM, DMA reduced IL-6 secretion to baseline levels (Figure 3).
DMA Inhibits Secretion of Proinflammatory Cytokines and Chemokines in Human Placental Explants
The effect of DMA on secretion of proinflammatory cytokines and other mediators in placental explants (n = 7) stimulated with LPS was evaluated. LDH levels in the supernatant of each treatment group were first determined, in order to test whether the treatment conditions affected tissue viability. Results revealed similar LDH levels in all treatment groups (Supplementary Figure S4). LPS significantly enhanced secretion of TNFα, IL-6, IL-8 and GM-CSF in placental explant supernatants as compared with the controls (p <0.0001) (Figures 4A–E). LPS did not significantly alter the levels of IL-10. The NF-κB inhibitor BAY 11-7082 did not significantly affect secretion of TNFα, IL-6, IL-8 or GM-CSF in the explants (data not shown). However, SB202190, an inhibitor of p38 α and p38 β MAPKs, significantly reduced secretion of all the aforementioned cytokines (Figures 4A–E).
Figure 4.

DMA inhibits cytokine and chemokine secretion in LPS-stimulated human placental explants. ELISA was performed to determine levels of (A) TNFα, (B) IL-6, (C) IL-10, (D) IL-8 and (E) GM-CSF after incubation with DMA or SB 202190 in the presence of LPS after 20 h. Data shown are means ± SEM of experiments done in duplicate (n = 7). * P ≤ 0.05; ** P ≤ 0.01; *** P ≤ 0.001; **** P ≤ 0.0001.
Interestingly, a similar pattern to that of the in vitro experiments emerged when we examined the effect of DMA on the secretion of various cytokines in the human placental explants. DMA, at the 10 mM concentration only, significantly reduced secretion of TNFα, IL-6, IL-8 and GM-CSF (Figures 4A–D). DMA also inhibited IL-10 secretion at 10 mM; however, this was not found to be significant (Figure 4E).
DMA Inhibits IκBα Degradation
To determine the mechanism by which DMA suppresses cytokine secretion, we determined how it affects degradation of the NF-κB inhibitor IκBα. We first conducted a time-profile experiment, using whole lysates from RAW 264.7 cells to determine the profile of IκBα degradation after exposure to LPS (1 μg mL−1) at various time points (Figure 5A). Western blotting showed that IκBα was degraded 15 min after addition of LPS. The IκBα bands were noted to reappear at 30 min, and their intensity increased with time. We then evaluated whether DMA inhibits IκBα degradation after 15 min exposure to LPS (Figure 5B). As expected, LPS significantly induced degradation of IκBα (p < 0.0001), as manifested by a decrease in IκBα levels in the LPS control as compared with the untreated group. However, we found that the levels of IκBα increased with increasing concentrations of DMA (Figure 5C). Data were normalized using β-actin, and statistical analysis of the relative densities of IκBα revealed that the highest concentration of DMA (10 mM) significantly (p <0.01) inhibited IκBα degradation relative to the LPS control (Figure 5C). We performed a similar experiment in human placental explants, aimed at testing the effect of varying concentrations of DMA on LPS-induced IκBα degradation. Although LPS produced only a slight decrease in IκBα levels detected in placental lysates by immunoblotting, levels trended back up with increasing concentrations of DMA (Figure S5).
Figure 5.

DMA prevents LPS-stimulated IκBα degradation but does not affect levels of MAPK pathway proteins in RAW 264.7 cells. (A) Time profile analysis of IkBα degradation was determined by immunoblot of IkBα from whole-cell lysate of RAW 264.7 cells at various time points after incubation with LPS (1 μg mL−1). (B) The effect of various concentrations of DMA on IkBα degradation was determined in the presence or absence of LPS for 15 min. (C) Graphical summary of the relative densities of IkBα expression in panel B quantified using ImageJ software. (D) Immunoblots of downstream proteins of the MAPK pathway after incubation with DMA in the presence of LPS. Representative immunoblots are shown and data are means ± SEM from three independent experiments. ** P ≤ 0.01; **** P ≤ 0.0001.
The effect of DMA on phosphorylation of downstream proteins in the MAPK signaling pathway was evaluated in LPS-induced RAW 264.7 cells by Western blotting of the phosphorylated and native forms of JNK, ERK1/2 and p38 (Figure 5D). Bands for phosphorylated JNK, ERK1/2 and p38 MAPK (p-JNK, p-ERK1/2 and p-p38 MAPK) were seen 15 min after exposure to LPS (Figure 5D, Lane 2). In contrast, bands were barely detected in the untreated controls (Figure 5D, Lane 1), with the exception of p-p38 MAPK, where bands were seen. Incubation of cells with increasing concentrations of DMA in the presence of LPS did not significantly change the expression of p-JNK, p-ERK1/2 or p-p38 MAPK (Figure 5D, Lanes 3–5) as compared with the controls. Similarly, the expression levels of the native forms of these proteins were not affected by DMA, as manifested by the uniform band densities across the treatment groups (Figure 5D, Lanes 3–5).
DMA Inhibits the Transcriptional Activity of NF-κB but Not AP-1 or C/EBP
The transcriptional activity of NF-κB and AP-1, but not C/EBP, was significantly stimulated by LPS, based on their respective luciferase reporter assays (data not shown). DMA, at 10 mM, significantly inhibited (p <0.0001) NF-κB transcriptional activity in the presence of LPS (Figure 6A). It did not affect AP-1–driven transcription (Figure 6B), but significantly enhanced transcriptional activity of C/EBP (Figure 6C). As expected, BAY 11-7082 (5 μM) significantly inhibited transcription of the NF-κB luciferase reporter (p <0.001) (Figure 6A) but enhanced AP-1 and C/EBP transcriptional activities (Figures 6B–C). DMSO, the vehicle for BAY 11-7082, had no significant effect on the transcriptional activity of NF-κB, AP-1 or C/EBP (Figures 6A–C).
Figure 6.

DMA decreases transcriptional activity of NF-κB but not AP-1 or C/EBP. Luciferase activity was evaluated using the DualGlo luciferase reagent system after DMA and BAY 11-7082 in LPS-stimulated HEK 293/TLR4 cells transiently transfected with (A) NF-κB-Luc, (B) AP-1-Luc and (C) C/EBP-Luc reporter plasmids. The activity of Renilla-Luc was used to normalize the data. DMSO was included as a vehicle control. Data shown are means ± SEM of at least three independent experiments done in triplicate. ** P ≤ 0.01; *** P ≤ 0.001; **** P ≤ 0.0001.
DISCUSSION
Inflammation-associated preterm birth continues to account for most cases of perinatal morbidity and mortality in the developed world. Safe and effective pharmacotherapy to prevent inflammation-driven preterm labor has remained elusive, partly because of toxic or teratogenic effects of potential therapies. Prior to using DMA in these experiments, we determined the highest concentration of DMA that did not reduce cell viability in the presence of LPS. Our experiments revealed that DMA does not cause significant toxicity in any of the three cell lines we used in this work at concentrations up to 10 mM. This concentration is equivalent to 871 mg/L of DMA. Although there is no reliable way to convert this in vitro concentration to an in vivo dose, we can approximate based on the presumption of a one-compartment model that this concentration is equivalent to a dose of 0.87 g/kg in mice or 0.07 g/kg in humans. This estimated amount is much lower than the reported oral LD50 of 4.6 g/kg in mice (40). In addition, this estimated concentration is also lower than what we previously used (1.6 g/kg) in our in vivo experiments, which successfully showed the positive effect of DMA without producing toxic effects in either mice dams or fetuses (35). Those previous results, together with our cell viability assays, served as the basis for using 10 mM as the highest concentration in the in vitro experiments here.
Investigation of antiinflammatory activity in in vitro models is commonly performed using various cell lines, including human peripheral blood mononuclear cells (PBMCs) and murine macrophage-like RAW 264.7 cells (41,42). In the present work, we first determined the effect of DMA on NO−2 secretion as an initial evaluation of its antiinflammatory potential in LPS-stimulated RAW 264.7 cells (43-45). Our data reveal that DMA at 10 mM results in a 21% reduction in NO−2 levels in LPS-induced RAW 264.7 cells, as compared with the LPS control. Because DMA is primarily metabolized in vivo into MMA (46), we explored the possibility that MMA might be contributing to the effect of DMA on NO secretion. However, the data from our experiments using MMA suggest otherwise. In fact, MMA resulted in a slightly higher level of NO−2 as compared with the LPS control, although this was not found to be statistically significant.
NO is an important inflammatory mediator produced by macrophages and other cells in response to bacterial products like LPS (47–49). In RAW 264.7 cells, NO is enzymatically produced in the presence of inflammatory stimuli through the induction of iNOS (45,50). We next determined whether DMA affects expression of LPS-induced iNOS and, as expected, and in agreement with its effect on NO secretion, iNOS expression was reduced by 10 mM DMA. To our knowledge, this is the first time that DMA is reported to inhibit iNOS expression in LPS-induced RAW 264.7 cells. We did not find detectable levels of NO−2 in the cell culture supernatants of JEG-3 cells or human placental explants after incubation with LPS. In JEG-3 cells, we tried increasing the concentration of LPS to 10 μg mL−1 and prolonging the exposure to 72 h, but the findings were similar. This could be explained by the absence of iNOS in human trophoblast cells, as previously reported (51). In another study, the presence of iNOS was reported in human placental villous tissue, specifically localized in Hofbauer cells, and iNOS was found to be expressed in syncytiotrophoblast and villous vascular endothelium (52).
We have previously shown that DMA regulates proinflammatory cytokine and chemokine expression in mouse placental tissue (35). Now, for the first time, we report DMA’s effect on regulating secreted proinflammatory cytokines and chemokines in RAW 264.7 and JEG-3 cells. Specifically, DMA (10 mM) was found to reduce TNFα, IL-6, IL-10 and GM-CSF secretion in LPS-induced RAW 264.7 cells. With respect to JEG-3 cells, we found that DMA (0.01–10 mM) reduced TNFα-induced IL-6 secretion. These findings, along with our data on this molecule’s effect on NO and iNOS expression, are an important indication of the antiinflammatory activity of DMA. Importantly, DMA suppressed cytokine secretion in cells stimulated by both LPS and TNFα, which rules out the possibility of DMA’s simply inactivating LPS by some direct interaction. Our results indicate that DMA (10 mM) significantly inhibits secretion of TNFα, IL-6, IL-8 and GM-CSF in LPS-stimulated human placental explants. Remarkably, the effects of DMA in reducing TNFα, IL-8 and GM-CSF were found comparable to that of SB202190, a selective p38 MAPK inhibitor (35), used here as a control. In addition, DMA was also shown to have a modest but significant inhibitory effect on IL-6 secretion and a trend toward inhibiting IL-10 inhibition, which was not found to be statistically significant.
The activity of LPS is mediated by TLR4, which subsequently activates both the NF-κB and MAPK pathways (13,53,54). It is not surprising then to see that SB202190 was able to suppress secretion of these inflammatory cytokines and mediators. Shoji et al. previously reported involvement of the MAPK pathway in the production of proinflammatory cytokines in the choriodecidua (55–57). What was unexpected was that BAY 11-7082, an NF-κB inhibitor, did not have a significant effect on the secretion of these cytokines in our explant model (data not shown). This might be explained by the concentration of BAY 11-7082 we used in the experiments, only 5 and 10 μM. Others have reported significant inhibition of the NF-κB-dependent production of proinflammatory cytokines using 50 μM of BAY 11-7082 (58,59). We found this concentration to be cytotoxic to RAW 264.7 and JEG-3 cells, as determined by our MTT assay (data not shown). In addition, it has been reported that BAY 11-7082 is cytotoxic at 30 μM (60).
In our PTB mouse model, we showed that DMA downregulates TNFα and IL-6 in the placenta, delaying parturition and decreasing the number of pups dropped (35). These cytokines are involved in transforming the uterus from a quiescent to an active state (61–63). In addition, our current data show that DMA inhibits IL-8 secretion. IL-8 is an important paracrine mediator of inflammation that amplifies inflammatory signals by demargination, activation and chemotaxis of inflammatory cells to the site of injury (64,65). Interestingly, we also demonstrated an increase in intravascular polymorphonuclear (PMN) cells in the murine placenta and uterine tissues in the presence of LPS in vivo, which was significantly reduced by DMA (35). Thus, these new data from our placental explant model showing the same reduction in TNFα, IL-6 and IL-8 corroborate our previous findings and further provide strong evidence of DMA’s antiinflammatory activity and its potential value in the treatment of PTB.
Another exciting finding in the current work is the inhibitory effect of DMA on GM-CSF secretion in both RAW 264.7 cells and human placental explants. GM-CSF is an important cytokine produced in the choriodecidua (65). GM-CSF has been reported to be involved in fetal membrane weakening (66) and in regulating the maturation and function of uterine dendritic cells and macrophages as antigen-presenting cells (67). In a review by van Nieuwenhuijze et al., investigators highlighted the potential of GM-CSF as a therapeutic target in various inflammatory diseases such as arthritis and Alzheimer’s disease (68). Further, Bozinovski et al. showed that LPS-induced lung inflammation is ameliorated by neutralizing GM-CSF (69). Finally, elevated levels of GM-CSF, in addition to IL-6 and IL-8, were recently found in amniotic fluid in the setting of infection (70). Thus, we are excited to find that DMA has an inhibitory effect on GM-CSF secretion in the presence of LPS, which could be further explored to establish DMA’s potential as an antiinflammatory agent in various settings.
IL-10 is a key antiinflammatory cytokine primarily produced by macrophages and monocytes. It inhibits production of proinflammatory cytokines such as TNFα, IL-1β, IL-6 and IL-8 (71,72). In the present study, we observed a decrease in IL-10 after treating RAW 264.7 cells with DMA in the presence of LPS. A similar trend was seen in the placental explants, although the results did not reach statistical significance. This is opposite to what we expected and also contrary to what we have observed in vivo (35). We speculate that this unexpected result might be due to the stark difference in the samples used, wherein lysates from whole placental tissues from our in vivo model were analyzed in our previous study, while 264.7 cell supernatants were tested in the current work. Another factor is the complexity of the in vivo system, where the effects of other organs, tissues and cells may contribute to the observed effect, as compared with the more controlled setting of a specific cell or tissue.
In the present study, we demonstrate that DMA significantly inhibits IκBα degradation in a concentration-dependent manner. As illustrated in Figure 7, increased intracellular concentrations of IκBα, in turn, inhibit the nuclear translocation of the NF-κB dimer and subsequent transcription of specific inflammatory mediators, thus explaining the decrease in proinflammatory cytokine secretion observed in both the in vitro and ex vivo experiments reported here. In addition, we demonstrated previously that DMA inhibits nuclear translocation of NF-κB using immunofluorescence (35). DMA’s lack of effect on expression levels of JNK, p-JNK, ERK 1/2, p-ERK 1/2, p38 MAPK and p-p38 MAPK indicates that it specifically suppresses the NF-κB pathway. The unchanged levels of MAPK pathway proteins further show that the effects of DMA reported here cannot be explained by DMA’s simply decreasing cell viability. The precise mechanism by which DMA prevents IκBα degradation—;ie, whether it inhibits IKK and prevents IκBα from being phosphorylated, whether it affects degradation of phosphorylated IκBα by the proteasome, or both—;remains to be determined. We speculate that DMA inhibits phosphorylation of IκBα by IKKs, which in turn leads to inhibition of IκBα degradation. But why DMA selectively inhibits these kinases and does not affect the phosphorylation of JNK, ERK1/2 and p38 MAPK remains to be determined. Further investigation is warranted to answer this important question. Significantly, inhibition of IκBα and IKKs has been implicated as a valuable therapeutic approach for various conditions including cancer (73, 74), chronic kidney disease (75), metabolic diseases (76), airway inflammatory disease (77), arthritis (78), and, in line with our findings in previous and current work, PTB (79).
Figure 7.

DMA prevents p50/Rel driven transcription of inflammatory cytokine genes by inhibiting IκB degradation.
Interestingly, our data do not support any significant effect of DMA on the MAPK pathway. Phosphorylation of JNK, ERK1/2 and p38 MAPK induced by LPS was not affected by DMA, as seen in the Western blots that we performed. To further test whether DMA affects the NF-κB and/or MAPK pathways, we evaluated its effect on NF-κB and AP-1 transcriptional activity in LPS-stimulated HEK 293 cells overexpressing TLR4 transfected with NF-κB–Luc, AP-1–Luc or C/EBP–Luc reporter genes. Our data indicate that DMA inhibits NF-κB–driven transcription, thereby shutting down NF-κB dimer–driven inflammation, but not transcription driven by AP-1 or C/EBP. This corroborates our Western blot data, where DMA was found not to affect the level of phosphorylated JNK, ERK1/2 and p38 MAPKs in LPS-stimulated RAW 264.7 cells.
CONCLUSION
DMA inhibits production of NO and proinflammatory cytokines and chemokines in vitro and ex vivo. Further, we show for the first time that this effect is brought about by DMA’s ability to block IkBα degradation and NF-κB–driven transcription. Taken together, our previous in vivo PTB data and the in vitro, ex vivo and in vivo data presented here establish DMA as a novel antiinflammatory agent and further strengthen its potential use not only in PTB, but in the management of a wide array of human diseases.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant from the National Institutes of Health (1R01NS069577) and by St. John’s University Seed Grants to SER. We are very grateful to Dr. Francine Einstein and the staff of the New York Cord Blood Bank at Albert Einstein College of Medicine for providing the placentas. We are also indebted to Ms. Helen Scaramell and the entire St. John’s University animal facility staff for their assistance with mating and maintaining the mice.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
SER and CRA Jr. have a patent pending (US 13/536,946) on the use of N, N-dimethylacetamide for inflammatory disorders.
Cite this article as: Pekson R, et al. (2016) N,N-Dimethylacetamide significantly attenuates LPS- and TNFα-induced proinflammatory responses via inhibition of the nuclear factor kappa B pathway. Mol. Med. 22:747–58.
REFERENCES
- Jialal I, Divaraj S. Anti-inflammatory strategies to prevent diabetic cardiovascular disease. Clin Pharm Ther. 2015;98:121–23. doi: 10.1002/cpt.105. [DOI] [PubMed] [Google Scholar]
- Ward RJ, Dexter DT, Crichton RR. Ageing, neuroinflammation and neurodegeneration. Front Biosci. 2015;7:189–204. doi: 10.2741/S433. [DOI] [PubMed] [Google Scholar]
- Burdet J, et al. Inflammation, infection and preterm birth. Curr Pharm Design. 2014;20:4741–48. doi: 10.2174/1381612820666140130202224. [DOI] [PubMed] [Google Scholar]
- Romero R, Dey SK, Fisher SJ. Preterm labor: one syndrome, many causes. Science. 2014;345:760–65. doi: 10.1126/science.1251816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero R, Espinoza J, Chaiworapongsa T, Kalache K. Infection and prematurity and the role of preventive strategies. Sem Neonatol. 2002;7:259–74. doi: 10.1016/s1084-2756(02)90121-1. [DOI] [PubMed] [Google Scholar]
- Liu L, et al. Global, regional and national causes of child mortality: an updated systematic analysis for 2010 with time trends since 2000. Lancet. 2012;379:2151–61. doi: 10.1016/S0140-6736(12)60560-1. [DOI] [PubMed] [Google Scholar]
- Blencowe H, et al. National, regional, and worldwide estimates of preterm birth rates in the year 2010 with time trends since 1990 for selected countries: A systematic analysis and implications. Lancet. 2012;379 :2162–72. doi: 10.1016/S0140-6736(12)60820-4. [DOI] [PubMed] [Google Scholar]
- Elovitz MA, Mrinalini C. Animal models of preterm birth. Trends Endocrin Met. 2004;15:479–87. doi: 10.1016/j.tem.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Clark DA. The use and misuse of animal analog models of human pregnancy disorders. J Reprod Immunol. 2014;103:1–8. doi: 10.1016/j.jri.2014.02.006. [DOI] [PubMed] [Google Scholar]
- Coll RC, O’Neill LA. New insights into the regulation of signalling by toll-like receptors and nod-like receptors. J Innate Immun. 2010;2:406–21. doi: 10.1159/000315469. [DOI] [PubMed] [Google Scholar]
- Lappas M. NOD1 and NOD2 regulate proinflammatory and prolabor mediators in human fetal membranes and myometrium via nuclear factor-kappa B. Biol Reprod. 2013;89:1–11. doi: 10.1095/biolreprod.113.110056. [DOI] [PubMed] [Google Scholar]
- Mitchell JA, Paul-Clark MJ, Clarke GW, McMaster SK, Cartwright N. Critical role of toll-like receptors and nucleotide oligomerisation domain in the regulation of health and disease. J Endocrinol. 2007;193:323–30. doi: 10.1677/JOE-07-0067. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. Signaling to NF-kappa B by toll-like receptors. Trends Mol Med. 2007;13:460–69. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Kondo T, Kawai T, Akira S. Dissecting negative regulation of toll-like receptor signaling. Trends Immunol. 2012;33:449–58. doi: 10.1016/j.it.2012.05.002. [DOI] [PubMed] [Google Scholar]
- Wang H, Hirsch E. Bacterially induced preterm labor and regulation of prostaglandin metabolizing enzyme expression in mice: The role of toll-like receptor 4. Biol Reprod. 2003;69:1957–63. doi: 10.1095/biolreprod.103.019620. [DOI] [PubMed] [Google Scholar]
- Chau TL, et al. Are the IKKs and IKK-related kinases TBK1 and IKK-epsilon similarly activated? Trends Biochem Sci. 2008;33:171–80. doi: 10.1016/j.tibs.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Lim KH, Staudt LM. Toll-like receptor signaling. Cold Spring Harb Perspect Biol. 2013;5:a011247. doi: 10.1101/cshperspect.a011247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas I, et al. Viral infection of the placenta leads to fetal inflammation and sensitization to bacterial products predisposing to preterm labor. J Immunol. 2010;185:1248–57. doi: 10.4049/jimmunol.1000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch E, Wang H. The molecular pathophysiology of bacterially induced preterm labor: Insights from the murine model. J Soc Gynecol Investig. 2005;12:145–55. doi: 10.1016/j.jsgi.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Salminen A, et al. Maternal endotoxin-induced preterm birth in mice: Fetal responses in toll-like receptors, collectins, and cytokines. Pediatr Res. 2008;63:280–86. doi: 10.1203/PDR.0b013e318163a8b2. [DOI] [PubMed] [Google Scholar]
- Thaxton JE, Nevers TA, Sharma S. TLR-mediated preterm birth in response to pathogenic agents. Infect Dis Obstet Gynecol. 2010:1–8. doi: 10.1155/2010/378472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, et al. Diagnostic value of the matrix metalloproteinase-8 rapid test for detecting microbial invasion of the amniotic cavity. Eur J Clin Microbiol Infect Dis. 2008;27:1257–60. doi: 10.1007/s10096-008-0566-7. [DOI] [PubMed] [Google Scholar]
- Romero R, et al. Interleukin-1 alpha and interleukin-1 beta in preterm and term human parturition. Am J Reprod Immunol. 1992;27:117–23. doi: 10.1111/j.1600-0897.1992.tb00737.x. [DOI] [PubMed] [Google Scholar]
- Romero R, et al. Tumor necrosis factor in preterm and term labor. Am J Obstet Gynecol. 1992;166:1576–87. doi: 10.1016/0002-9378(92)91636-o. [DOI] [PubMed] [Google Scholar]
- Dudley DJ, Collmer D, Mitchell MD, Trautman MS. Inflammatory cytokine mRNA in human gestational tissue: implications for term and preterm labor. J Soc Gynecol Investig. 1996;3:328–35. [PubMed] [Google Scholar]
- Challis JR, et al. Understanding preterm labor. Ann NY Acad Sci. 2001;943:225–34. doi: 10.1111/j.1749-6632.2001.tb03804.x. [DOI] [PubMed] [Google Scholar]
- Challis JR, et al. Inflammation and pregnancy. Reprod Sci. 2009;16:206–15. doi: 10.1177/1933719108329095. [DOI] [PubMed] [Google Scholar]
- Wang JJ, Jenkinson CP, Griscavage JM, et al. Co-induction of arginase and nitric oxide synthase in murine macrophages activated by lipopolysaccharide. Biochem Biophys Res Commun. 1995;210:1009–16. doi: 10.1006/bbrc.1995.1757. [DOI] [PubMed] [Google Scholar]
- Elovitz MA, Wang Z, Chien EK, Rychlik DF, Phillippe M. A new model for inflammation-induced preterm birth: The role of platelet-activating factor and toll-like receptor-4. Am J Pathol. 2003;163:2103–11. doi: 10.1016/S0002-9440(10)63567-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier SL, et al. The relationship of amniotic fluid cytokines and preterm delivery, amniotic fluid infection, histologic chorioamnionitis, and chorioamnion infection. Obstet Gynecol. 1993;81:941–48. [PubMed] [Google Scholar]
- Peltier MR. Immunology of term and preterm labor. Reprod Biol Endocrinol. 2003;1:122. doi: 10.1186/1477-7827-1-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappas M, Permezel M, Georgiou HM, Rice GE. Nuclear factor kappa B regulation of proinflammatory cytokines in human gestational tissues in vitro. Biol Reprod. 2002;67:668–73. doi: 10.1095/biolreprod67.2.668. [DOI] [PubMed] [Google Scholar]
- Belt AR, Baldassare JJ, Molnar M, Romero R, Hertelendy F. The nuclear transcription factor NF-kappa B mediates interleukin-1beta induced expression of cyclooxygenase-2 in human myometrial cells. Am J Obstet Gynecol. 1999;181:359–66. doi: 10.1016/s0002-9378(99)70562-4. [DOI] [PubMed] [Google Scholar]
- Lindstrom TM, Bennett PR. The role of nuclear factor kappa B in human labour. Reprod. 2005;130:569–81. doi: 10.1530/rep.1.00197. [DOI] [PubMed] [Google Scholar]
- Sundaram S, et al. N,N-Dimethylacetamide regulates the proinflammatory response associated with endotoxin and prevents preterm birth. Am J Path. 2013;183:422–30. doi: 10.1016/j.ajpath.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelm M. Nitric oxide metabolism and breakdown. Biochim Biophys Acta. 1999;1411:273–89. doi: 10.1016/s0005-2728(99)00020-1. [DOI] [PubMed] [Google Scholar]
- Che Q, et al. Activation of a positive feedback loop involving IL-6 and aromatase promotes intratumoral 17b-estradiol biosynthesis in endometrial carcinoma microenvironment. Int J Cancer. 2014;135:282–94. doi: 10.1002/ijc.28679. [DOI] [PubMed] [Google Scholar]
- Manthey CL, Wang S-W, Kinney SD, Yao Z. SB202190, a selective inhibitor of p38 mitogen-activated protein kinase, is a powerful regulator of LPS-induced mRNAs in monocytes. Biochem Biophys Res Commun. 1999;263:825–31. doi: 10.1002/jlb.64.3.409. [DOI] [PubMed] [Google Scholar]
- Barnes JE, Ranta KE. The metabolism of dimethylformamide and dimethylacetamide. Tox App Pharm. 1972;23:271–76. doi: 10.1016/0041-008x(72)90190-1. [DOI] [PubMed] [Google Scholar]
- Menegola E, Broccia ML, Prati M, Giavini E. In vitro embryotoxicity study of n, n-dimethylacetamide and its main metabolite N-monomethylacetamide. Toxicol In Vitro. 1999;13:409–15. doi: 10.1016/s0887-2333(99)00006-5. [DOI] [PubMed] [Google Scholar]
- Tweedie D, et al. A cellular model of inflammation for identifying TNF-α synthesis inhibitors. J Neurosci Meth. 2009;183:182–87. doi: 10.1016/j.jneumeth.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raschke WC, Baird S, Ralph P, Nakoinz I. Functional macrophage cell lines transformed by Abelson leukemia virus. Cell. 1978;15:261–67. doi: 10.1016/0092-8674(78)90101-0. [DOI] [PubMed] [Google Scholar]
- Haskó G, et al. Isoproterenol inhibits IL-10, TNF-α, and nitric oxide production in RAW 264.7 macrophages. Brain Res Bull. 1998;45:183–87. doi: 10.1016/s0361-9230(97)00337-7. [DOI] [PubMed] [Google Scholar]
- Lee AK, Sung SH, Kim YC, Kim SG. Inhibition of lipopolysaccharide-inducible nitric oxide synthase, TNF-α and COX-2 expression by sauchinone effects on I-κBα phosphorylation, C/EBP and AP-1 activation. Br J Pharmacol. 2003;139:11–20. doi: 10.1038/sj.bjp.0705231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TH, Jung M, Bang M, Chung DK, Kim J. Inhibitory effects of a spinasterol glycoside on lipopolysaccharide-induced production of nitric oxide and proinflammatory cytokines via down-regulating MAP kinase pathways and NF-κB activation in RAW 264.7 macrophage cells. Int Immunopharmacol. 2012;13:264–70. doi: 10.1016/j.intimp.2012.05.005. [DOI] [PubMed] [Google Scholar]
- Kennedy Jr GL. Biological effects of acetamide, formamide, and their mono and dimethyl derivatives: an update. CRC Crit Rev Toxicol. 2001;31:139–222. doi: 10.1080/200140911116861. [DOI] [PubMed] [Google Scholar]
- Green SJ, Mellouk S, Hoffman SL, Meltzer MS, Nacy CA. Cellular mechanisms of nonspecific immunity to intracellular infection: Cytokine induced synthesis of toxic nitrogen oxides from L-arginine by macrophages and hepatocytes. Immunol Lett. 1990;25:15–19. doi: 10.1016/0165-2478(90)90083-3. [DOI] [PubMed] [Google Scholar]
- Green SJ, et al. Nitric oxide: Cytokine regulation of nitric oxide in host resistance to intracellular pathogens. Immunol Lett. 1994;43:87–94. doi: 10.1016/0165-2478(94)00158-8. [DOI] [PubMed] [Google Scholar]
- Hibbs JB, Taintor RR, Vavrin Z, Rachlin EM. Nitric oxide: A cytotoxic activated macrophage effector molecule. Biochem Biophys Res Commun. 1988;157:87–94. doi: 10.1016/s0006-291x(88)80015-9. [DOI] [PubMed] [Google Scholar]
- Archer S. Measurement of nitric oxide in biological models. FASEB J. 1993;7:349–60. doi: 10.1096/fasebj.7.2.8440411. [DOI] [PubMed] [Google Scholar]
- Das C, Sanyal M, Chandra Nag T. Localization of nitric oxide synthase in human trophoblast cells: role of nitric oxide in trophoblast proliferation and differentiation. Am J Reprod Immunol. 2000;43:70–77. doi: 10.1111/j.8755-8920.2000.430202.x. [DOI] [PubMed] [Google Scholar]
- Myatt L, et al. Inducible (type II) nitric oxide synthase in human placental villous tissue of normotensive, pre-eclamptic and intrauterine growth-restricted pregnancies. Placenta. 1997;18:261–68. doi: 10.1016/s0143-4004(97)80060-4. [DOI] [PubMed] [Google Scholar]
- Li X, Jiang S, Tapping RI. Toll-like receptor signaling in cell proliferation and survival. Cytokine. 2010;49:1–9. doi: 10.1016/j.cyto.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SH, Hwang D. Murine Toll-like receptor 4 confers lipopolysaccharide responsiveness as determined by activation of NF kappa B and expression of the inducible cyclooxygenase. J Biol Chem. 2000;275:34035–40. doi: 10.1074/jbc.M007386200. [DOI] [PubMed] [Google Scholar]
- Shoji T, et al. Involvement of p38 MAP kinase in lipopolysaccharide-induced production of pro- and anti-inflammatory cytokines and prostaglandin E 2 in human choriodecidua. J Reprod Immunol. 2007;75:82–90. doi: 10.1016/j.jri.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Lappas M, Permezel M, Rice GE. Mitogen-Activated Protein Kinase Proteins Regulate LPS-Stimulated Release of Pro-inflammatory Cytokines and Prostaglandins from Human Gestational Tissues. Placenta. 2007;28:936–45. doi: 10.1016/j.placenta.2007.02.009. [DOI] [PubMed] [Google Scholar]
- MacIntyre DA, et al. Activator protein 1 is a key terminal mediator of inflammation-induced preterm labor in mice. FASEB J. 2014;28:2358–68. doi: 10.1096/fj.13-247783. [DOI] [PubMed] [Google Scholar]
- Lappas M. Visfatin regulates the terminal processes of human labour and delivery via activation of the nuclear factor-kappa B pathway. Mol Cell Endocrinol. 2012;348:128–34. doi: 10.1016/j.mce.2011.07.048. [DOI] [PubMed] [Google Scholar]
- Lappas M, Yee K, Permezel M, Rice GE. Lipopolysaccharide and TNF-α activate the nuclear factor-kappa B pathway in the human placental JEG-3 cells. Placenta. 2006;27:568–75. doi: 10.1016/j.placenta.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Rauert-Wunderlich H, et al. The IKK inhibitor Bay 11–7082 induces cell death independent from inhibition of activation of NF-κB transcription factors. PloS One. 2013;8:e59292. doi: 10.1371/journal.pone.0059292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider S, Knöfler M. Human Tumour Necrosis Factor: Physiological and Pathological Roles in Placenta and Endometrium. Placenta. 2009;30:111–23. doi: 10.1016/j.placenta.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornblom SA, et al. Non-infected preterm parturition is related to increased concentrations of IL-6, IL-8 and MCP-1 in human cervix. Reprod Biol Endocrinol. 2005;3:39. doi: 10.1186/1477-7827-3-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch E, Wang H. The molecular pathophysiology of bacterially induced preterm labor: insights from the murine model. J Soc Gynecol Investig. 2005;12:145–55. doi: 10.1016/j.jsgi.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Miyamoto K, Matsukawa A, Ohkawara S, Takagi K, Yoshinaga M. IL-8 is involved in homologous TNFα-, but not in IL-1β-induced neutrophil infiltration in rabbits. Inflamm Res. 1997;46:472–77. doi: 10.1007/s000110050227. [DOI] [PubMed] [Google Scholar]
- Kloesch B, Liszt M, Broell J, Steiner G. Dimethyl sulphoxide and dimethyl sulphone are potent inhibitors of IL-6 and IL-8 expression in the human chondrocyte cell line C-28/I2. Life Sci. 2011;89:473–78. doi: 10.1016/j.lfs.2011.07.015. [DOI] [PubMed] [Google Scholar]
- Kumar D, et al. Decidual GM-CSF is a critical common intermediate necessary for thrombin and TNF induced in vitro fetal membrane weakening. Placenta. 2014;35:1049–56. doi: 10.1016/j.placenta.2014.10.001. [DOI] [PubMed] [Google Scholar]
- Moldenhauer L, Keenihan S, Hayball J, Robertson S. GM-CSF regulates uterine dendritic cell and macrophage maturation and antigen presentation. J Reprod Immunol. 2011;90:134–35. [Google Scholar]
- van Nieuwenhuijze A, et al. GM-CSF as a therapeutic target in inflammatory diseases. Mol Immunol. 2013;56:675–82. doi: 10.1016/j.molimm.2013.05.002. [DOI] [PubMed] [Google Scholar]
- Bozinovski S, et al. Innate immune responses to LPS in mouse lung are suppressed and reversed by neutralization of GM-CSF via repression of TLR-4. Am J Physiol Lung Cell Mol Physiol. 2004;286:L877–85. doi: 10.1152/ajplung.00275.2003. [DOI] [PubMed] [Google Scholar]
- Kacerovsky M, et al. Amniotic fluid protein profiles of intraamniotic inflammatory response to Ureaplasma spp. and other bacteria. PloS One. 2013;8:e60399. doi: 10.1371/journal.pone.0060399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanteux H, Guisset AC, Pilette C, Sibille Y. LPS induces IL-10 production by human alveolar macrophages via MAPKinase- and Sp1-dependent mechanisms. Respir Res. 2007;8:71. doi: 10.1186/1465-9921-8-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato TA, Keelan JA, Mitchell MD. Critical paracrine interactions between TNF-alpha and IL-10 regulate lipopolysaccharide-stimulated human choriodecidual cytokine and prostaglandin E2 production. J Immunol. 2003;170:158–66. doi: 10.4049/jimmunol.170.1.158. [DOI] [PubMed] [Google Scholar]
- Anchoori RK, Harikumar KB, Batchu VR, Aggarwal BB, Khan SR. Inhibition of IkB kinase and NF-κB by a novel synthetic compound SK 2009. Bioorg Med Chem. 2010;18:229–35. doi: 10.1016/j.bmc.2009.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiDonato JA, Mercurio F, Karin M. NF-κB and the link between inflammation and cancer. Immunol Rev. 2012;246:379–400. doi: 10.1111/j.1600-065X.2012.01099.x. [DOI] [PubMed] [Google Scholar]
- Ruiz S, Pergola PE, Zager RA, Vaziri ND. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int. 2013;83:1029–41. doi: 10.1038/ki.2012.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RG, Hayden MS, Ghosh S. NF-κB, inflammation, and metabolic disease. Cell Metab. 2011;13:11–22. doi: 10.1016/j.cmet.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catley MC, Chivers JE, Holden NS, Barnes PJ, Newton R. Validation of IKKβ as therapeutic target in airway inflammatory disease by adenoviral-mediated delivery of dominant-negative IKKβ to pulmonary epithelial cells. Br J Pharmacol. 2005;145:114–22. doi: 10.1038/sj.bjp.0706170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman-Blas JA, Jimenez SA. Targeting NF-κB: a promising molecular therapy in inflammatory arthritis. Int Rev Immunol. 2008;27:351–74. doi: 10.1080/08830180802295740. [DOI] [PubMed] [Google Scholar]
- Stinson LF, et al. Effects of cytokine-suppressive anti-inflammatory drugs on inflammatory activation in ex vivo human and ovine fetal membranes. Reproduction. 2014;147:313–20. doi: 10.1530/REP-13-0576. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.