

Abstract
Gastric cancer (GC) is characterized by amplifications of receptor tyrosine kinases (RTK) and KRAS, therefore, targeting of the RTK/KRAS downstream pathways could help to broaden the applicability of molecular targeted therapy for GC. We assembled a panel of 48 GC cell lines and screened predictors of responsiveness to inhibition of the RAF/MEK/ERK pathway, one of the RTK/KRAS downstream pathways. We found that GC cells with MET amplification or KRAS mutation, but not amplification, tended to be sensitive to MEK inhibition. However, several cell lines without RTK/KRAS alterations also showed high sensitivity to MEK inhibition. We then focused on the phosphorylation of RTK/KRAS downstream molecules to screen for predictors’ sensitivity to MEK inhibition. We found that the phosphorylation level of mammalian target of rapamycin complex 1 (mTORC1) downstream molecules, including p70S6K, 4EBP1, and S6, was significantly associated with sensitivity to MEK inhibition in GC cells (P < 0.05), suggesting that mTORC1 activity is related to the sensitivity to MEK inhibition. Furthermore, the change in mTORC1 activity after MEK inhibition was also significantly associated with this sensitivity (P < 0.001). Among the mTORC1 downstream molecules, the change in S6 phosphorylation (pS6) showed the most significant correlation with sensitivity. Using xenograft models derived from highly sensitive and resistant cell lines, we found specific reduction of pS6 in xenografts from highly sensitive cell lines after 6 h of treatment with an MEK inhibitor. Thus, our data suggest the potential clinical applicability of an MEK inhibitor for a proportion of GC patients who could be selected on the basis of pS6 change after MEK inhibition.
Keywords: Gastric cancer, MEK inhibitor, mTORC1, receptor tyrosine kinase, ribosomal protein S6
Gastric cancer remains one of the most common malignant diseases, with an estimated annual mortality of 700 000 worldwide, ranking third after lung and liver cancer (GLOBOCAN 2012).1 Despite a number of chemotherapy regimens using cytotoxic drugs,2, 3, 4 the prognosis of patients with advanced disease is still disappointing.
In addition to conventional chemotherapies, molecular targeted therapies have been applied successfully for treatment of various tumors, including GC.5, 6, 7, 8, 9, 10 A recent phase III randomized study (ToGA) has revealed that trastuzumab, an anti‐ERBB2 targeting antibody, improved the survival of GC patients with ERBB2 amplification when combined with cytotoxic drugs.10 However, patients with ERBB2‐positive GC represent only a small fraction (7–17%) of those with advanced GC.11, 12, 13 Therefore, identification of other targets is needed in order to increase the number of GC patients who would benefit from molecular targeted therapies.
It has been reported that genomic alterations in RTKs and KRAS occur in a mutually exclusive manner among GC patients,11, 14 suggesting that activation of the pathways downstream of RTK/KRAS, such as the RAF/MEK/ERK and PI3K/AKT/mTOR pathways, plays an important role in the proliferation and survival of GC cells. Therefore, targeting the pathways downstream of RTK/KRAS might broaden the applicability of molecular targeted therapy for GC patients. To verify this hypothesis, a large panel of GC cell lines, including a proportion harboring each of the RTK/KRAS alterations, would be required. However, drug sensitivity screening based on this approach has not yet been achieved for GC.
In the present study, we assembled a panel of 48 GC cell lines to examine the drug sensitivity profile of GC. To identify predictors of responsiveness to RAF/MEK/ERK pathway inhibition in GC, we evaluated the sensitivities of GC cells to MEK inhibition and compared them with the RTK/KRAS alteration status and phosphorylation levels of RTK/KRAS downstream molecules.
Materials and Methods
Details of short tandem repeat analysis, CGH, KRAS mutation analysis, and Bio‐Plex phosphoprotein assay (Bio‐Rad Laboratories, Hercules, CA, USA) are provided in Appendix S1.
Cell lines
We assembled a panel of 48 GC cell lines, purchased from cell repository banks (JCRB, RIKEN, ATCC, KCLB, and IBL) or provided by laboratories.15, 16, 17, 18, 19 The sources, culture conditions, differentiation statuses, and TP53 mutation statuses of the cell lines are listed in Table S1. HSC‐57, SH101‐P4, HSC‐64, HSC‐58, HSC‐60, HSC‐39, HSC‐43, HSC‐44PE, and HSC‐59 were established by one of the authors (K.Y.) previously.15, 16, 17, 18 Cell lines were obtained directly from the original providers, except for TMK‐1, which was provided by Dr. H. Ito (Tottori Prefectural Kousei Hospital, Tottori, Japan) with permission from an original provider, Dr. A. Ochiai (National Cancer Center, Kashiwa, Japan).19 Within 1 month of receipt, the cell lines were grown for several passages, and aliquots of each were frozen. For experiments, the cells were thawed and cultured for no more than 1 month before experimental use. All cell lines were cultured under conditions recommended by the providers and authenticated by short tandem repeat analysis with a PowerPlex16 HS System and PowerPlex Matrix Standards 3100/3130 kits in November 2014, in accordance with the manufacturer's instructions with slight modifications (Promega, Madison, WI, USA; Table S2).
Drug sensitivity assay
Cells were seeded at densities of 0.8–7.0 × 103 cells/well in 96‐well plates, depending on their growth speed, to reach subconfluency at 96 h after plating. They were incubated for 24 h and then treated with DMSO or the serially diluted compound (0.01, 0.1, 1.0, and 10 μM) for 72 h. The growth‐inhibitory effect of the MEK inhibitor PD0325901 (LC Laboratories, Woburn, MA, USA) was analyzed by CellTiter96 aqueous one solution cell proliferation assay (Promega) in quadruplicate and expressed as the IC50 value (Fig. S1), which was calculated using the linear relationship between the percentage inhibition and log concentration. The growth‐inhibitory effect of the PI3K/mTOR dual inhibitor PF04691502 (Selleck Chemicals, Houston, TX, USA) at a concentration of 1 μM was also analyzed in HGC‐27 cells.
Bio‐Plex phosphoprotein assay
Basal phosphorylation levels of ERK1/2 (Thr202/Tyr204 and Thr185/Tyr187 in ERK1 and ERK2, respectively), MEK1 (Ser217/Ser221), p90RSK (Thr359/Ser363), p38MAPK (Thr180/Tyr182), CREB (Ser133), ATF2 (Thr71), JNK (Thr183/Tyr185), c‐Jun (Ser63), AKT (Ser473), IRS1 (Ser636/Ser639), GSK3α/β (Ser9/Ser21), p70S6K (Thr421/Ser424), SRC (Tyr416), and EGFR (Tyr) were determined using customized panels of the Bio‐Plex Phosphoprotein Assay, Bio‐Plex Phosphoprotein Reagent, and Bio‐Plex Cell Lysis kits in accordance with the manufacturer's instructions (Bio‐Rad Laboratories). Details are provided in Appendix S1.
Western blot analysis
Western blotting was carried out as described previously.20 Cells were lysed on ice for 20 min in SDS‐modified RIPA buffer containing protease and phosphatase inhibitor cocktails (cOmplete Mini and PhosSTOP EASYpack; Roche Diagnostics, Mannheim, Germany), and then centrifuged at 17 400 g at 4°C for 20 min. The resulting cell lysates (20‐μg samples) were boiled with Laemmli sample buffer and subjected to SDS‐PAGE. The samples were transferred to a PVDF membrane (Millipore, Darmstadt, Germany), which was blocked for 1 h in Block Ace (DS Pharma, Osaka, Japan) at room temperature, then incubated overnight at 4°C with primary antibodies against pAKT (S473), pERK1/2 (T202/Y204), p‐p70S6K (T389), pS6 (S235/S236), p4EBP1 (S65), AKT, ERK, p70S6K, S6, and 4EBP (1:1000–2000; Cell Signaling Technology, Danvers, MA, USA), and GAPDH (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA). After washing with 1× TBS containing 0.1% Tween 20, membranes were incubated for 1 h at RT with appropriate secondary antibodies, followed by rewashing. Finally, the signals were detected using an ECL Western blotting analysis system (GE Healthcare, Piscataway, NJ, USA) in accordance with the manufacturer's instructions.
[35S]Methionine incorporation assay
To determine the rate of protein synthesis, cells treated with DMSO or 1 μM PD0325901 were incubated with methionine‐free RPMI‐1640 for 10 min, and then labeled with 2 MBq/mL [35S]methionine (PerkinElmer Japan, Tokyo, Japan) for 20 min. After a wash with PBS, the cells were lysed with a Total Protein Extraction Kit (BioChain, Hayward, CA, USA) in accordance with the manufacturer's instructions and centrifuged at 17 400 g for 20 min. The resulting cell lysates (40 μg/sample) were resuspended in Laemmli sample buffer and subjected to SDS‐PAGE using 10% gel. Radiolabeled proteins were visualized using a Typhoon FLA7000 laser scanner (GE Healthcare).
Xenograft model
A total of 1 × 107 cells suspended in 200 μL OPTI‐MEM (Life Technologies, Gaithersburg, MD, USA) were injected s.c. into the left flank of 7–10‐week‐old male NOD‐SCID mice (Charles River Laboratories, Yokohama, Japan). To determine the efficacy of the MEK inhibitor, PD0325901, the mice were divided into two groups 4 weeks after injection and then treated with oral PD0325901 (12.5 mg/kg/day, 5 consecutive days/week) dissolved in 200 μL vehicle (0.5% hydroxypropyl methylcellulose solution with 0.2% Tween 80) or an equal volume of vehicle only. Tumor volume was calculated twice a week using the formula: (width × length × height) / 2. Tumor volume data are presented as the mean tumor volume ± SE, and were analyzed by Student's t‐test. To determine the effect of MEK inhibition on pS6 expression, the mice were given PD0325901 orally (12.5 mg/kg) dissolved in 200 μL vehicle or an equal volume of vehicle at 4 weeks after the injection. Six hours after treatment, the tumors were removed and subjected to immunohistochemical analysis. All protocols for animal studies were approved by the animal ethics committee at Oita University (approval no. K008001).
Immunohistochemistry
We carried out immunohistochemistry on paraffin‐embedded blocks of xenografted tumor tissues using rabbit polyclonal antibodies against pERK diluted 1:100 (Cell Signaling Technology) and pS6 diluted 1:400 (Cell Signaling Technology), as described previously (ref: GC CGH), with slight modifications. Briefly, after antigen retrieval and inactivation of endogenous peroxidase, tissue sections were blocked with 2.5% horse serum (ImmPRESS reagent kit; Vector Laboratories, Burlingame, CA, USA) at room temperature for 30 min and then incubated at 4°C overnight with antibodies against pERK and pS6 diluted in SignalStain Antibody Diluent (Cell Signaling Technology) and 1% BSA/PBS, respectively. After washing in PBS, the sections were incubated at room temperature for 30 min with peroxidase‐conjugated horse anti‐rabbit Ig (ImmPRESS reagent kit; Vector Laboratories). Peroxidase activity was detected using ImmPACT DAB Peroxidase Substrate (Vector Laboratories).
Statistical analysis
Differences were analyzed by Mann–Whitney U‐test or two‐sided Student's t‐test by StatView (SAS Institute, Cary, NC, USA) and considered statistically significant at P < 0.05.
Results
Sensitivity of a panel of 48 GC cell lines to MEK inhibition
First, we investigated the status of RTK/KRAS amplifications and KRAS mutation in the GC cell lines using array CGH and sequencing, respectively (Fig. 1). Consistent with the clinical GC samples,11, 14 alterations related to RTK/KRAS signaling, including amplification of MET, FGFR2, ERBB2, and KRAS, were found to occur in a mutually exclusive manner (Fig. 1). Oncogenic mutation of KRAS, which is detected in 7.6% of all GC cases according to The Cancer Genome Atlas (http://cancergenome.nih.gov/),21 was also mutually exclusive to RTK amplifications in the cell lines (Fig. 1). These results suggested that the panel of 48 GC cell lines might be suitable for investigating the molecular basis of inhibition in GC. Next, we evaluated the sensitivity of a panel of 48 GC cell lines to PD0325901, an allosteric inhibitor of MEK, and divided them into sensitive (n = 22) and resistant (n = 26) groups at a cut‐off value of 1 μM (Fig. S1). We did not find any statistically significant relationships between sensitivity to MEK inhibition and the histology of the original tissues of each cell line (data not shown). As expected from previous reports, the cell lines with KRAS oncogenic mutations, but not amplifications, were sensitive to PD0325901,22, 23 except for one cell line, SH‐10‐TC (Fig. 1). Among the cell lines with RTK amplifications, MET‐amplified cells were relatively more sensitive to PD0325901 than ERBB2‐ or FGFR2‐amplified cells (Fig. 1). These results suggested that KRAS mutation and MET amplification are related to the responsiveness of GC cells to PD0325901. It was noteworthy that HSC‐64, NUGC‐4, OCUM‐1, and SNU‐719 showed the highest sensitivities to PD0325901, despite their lack of RTK/KRAS amplifications or KRAS mutation.
Figure 1.
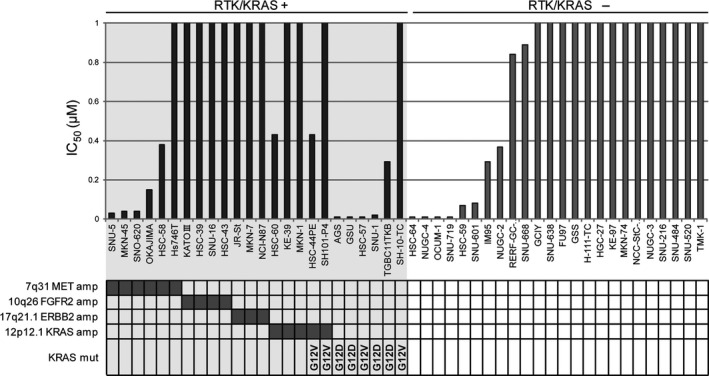
Relationship between sensitivity to PD0325901 and RTK/KRAS alterations in the panel of 48 gastric cancer cell lines. Cells were treated with 0.01–10 μM PD0325901 for 72 h and IC50 values were determined using the MTS assay. The amplification (amp) status of MET, FGFR2, ERBB2, and KRAS as well as mutation (mut) status of KRAS are shown below the cell line names.
Lower phosphorylation of mTORC1 downstream molecules correlates with susceptibility to MEK inhibition
Next, we compared the sensitivities of GC cells to MEK inhibition with the basal phosphorylation levels of the molecules downstream of RTK/KRAS, which were measured in 40 GC cell lines using the Bio‐Plex suspension array system (Fig. S2). As shown in Figure 2a, basal levels of pMEK and pERK were not correlated with PD0325901 sensitivity. Interestingly, although the basal level of pAKT showed only a slight tendency to correlate with PD0325901 sensitivity (P = 0.062), a considerably lower level of p‐p70S6K, which is regulated by the PI3K/AKT/mTOR pathway through mTORC1‐dependent phosphorylation, was observed in sensitive cells (P = 0.0041) (Fig. 2a). Such an association between the basal level of p‐p70S6K and sensitivity to PD0325901 was also observed by Western blotting (P = 0.0393; Fig. 2b). Furthermore, the PD0325901‐sensitive cells also showed a significantly lower level of p4EBP1 (P = 0.0017; Fig. 2b), another target of mTORC1, and pS6 (P = 0.0393; Fig. 2b), a direct target of p70S6K, suggesting that basal activity of mTORC1 is related to PD0325901 sensitivity in GC cells.
Figure 2.
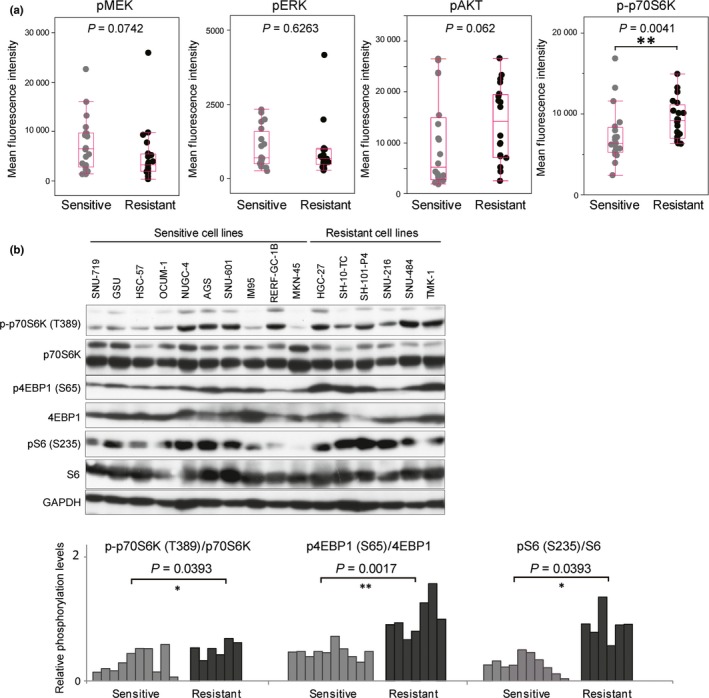
Lower basal levels of phosphorylated (p‐)p70S6K, p4EBP, and pS6 are related to sensitivity to MEK inhibition. (a) Basal levels of pMEK, pERK, pAKT, and p‐p70S6K were analyzed by Bio‐Plex phosphoprotein assay in 40 gastric cancer cell lines, and compared between the sensitive (20 cell lines) and resistant (20 cell lines) groups. (b) Basal levels of p‐p70S6K, p70S6K, p4EBP1, 4EBP1, pS6, S6, and GAPDH were analyzed by Western blotting in 16 gastric cancer cell lines (upper panel). Levels of p‐p70S6K/p70S6K, p4EBP1/4EBP1, and pS6/S6 were measured by quantification of the band intensities with ImageJ software (lower panel) and compared between the sensitive (10 cell lines) and resistant (6 cell lines) groups. Differences were calculated by the Mann–Whitney U‐test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Gastric cancer cell lines sensitive to MEK inhibitor show significant reduction of mTORC1 activity after MEK inhibition
Next, to further determine whether mTORC1 activity is related to PD0325901 sensitivity, we investigated changes in the phosphorylation levels of ERK, AKT, and downstream molecules of mTORC1, including p70S6K, 4EBP1, and S6, resulting from PD0325901 treatment and compared them between sensitive (n = 21) and resistant (n = 26) groups. As expected, pERK was effectively reduced by the PD0325901 treatment (Fig. 3a), and no significant difference in the degree of reduction was observed between the two groups (Fig. 3b), suggesting that sensitivity to PD0325901 is independent of the level of pERK suppression. The level of pAKT was modestly affected in some cell lines by the treatment (Fig. 3a). Overall, the resistant group showed a slight, but not significant, increase in the level of pAKT (P = 0.0627; Fig. 3b). In contrast, striking decreases in the levels of p‐p70S6K, p4EBP1, and pS6 were observed specifically in sensitive cells after MEK inhibition (Figs 3a,S3), and the ratios of the decrease were much greater in the sensitive group (P < 0.001; Fig. 3b), suggesting that reduction of mTORC1 activity after MEK inhibition may be closely associated with PD0325901 sensitivity. Indeed, the changes in p‐p70S6K and p4EBP1 expression, both of which are direct targets of mTORC1, after the PD0325901 treatment were significantly correlated (Pearson's r = 0.59, P < 0.0001; Fig. 3c). Among the three downstream molecules of mTORC1, pS6 showed the most significant association with sensitivity, even when we changed the cut‐off values to 0.01 μM and 10 μM to distinguish highly sensitive and resistant cell lines (Fig. S4). Furthermore, the strongest correlation was observed between the ratios of the change in pS6 and antiproliferative activity following treatment with 1 μM PD0325901 (Pearson's r = 0.73, P < 0.0001; Fig. S5). These results suggest that the change in pS6 resulting from PD0325901 treatment could be a better predictor of the sensitivity to PD0325901 than other mTORC1 downstream molecules.
Figure 3.
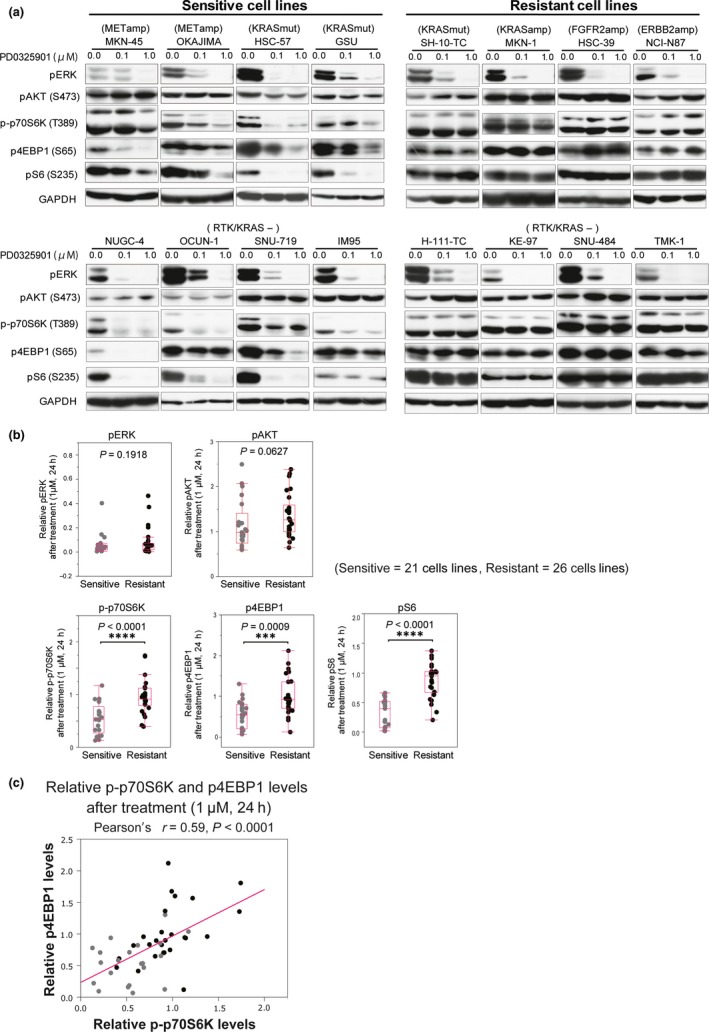
Suppression of phosphorylated (p‐)p70S6K, p4EBP, and pS6 after MEK inhibition is related to sensitivity to MEK inhibition. (a) Sensitive (MKN‐45, OKAJIMA, HSC‐57, GUS, NUGC‐4, OCUM‐1, SNU‐719, and IM95) and resistant (SH‐10‐TC, MKN‐1, HSC‐39, NCI‐N87, H‐111‐TC, KE‐97, SNU‐484, and TMK‐1) cell lines were treated with DMSO or PD0325901 (0.1 or 1 μM) for 24 h and then subjected to Western blotting using antibodies against pERK, pAKT, p‐p70S6K, p4EBP1, pS6, and GAPDH. (b) Sensitive (n = 21) and resistant (n = 26) cell lines were treated with DMSO or PD0325901 (1 μM) for 24 h and subjected to Western blotting using antibodies against pERK, pAKT, p‐p70S6K, p4EBP1, pS6, and GAPDH. Band intensities of phosphoproteins were normalized against that of GAPDH. Differences in the phosphorylation levels of PD0325901‐treated cells, expressed relative to those in DMSO‐treated cells, between the sensitive and resistant groups were calculated by the Mann–Whitney U‐test. (c) The correlation between p‐p70S6K and p4EBP1 after PD0325901 treatment (1 μM) for 24 h.
As mTORC1 mainly functions in translational control through p70S6K, 4EBP1, and pS6,24, 25, 26 we investigated the effect of the decrease in their phosphorylation levels on global protein synthesis after PD0325901 treatment. As shown in Figure 4a, sensitive cells showed significant reductions in the incorporation of [35S]methionine as a result of the treatment, whereas resistant cells did not, suggesting that reduced mTORC1 activity is associated with the rate of decrease in protein synthesis. Interestingly, treatment with 1 μM PF04691502, which inhibits the activities of both PI3K and mTOR, markedly reduced the viability of HGC‐27 (Fig. 4b), a PD0325901‐resistant cell line (Figs 1,4a), accompanied by a decrease of p‐p70S6K, p4EBP1, pS6, and protein synthesis (Fig. 4b). Therefore, it is possible that, in a proportion of PD0325901‐resistant cells, mTORC1 activity also plays a pivotal role in viability by sustaining the translation through p‐p70S6K, p4EBP1, and S6.
Figure 4.
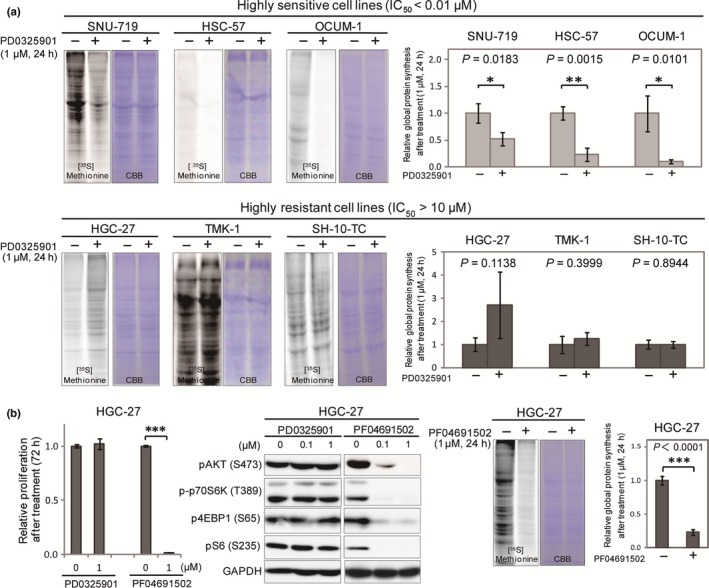
Suppression of global protein synthesis after MEK inhibition is involved in sensitivity to MEK inhibition. (a) Sensitive (SNU‐719, HSC‐57, and OCUM‐1) and resistant (HGC‐27, TMK‐1, and SH‐10‐TC) cell lines were treated with DMSO or 1 μM PD0325901 for 24 h and then subjected to [35S]methionine incorporation assay. Equal loading of the proteins was shown by staining with Coomassie brilliant blue. Intensities of protein bands labeled with [35S]methionine were quantified by ImageJ software, and normalized against those labeled with Coomassie brilliant blue. Representative band images are shown on the left. Differences in incorporated [35S]methionine between cells treated with DMSO (n = 3) and 1 μM PD0325901 (n = 3) were analyzed by Student's t‐test and shown on the right. (b) HGC‐27 cells were treated with DMSO, PD0325901, or PF04691502 (0.1 or 1.0 μM) and subjected to cell proliferation analyses and Western blotting with antibodies against pAKT, p‐p70S6K, p4EBP1, and GAPDH (left). HGC‐27 cells treated with DMSO or 1 μM PF042691502 were also subjected to [35S]methionine incorporation assay (right). *P < 0.05, **P < 0.01, ****P < 0.0001.
Prediction of sensitivity to MEK inhibition by immunohistochemical detection of pS6 in xenograft models
To determine whether the change in pS6 after MEK inhibition can be used to predict sensitivity to MEK inhibition in vivo, we established xenograft models using in vitro sensitive (HSC‐57) and resistant (HGC‐27) cell lines (Fig. 5). Consistent with the in vitro results (Fig. 1), HSC‐57 and HGC‐27 derived xenografts were found to be highly sensitive and resistant to the PD0325901 treatment, respectively (Fig. 5). As shown in Figure 5c, we found that a single treatment with PD0325901 for 6 h suppressed pS6 as well as pERK in HSC‐57 derived xenografts. We determined this treatment period based on the observation that pS6 was completely suppressed in highly sensitive cells in vitro at 6 h after treatment (Fig. S6). In contrast, treatment with PD0325901 did not affect the level of pS6 in HGC‐27 xenografts, despite the complete suppression of pERK (Fig. 5d). We also found that treatment with PD0325901 for 6 h caused suppression of pS6 in in vitro xenografts derived from the highly sensitive cell lines, NUGC‐4 and GSU, whereas pS6 was not affected in in vitro xenografts derived from the highly resistant cell lines, SH‐10‐TC, TMK‐1, and MKN‐1 (Fig. S7). Although the number of xenograft models was limited, these results suggested that the change in pS6 resulting from MEK inhibition would allow distinction between highly sensitive and resistant tumors in vivo.
Figure 5.
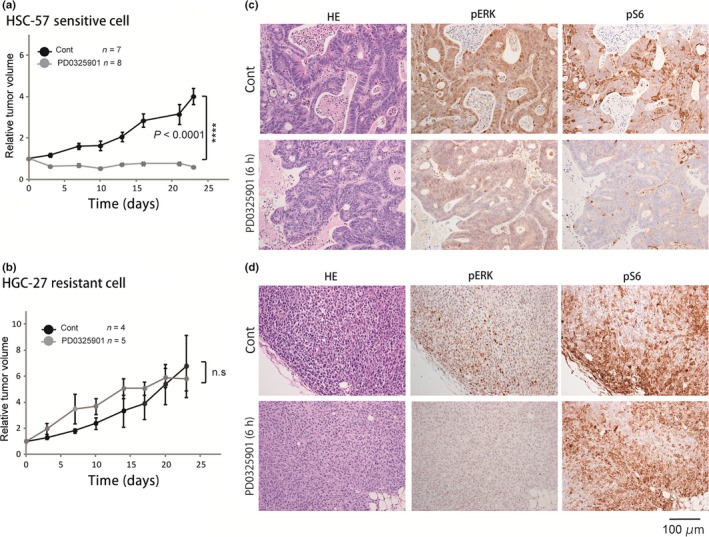
Efficacy of MEK inhibition on xenograft models established from HSC‐57 and HGC‐27 cell lines. (a, b) Tumor xenografts established from HSC‐57 (highly sensitive) (a) and HGC‐27 (highly resistant) (b) cell lines were treated with vehicle (Cont) or PD0325901 (12.5 mg/kg) for 5 consecutive days/week for 23 days. The mean changes in tumor volume relative to initial tumor volume are shown. Error bars represent mean ± SE. (c, d) Representative images of immunohistochemistry using antibodies against phosphorylated (p)ERK and pS6 in HSC‐57 (highly sensitive) (c) and HGC‐27 (highly resistant) (d) tumor xenografts from mice killed 6 h after treatment with a single dose of vehicle (Cont) or PD0325901 (12.5 mg/kg).
Discussion
A primary finding of this study was that the change in mTORC1 activity after MEK inhibition was able to predict the responsiveness of gastric cancer cells to MEK inhibition. In general, genomic alterations, such as mutations or amplifications, tend to be utilized as predictors of responsiveness to molecular targeting drugs. However, in GC, it seems reasonable to use factors other than genomic alterations for prediction of responsiveness to MEK inhibition, for the following reasons. First, the RAF/MEK/ERK pathway in GC can be activated by multiple types of genomic alterations, such as RTK amplification, KRAS amplification/mutation, and MEK mutation.11, 21, 27, 28, 29, 30 Second, even when cancer cells harbor genetic alterations, such as KRAS mutation, that are highly associated with sensitivity to MEK inhibition, they often show resistance to MEK inhibition due to signaling cross‐talk or feedback loops at the protein level.31, 32, 33, 34 In this context, we hypothesized that the response of the signaling pathway to drug treatment could be a better predictor than conventional genomic alterations in GC. In fact, we showed that the change in mTORC1 activity after MEK inhibition was well correlated with responsiveness to MEK inhibition, even among cell lines harboring KRAS mutation. Moreover, highly sensitive cell lines without RTK/KRAS alterations, such as HSC‐64, NUGC‐4, OCUM‐1, and SNU‐719, also showed reduction of mTORC1 activity after MEK inhibition, suggesting that reduced mTORC1 activity might predict responsiveness to MEK inhibition even in patients without conventional genetic predictors. Thus, our data suggest that reduced mTORC1 activity might have potential utility as a predictor of MEK inhibition, independent of genetic predictors. By combining conventional genetic predictors, such as KRAS mutation, with the change in mTORC1 activity after MEK inhibition, it might be possible to establish a more accurate system for prediction of responsiveness to MEK inhibition.
The mechanism whereby MEK inhibition leads to suppression of mTORC1 activity remains largely unknown. The majority of previous studies have suggested that the PI3K/AKT pathway is a dominant regulator of mTORC1.25, 35 However, at least in GCs, the pathway seems unrelated to MEK inhibitor‐mediated regulation of mTORC1 activity, because the change in pAKT was not as significantly associated with sensitivity to MEK inhibition compared with the changes in p‐p70S6K, p4EBP1, and pS6. Importantly, similarly to previous reports that mTORC1 activity mediated by PI3K/AKT signaling is important for protein synthesis and cell proliferation,35, 36 the changes in mTORC1 activity after MEK inhibition shown in our present study are also strongly associated with the translation rate and antiproliferative effect in the highly sensitive and resistant GC cells. Interestingly, even in an MEK inhibition‐resistant cell line, HGC‐27, suppression of mTORC1 activity by treatment with PF04691502 caused a significant antiproliferative effect accompanied by a decrease in protein synthesis, although MEK inhibition had little effect on mTORC1 activity in this cell line. Thus, our data suggest that mTORC1 activity may serve as not only a predictor of responsiveness to MEK inhibition but also a pivotal survival factor in MEK inhibition‐sensitive and some‐resistant GC cells. Further studies will be required to determine whether mTORC1 inhibition could be effective in GC cells.
The reason why HSC‐64, NUGC‐4, OCUM‐1, and SNU‐719 cells showed high sensitivity to MEK inhibition remains unclear. One possibility is that these cell lines harbored alterations of molecules in the RTK/KRAS/ERK pathway that were undetectable by the methods used in this study. Indeed, Sogabe et al. reported that GC cell lines with MEK mutation, including OCUM‐1, show high sensitivity to MEK inhibition.28 Considering that sensitivity to MEK inhibition is highly associated with mTORC1 activity, alterations of the regulators of mTORC1 activity, such as negative and positive regulators, might also be plausible. An understanding of the mechanisms underlying the high sensitivity of these cell lines to MEK inhibition would be beneficial for identification of novel predictors of MEK inhibition.
In order to predict the sensitivity to MEK inhibition based on the response to drug treatment in a clinical setting, the response should be dynamic and easy to detect using conventional methods. Our data suggest that the change in pS6 might be most suitable for prediction of responsiveness to MEK inhibition because the change in pS6 after MEK inhibition showed the highest correlation with the antiproliferative effect of MEK inhibition among the three mTORC1 downstream molecules. Furthermore, the degree of difference between the median values of the sensitive and resistant groups was the greatest in pS6 among the mTORC1 downstream molecules (median values for p‐p70S6K, p4EBP1, and pS6 among the sensitive versus resistant cell lines were 0.53 vs 0.92, 0.55 vs 0.92, and 0.40 vs 0.95, respectively; Fig. 3b). These differences were more evident when we used the cut‐off values 0.01 μM and 10 μM to distinguish highly sensitive and resistant cell lines (Fig. S4). This feature is preferable as a predictor because selection of super‐responsive or highly resistant patients is crucial for the clinical application of molecular targeting drugs. In the present study, we showed that the change in pS6 was easy to detect by immunohistochemistry with HSC‐57 and HGC‐27 xenograft models, and sufficiently dynamic to allow distinction between HSC‐57 xenograft models with and without MEK inhibition. Thus, our data suggest that patients with MEK inhibition‐sensitive GC would be distinguishable by evaluating the change in pS6 after treatment with an MEK inhibitor.
In order to achieve molecular targeting therapy for genetically heterogeneous GC, we propose a new concept that would allow patients to be selected on the basis of the phosphorylation level of signaling molecules and targeting of a common pathway downstream from various types of genomic alterations. Currently, trastuzumab, an ERBB2 targeting drug, is the only molecular targeting drug that has been shown to be effective for GC patients.10 However, patients with ERBB2 amplification represent only a small fraction of GC patients (7–17%).11, 12, 13 Interestingly, we found that none of the cells that were highly sensitive to MEK inhibition harbored ERBB2 amplification, suggesting that use of MEK targeting drugs might increase the proportion of GC patients who would benefit from molecular targeted therapy. Furthermore, our data suggest that it would be possible to distinguish GC patients with high sensitivity to MEK inhibitor by comparison of pS6 before and after treatment with an MEK inhibitor. Although currently it would be difficult to obtain paired clinical samples of GC before and after chemotherapy, this could be a feasible and worthwhile approach, as immunohistochemical analysis of serial biopsy samples from melanoma patients before and after RAF/MEK/ERK pathway inhibition has shown a tight correlation between a reduced level of pS6 after treatment and subsequent responsiveness to RAF and/or MEK inhibition.37 Currently, clinical use of MEK inhibitor is restricted only to melanoma patients. In future, through selection of the patients based on the change in pS6, clinical trials of MEK inhibitors might be feasible for GC. Further studies to investigate the association between sensitivity to MEK inhibition and the change in pS6 using larger number of models will be required.
Disclosure Statement
The authors have no conflict of interest.
Abbreviations
- AKT
protein kinase B
- CGH
array‐comparative genomic hybridization
- ERBB2
Erb‐B2 receptor tyrosine kinase 2
- FGFR2
fibroblast growth factor receptor 2
- GC
gastric cancer
- mTOR
mammalian target of rapamycin
- mTORC1
mammalian target of rapamycin complex 1
- p
phosphorylated
- PI3K
phosphatidylinositol 3‐kinase
- pS6
phosphorylated ribosomal protein S6
- RTK
receptor tyrosine kinase
Supporting information
Fig. S1. IC50 values of 48 GC cell lines for PD0325901 treatment as assessed by MTS assay.
Fig. S2. Correlation between phosphorylation of RTK/KRAS downstream molecules and sensitivity to MEK inhibition.
Fig. S3. Phosphorylation levels of p70S6K, 4EBP1, and S6 after MEK inhibition.
Fig. S4. Comparison of mTORC1 activity after MEK inhibition between two groups of gastric cancer cell lines at a cut‐off value of 0.01 or 10 μM.
Fig. S5. Coefficient of correlation between phosphorylation levels downstream of mTORC1 and proliferative activity after MEK inhibition.
Fig. S6. Time course of reduction in pERK, p‐p70S6K, pS6, and p4EBP1 after MEK inhibition in highly sensitive cells.
Fig. S7. Prediction of sensitivity to MEK inhibition by immunohistochemical detection of pS6 in highly sensitive and resistant cell lines.
Table S1. Characteristics of the 48 gastric cancer cell lines.
Table S2. Short tandem repeat analysis of gastric cancer cell lines.
Appendix S1. Supplementary materials and methods.
Acknowledgments
We would like to thank Ms. Misuzu Taguchi, Ms. Kanako Yoshizumi, and Ms. Mami Kimoto for their excellent assistance with experiments. This work was supported by the Discretion of the President of Oita University, 2013 and 2014.
Cancer Sci 107 (2016) 1919–1928
Funding Information
Oita University
References
- 1. Ferlay J, Soerjomataram I, Dikshit R et al Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015; 136: E359–E386. [DOI] [PubMed] [Google Scholar]
- 2. Koizumi W, Narahara H, Hara T et al S‐1 plus cisplatin versus S‐1 alone for first‐line treatment of advanced gastric cancer (SPIRITS trial): a phase III trial. Lancet Oncol 2008; 9: 215–221. [DOI] [PubMed] [Google Scholar]
- 3. Narahara H, Iishi H, Imamura H et al Randomized phase III study comparing the efficacy and safety of irinotecan plus S‐1 with S‐1 alone as first‐line treatment for advanced gastric cancer (study GC0301/TOP‐002). Gastric Cancer 2011; 14(1): 72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boku N, Yamamoto S, Fukuda H, Shirao K, Doi T, Sawaki A et al Fluorouracil versus combination of irinotecan plus cisplatin versus S‐1 in metastatic gastric cancer: a randomised phase 3 study. Lancet Oncol 2009; 10: 1063–1069. [DOI] [PubMed] [Google Scholar]
- 5. Maemondo M, Inoue A, Kobayashi K et al Gefitinib or chemotherapy for non‐small‐cell lung cancer with mutated EGFR. N Engl J Med 2010; 362: 2380–2388. [DOI] [PubMed] [Google Scholar]
- 6. Chapman PB, Hauschild A, Robert C et al Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011; 364: 2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hortobagyi GN. Trastuzumab in the treatment of breast cancer. N Engl J Med 2005; 353: 1734–1736. [DOI] [PubMed] [Google Scholar]
- 8. Druker BJ, Guilhot F, O'Brien SG et al Five‐year follow‐up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 2006; 355: 2408–2417. [DOI] [PubMed] [Google Scholar]
- 9. Van Cutsem E, Kohne CH, Hitre E et al Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 2009; 360: 1408–1417. [DOI] [PubMed] [Google Scholar]
- 10. Bang YJ, Van Cutsem E, Feyereislova A et al Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2‐positive advanced gastric or gastro‐oesophageal junction cancer (ToGA): a phase 3, open‐label, randomised controlled trial. Lancet 2010; 376: 687–697. [DOI] [PubMed] [Google Scholar]
- 11. Deng N, Goh LK, Wang H et al A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co‐occurrence among distinct therapeutic targets. Gut 2012; 61: 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gravalos C, Jimeno A. HER2 in gastric cancer: a new prognostic factor and a novel therapeutic target. Ann Oncol 2008; 19: 1523–1529. [DOI] [PubMed] [Google Scholar]
- 13. Tanner M, Hollmen M, Junttila TT et al Amplification of HER‐2 in gastric carcinoma: association with Topoisomerase IIalpha gene amplification, intestinal type, poor prognosis and sensitivity to trastuzumab. Ann Oncol 2005; 16: 273–278. [DOI] [PubMed] [Google Scholar]
- 14. Das K, Gunasegaran B, Tan IB, Deng N, Lim KH, Tan P. Mutually exclusive FGFR2, HER2, and KRAS gene amplifications in gastric cancer revealed by multicolour FISH. Cancer Lett 2014; 353: 167–175. [DOI] [PubMed] [Google Scholar]
- 15. Yanagihara K, Tanaka H, Takigahira M et al Establishment of two cell lines from human gastric scirrhous carcinoma that possess the potential to metastasize spontaneously in nude mice. Cancer Sci 2004; 95: 575–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yanagihara K, Ito A, Toge T, Numoto M. Antiproliferative effects of isoflavones on human cancer cell lines established from the gastrointestinal tract. Cancer Res 1993; 53: 5815–5821. [PubMed] [Google Scholar]
- 17. Yanagihara K, Seyama T, Tsumuraya M, Kamada N, Yokoro K. Establishment and characterization of human signet ring cell gastric carcinoma cell lines with amplification of the c‐myc oncogene. Cancer Res 1991; 51(1): 381–386. [PubMed] [Google Scholar]
- 18. Yanagihara K, Kamada N, Tsumuraya M, Amano F. Establishment and characterization of a human gastric scirrhous carcinoma cell line in serum‐free chemically defined medium. Int J Cancer 1993; 54: 200–207. [DOI] [PubMed] [Google Scholar]
- 19. Ochiai A, Yasui W, Tahara E. Growth‐promoting effect of gastrin on human gastric carcinoma cell line TMK‐1. Jpn J Cancer Res 1985; 76: 1064–1071. [PubMed] [Google Scholar]
- 20. Moriyama M, Tsukamoto Y, Fujiwara M et al Identification of a novel human ankyrin‐repeated protein homologous to CARP. Biochem Biophys Res Commun 2001; 285: 715–723. [DOI] [PubMed] [Google Scholar]
- 21. Research CGA. N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014; 513: 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yeh JJ, Routh ED, Rubinas T et al KRAS/BRAF mutation status and ERK1/2 activation as biomarkers for MEK1/2 inhibitor therapy in colorectal cancer. Mol Cancer Ther 2009; 8: 834–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jing J, Greshock J, Holbrook JD et al Comprehensive predictive biomarker analysis for MEK inhibitor GSK1120212. Mol Cancer Ther 2012; 11: 720–729. [DOI] [PubMed] [Google Scholar]
- 24. Schalm SS, Blenis J. Identification of a conserved motif required for mTOR signaling. Curr Biol 2002; 12: 632–639. [DOI] [PubMed] [Google Scholar]
- 25. Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell 2007; 12: 9–22. [DOI] [PubMed] [Google Scholar]
- 26. Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. A unifying model for mTORC1‐mediated regulation of mRNA translation. Nature 2012; 485: 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Roberts PJ, Der CJ. Targeting the Raf‐MEK‐ERK mitogen‐activated protein kinase cascade for the treatment of cancer. Oncogene 2007; 26: 3291–3310. [DOI] [PubMed] [Google Scholar]
- 28. Sogabe S, Togashi Y, Kato H et al MEK inhibitor for gastric cancer with MEK1 gene mutations. Mol Cancer Ther 2014; 13: 3098–3106. [DOI] [PubMed] [Google Scholar]
- 29. Morishita A, Gong J, Masaki T. Targeting receptor tyrosine kinases in gastric cancer. World J Gastroenterol 2014; 20: 4536–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee SH, Lee JW, Soung YH et al BRAF and KRAS mutations in stomach cancer. Oncogene 2003; 22: 6942–6945. [DOI] [PubMed] [Google Scholar]
- 31. Sun C, Hobor S, Bertotti A et al Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep 2014; 7(1): 86–93. [DOI] [PubMed] [Google Scholar]
- 32. Temraz S, Mukherji D, Shamseddine A. Dual inhibition of MEK and PI3K pathway in KRAS and BRAF mutated colorectal cancers. Int J Mol Sci 2015; 16: 22976–22988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Van Schaeybroeck S, Kalimutho M, Dunne PD et al ADAM17‐dependent c‐MET‐STAT3 signaling mediates resistance to MEK inhibitors in KRAS mutant colorectal cancer. Cell Rep 2014; 7: 1940–1955. [DOI] [PubMed] [Google Scholar]
- 34. Lamba S, Russo M, Sun C et al RAF suppression synergizes with MEK inhibition in KRAS mutant cancer cells. Cell Rep 2014; 8: 1475–1483. [DOI] [PubMed] [Google Scholar]
- 35. Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci 2009; 122: 3589–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Silvera D, Formenti SC, Schneider RJ. Translational control in cancer. Nat Rev Cancer 2010; 10: 254–266. [DOI] [PubMed] [Google Scholar]
- 37. Corcoran RB, Rothenberg SM, Hata AN et al TORC1 suppression predicts responsiveness to RAF and MEK inhibition in BRAF‐mutant melanoma. Sci Transl Med 2013; 5: 196ra98. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. IC50 values of 48 GC cell lines for PD0325901 treatment as assessed by MTS assay.
Fig. S2. Correlation between phosphorylation of RTK/KRAS downstream molecules and sensitivity to MEK inhibition.
Fig. S3. Phosphorylation levels of p70S6K, 4EBP1, and S6 after MEK inhibition.
Fig. S4. Comparison of mTORC1 activity after MEK inhibition between two groups of gastric cancer cell lines at a cut‐off value of 0.01 or 10 μM.
Fig. S5. Coefficient of correlation between phosphorylation levels downstream of mTORC1 and proliferative activity after MEK inhibition.
Fig. S6. Time course of reduction in pERK, p‐p70S6K, pS6, and p4EBP1 after MEK inhibition in highly sensitive cells.
Fig. S7. Prediction of sensitivity to MEK inhibition by immunohistochemical detection of pS6 in highly sensitive and resistant cell lines.
Table S1. Characteristics of the 48 gastric cancer cell lines.
Table S2. Short tandem repeat analysis of gastric cancer cell lines.
Appendix S1. Supplementary materials and methods.