

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is the most representative form of pancreatic cancers. PDAC solid tumours are constituted of heterogeneous populations of cells including cancer stem cells (CSCs), differentiated cancer cells, desmoplastic stroma and immune cells. The identification and consequent isolation of pancreatic CSCs facilitated the generation of genetically engineered murine models. Nonetheless, the current models may not be representative for the spontaneous tumour occurrence. In the present study, we show the generation of a novel pancreatic iPSC-converted cancer stem cell lines (CSCcm) as a cutting-edge model for the study of PDAC. The CSCcm lines were achieved only by the influence of pancreatic cancer cell lines conditioned medium and were not subjected to any genetic manipulation. The xenografts tumours from CSCcm lines displayed histopathological features of ADM, PanIN and PDAC lesions. Further molecular characterization from RNA-sequencing analysis highlighted primary culture cell lines (1st CSCcm) as potential candidates to represent the pancreatic CSCs and indicated the establishment of the pancreatic cancer molecular pattern in their subsequent progenies 2nd CSCcm and 3rd CSCcm. In addition, preliminary RNA-seq SNPs analysis showed that the distinct CSCcm lines did not harbour single point mutations for the oncogene Kras codon 12 or 13. Therefore, PDAC-CSCcm model may provide new insights about the actual occurrence of the pancreatic cancer leading to develop different approaches to target CSCs and abrogate the progression of this fatidic disease.
Keywords: Cancer stem cells, iPSCs, pancreatic cancer, PDAC, CSCcm, conditioned medium
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the most highly desmoplastic tumours which unfortunately due to its aggressiveness and rapid dissemination together with the strong resistance to the radiation therapy and chemotherapy contributes to the dismal prognosis. Over the past decades the strategies to find new diagnostic approaches at early stages along with the effective treatments has not improved significantly [1,2]. Therefore, this highlights the urgent need to find novel models to study the origin as well as the progression of the disease.
PDAC solid tumours are comprised of a wide range of heterogeneous populations of cells including cancer stem cells (CSCs), the actual differentiated cancer cells together with desmoplastic stroma and immune cells which represent a high proportion of the tumour mass [3]. CSCs are considered as cells that possess stem cell properties and produce diverse lineages of cancer cells. Hence, CSCs have been associated with the tumour initiation and progression, and have been reported to be involved in tumour metastasis [4].
The isolation of pancreatic CSCs succeeded in providing new insights regarding the chemoresistance and the high metastatic ability in PDAC. Since CSCs and non-CSCs share an identical genetic background it is difficult to find appropriate in vitro and in vivo systems that allows to select reproducibly and exclusively enriched CSCs populations. Furthermore, these new approaches include particular markers that are also found in differentiated adult cells what makes questionable the identification of the CSCs [7]. Recent advances have been developed in targeting CSCs and their identification and isolation consequently facilitate the generation of new murine models [5,6]. However, the current models are genetically engineered and therefore may not be suitable for a better understanding of the spontaneous tumour occurrence.
As have been seen in regenerative medicine field, iPSCs when exposed to appropriate environments are able to directly differentiate into progenitor cells that lead to the latter matured form of cells. Hence, the signals found in the niche simultaneously regulate the differentiation as well as support the tissue homeostasis preserving the self-renewal potential from a minor but required stem cells number [8]. Based on this we previously hypothesized that CSCs might be considered as progenitor cells that are destined to differentiate into cancer cells and that consequently if the cell fate comes determined by the events and factors present in the niche, the tumour microenvironment should exert the same effects when healthy cells are exposed to it. Chen L and Kasai T et al. demonstrated the impact of the so-called cancerous niche when by exposing Nanog iPSCs to a Lewis Lung carcinoma conditioned medium (LLCcm) a malignant tumour was obtained exhibiting angiogenesis in vivo, capacity of self-renewal and expressed markers associated to stem cell properties and undifferentiated state such as Nanog, Rex1, Eras, Esg1 and Cripto. In contrast, when control Nanog iPSCs were implanted into Balb/c nude mice formed typical teratomas displaying contained differentiated tissues without metastasis. Thus a new model of CSC-like cells generated exclusively under the influence of the microenvironment was proposed [9].
In the present study, we show the generation of a novel pancreatic iPSC-converted cancer stem cell lines (CSCcm) together with the subsequent characterization of the tumours obtained as a result from the transplantation of the CSCcm lines in vivo demonstrating that CSCcm is a promising cutting-edge model for the study of PDAC occurrence and progression.
Material and methods
Cell culture
Human pancreatic carcinoma cell lines PK-8 and KLM-1 (RIKEN cell Bank, Japan) were cultured in RPMI 1640 Sigma, 10% FBS and 100 U/mL Penicillin. Prior to use, CM was centrifuged at 1000 rpm for 5 min and filtered using 0.22 µm diameter pore filter (Millipore, Ireland). Undifferentiated feeder-less Nanog iPSCs were seeded at 5×105 cells/mL and maintained with iPSCs medium (DMEM D5796 Sigma, 15% FBS, 2 mM L-Glutamine, 0.1 mM NEAA, 50 U/mL Penicillin and 50 U/mL Streptomycin, 0.1 mM 2-mercaptoethanol), without LIF and in combination with the CM from PK-8 and KLM-1 cell lines during 30 days. The medium was changed every 24 h. Primary cultures from 1st CSCcm, 2nd CSCcm and 3rd CSCcm of PK8CM and KLM-1CM mouse allografts were prepared as Chen L and Kasai T et al. [9]. After 24 h cultures were enriched with 1 mg/mL puromycin and medium was replaced every 24 h. Cells were maintained no longer than the 5th passage.
Sphere formation assay
CSCcm spheres were generated as previously described [9].
Animal experiments
Balb/c-nu/nu, female, 4-week old were purchased from Charles River, Japan. For subcutaneous transplantation cells were suspended in 100 µl of HBSS buffer. Pancreatic orthotopic transplantation was performed as described by Bruns et al. 1999 [40] with the only variation in the number of implanted cells (105/20 µl and 106/20 µl HBSS). The mice were housed in accordance with the plan of animal experiments reviewed and approved by the ethics committee for animal experiments of Okayama University under the IDs OKU-2008211, OKU-2009144, OKU-2010179 and OKU-2011-305.
Histologic analysis
Tumours were fixed in 4% PFA (WAKO, Japan), embedded in paraffin-wax and sectioned for histologic examination at 5 µm. Sections were stained with Hematoxylin (Sigma-Aldrich, MO; 0.5%) and Eosin Y (Sigma Aldrich, MO).
Immunohistochemistry (IHC)
Paraffin-embedded (FFPE) blocks were serially sectioned and IHC were performed using Ellite anti-rabbit and anti-mouse ABC staining Vectastain kit (Vector, MI). Primary antibodies and dilutions Rabbit polyclonal CD133 1:100 (#NB120-16518, Novus Biologicals, USA), Rabbit monoclonal GFP 1:200 (#2956, Cell Signaing, MA), Mouse monoclonal MUC1 1:100 (#ab15481, Abcam, UK), Mouse monoclonal MUC5aC 1:200 (#NCL-MUC-5AC, Novocastra, Leica Biosystems Newcastle, UK), Mouse monoclonal PTF1a 1:100 (#sc-393011, Santa Cruz Biotechnology INC., Europe).
RNA preparation and RT-qPCR
Total RNA was isolated by RNeasy Mini Kit (QIAGEN, Germany) and treated with DNase Amplification Grade (Invitrogen, CA). cDNA synthesis was performed using SuperScript III First strand kit (Invitrogen, CA). RT-qPCR was performed with Cycler 480 SYBR Green I Master mix (Roche, Switzerland). Primers used were as following, CD24a 5’-TTCTGGCACTGCTCCTACC-3’ and 5’-GCGTTACTTGGATTTGGGGAA-3’, CD133 5’-CCTTGTGGTTCTTACGTTTGTTG-3’ and 5’-CGTTGACGACATTCTCAAGCTG-3’, B-Catenin 5’-TCCCATCCACGCAGTTTGAC-3’ and 5’-TCCTCATCGTTTAGCAGTTTTGT-3’, PI3K 5’-TGGGACCTTTTTGGTACGAGA-3’ and 5’-AGCTAAAGACTCATTCCGGTAGT-3’, Akt 5’-GGCCCCTGACCAGACCTTA-3’ and 5’-GATAGCCCGCATCCACTCTTC-3’, Pdx1 5’-ATTCTTGAGGGCACGAGAGC-3’ and 5’-GGTCCGTATTGGAACGCTCA-3’, Foxa2 5’-TGGTCACTGGGGACAAGGGAA-3’ and 5-‘GCAACAACAGCAATAGAGAAC-3’, Hes1 5’-CGGCATTCCAAGCTAGAGAAGG-3’ and 5’-GGTAGGTCATGGCGTTGATCTG-3’, Kras 5’-CAAGAGCGCCTTGACGATACA-3’ and 5’-CCAAGAGACAGGTTTCTCCATC-3’, EpCAM 5’-CTGGCGTCTAAATGCTTGGC-3’ and 5’-CCTTGTCGGTTCTTCGGACTC-3’, Tg Klf4 5’-GCGAACTCACACAGGCGAGAAACC-3’ and 5’-TTATCGTCGACCACTGTGCTGCTG-3’, Tg c-Myc 5’-CAGAGGAGGAACGAGCTGAAGCGC-3’ and 5’-TTATCGTCGACCACTGTGCTGCTG-3’.
RNA-seq library construction and sequencing
Isolation of total RNA was performed using QIAGEN RNeasy kit. RNA samples were prepared for sequencing using Illumina TruSeq RNA Sample Preparation Kit and were sequenced in an Illumina HiSeq 2500. Sample preparation, RNA-sequencing and Bioinformatic analysis were carried out by Fligen, INC. (Novogene, Nagoya Japan).
Results
iPSCs-converted CSCs (CSCcm) display CSCs features
As described in the protocol established by Chen L and Kasai T et al., to generate a model of pancreatic CSCs, iPSCs were exposed to different conditioned medium (CM) from pancreatic carcinoma cell lines PK-8CM and KLM-1CM, a process named as conversion (Figure 1A) [9]. Similarly to the established protocol, iPSCs were maintained in feeder-less conditions and the Leukemia inhibitory factor (LIF) which is essential for their viability was removed from the medium and thereby restricting them to the effect exclusively of the conditioned medium. Whereas the iPSCs cultured only with iPSCs media without LIF expired after 7 to 10 days (Figure 1B), the viability and proliferation of the cells maintained in presence of conditioned medium was not affected. Nanog iPSCs cells were generated by the retrovirus-mediated introduction of the four factors Sox2, Oct3/4, Klf4 and c-Myc into the Nanog-GFP-IRES-Puror of mouse embryonic fibroblast [42], therefore iPSCs could be monitored throughout the conversion by the GFP expression as a validation of the stemness. The population of converted cells displayed a diverse pattern of differentiation harbour cells expressing strong to moderate GFP and other differentiated cells which the expression was null. Once the process was completed the cells were termed as CSCcm PK8 and CSCcm KLM-1 and their stem-like properties were tested through sphere suspension assay (Figure 1B).
Figure 1.
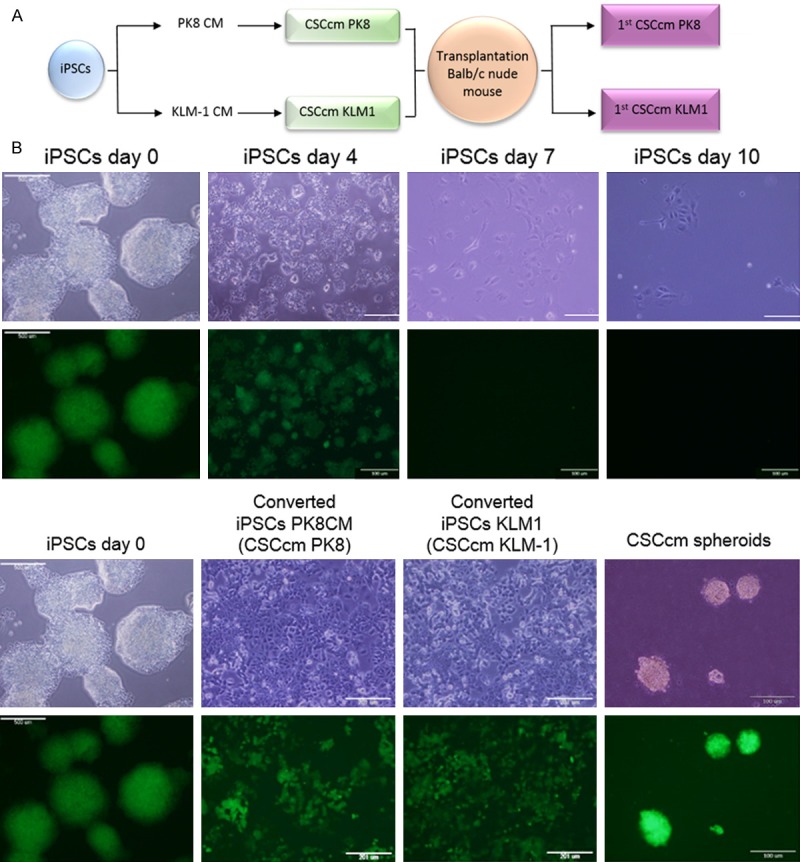
Conversion. A. Representative scheme of the conversion procedure. B. Viability of iPSCs maintained in control medium without LIF was no longer than 10 days. iPSCs underwent differentiated with no remaining GFP positive cells and eventually expired (upper panel). Stemness tracking during conversion by the presence of GFP protein. Self-renewing potential was validated by sphere formation assay previously to the subcutaneous implantation (lower panel). Original magnification 4× and 20×.
Since the CSCcm population was heterogeneous, in order to ensure a high frequency of cells displaying CSC-like properties, 104, 105 and 106 cells from the two new pancreatic CSCcm lines were subcutaneously transplanted into immunocompromised Balb/c nude mice. After 30 days, CSCcm engrafted and generated tumours in 9 out of 9 mice for each cell line indicating experimental reproducibility and demonstrating their tumorigenic potential. Chen and Kasai et al. previously shown that iPSCs cultured under control medium generated benign teratoma. Consistently, the xenograft tumours histology showed specific characteristics that resembled the actual pancreatic ductal adenocarcinoma phenotype. Primary tumours were rich in stroma and among the epithelial-like structures pancreatic intraepithelial neoplastic (PanIN) lesions were found together with moderate to poorly differentiated ductal structures (Figure 2A). To further validate the PDAC-like structures arose from CSCcm we explored the expression of GFP protein in primary tumour. GFP expressing cells were found all over the tissue samples and although it was predominantly located by undifferentiated cells few epithelial cells from ductal-like structures remained positive for its expression (Figure 2B). When the expression of specific CSC markers CD133, CD24a and EpCAM was evaluated, their up-regulation was already observed in CSCcm and was similarly detected or even enhanced in primary cultures (1st CSCcm) (Figure 2C) confirming their acquisition of CSCs features.
Figure 2.
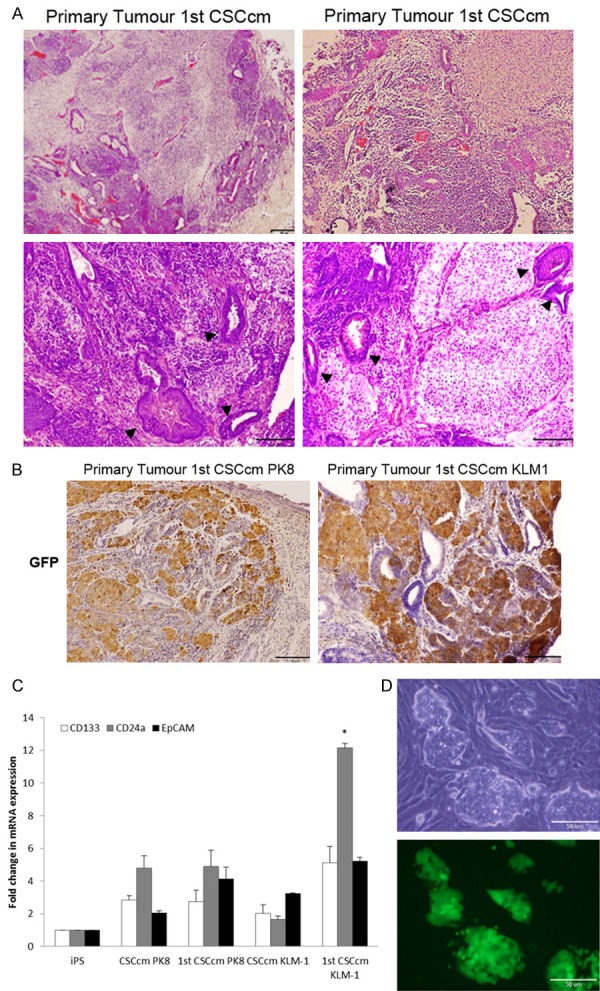
Malignant transformation and CSCs features. A. Histopathological features of 1st CSCcm primary tumours were evaluated by H&E staining. Specific PanIN lesions are indicated with arrowheads. Original magnification 10× and 20×. B. Lineage tracing by GFP protein showed that it was predominantly expressed in undifferentiated cells, however was also partially found in ductal-like structures. Original Magnification 10× and 20×. C. RT-qPCR analysis of CSCs markers CD133, CD24a and EpCAM in converted cells CSCcm and primary cultures 1st CSCcm. D. Cell distribution of primary cultures (1st CSCcm) after puromycin enrichment.
Primary cultures 1st CSCcm were enriched by puromycin obtaining the corresponding lineage from the top of the cell hierarchy system and avoiding the intrusion of cells from the host. As compared to CSCcm it was noteworthy that 1st CSCcm generated a specific cell distribution composed by well-defined colonies surrounded by myofibroblast-like cells most likely PSCs (pancreatic stellate cells). This particular feature of generating a spontaneous self-supporting system for the integrity and maintenance of the 1st CSCcm population was not observed neither in iPSCs control nor CSCcm cultures, therefore might be considered as an indicative of their enhanced malignant transformation (Figure 2D).
Serial transplantation leads to a more established PDAC phenotype
With the purpose to assess whether CSCcm display long-term tumorigenic potential we performed serial transplantation. Thus, cells from 1st CSCcm were transplanted into secondary and subsequently into a tertiary nude mouse (Figure 3A). Similarly to the primary culture the cells from secondary culture (2nd CSCcm) as well as tertiary cultures (3rd CSCcm) were enriched by puromycin. Thereby 1st CSCcm PK8 and 1st CSCcm KLM-1 were subcutaneously transplanted giving rise tumours within a short period of time of 25 days. Next, 2nd CSCcm PK8 and 2nd CSCcm KLM-1 were subsequently transplanted, however this time cells were orthotopically transplanted into the pancreas generating tumours within 15-20 days. Orthotopic tumours were remarkably bigger and specific liver metastasis was found (Table 1).
Figure 3.
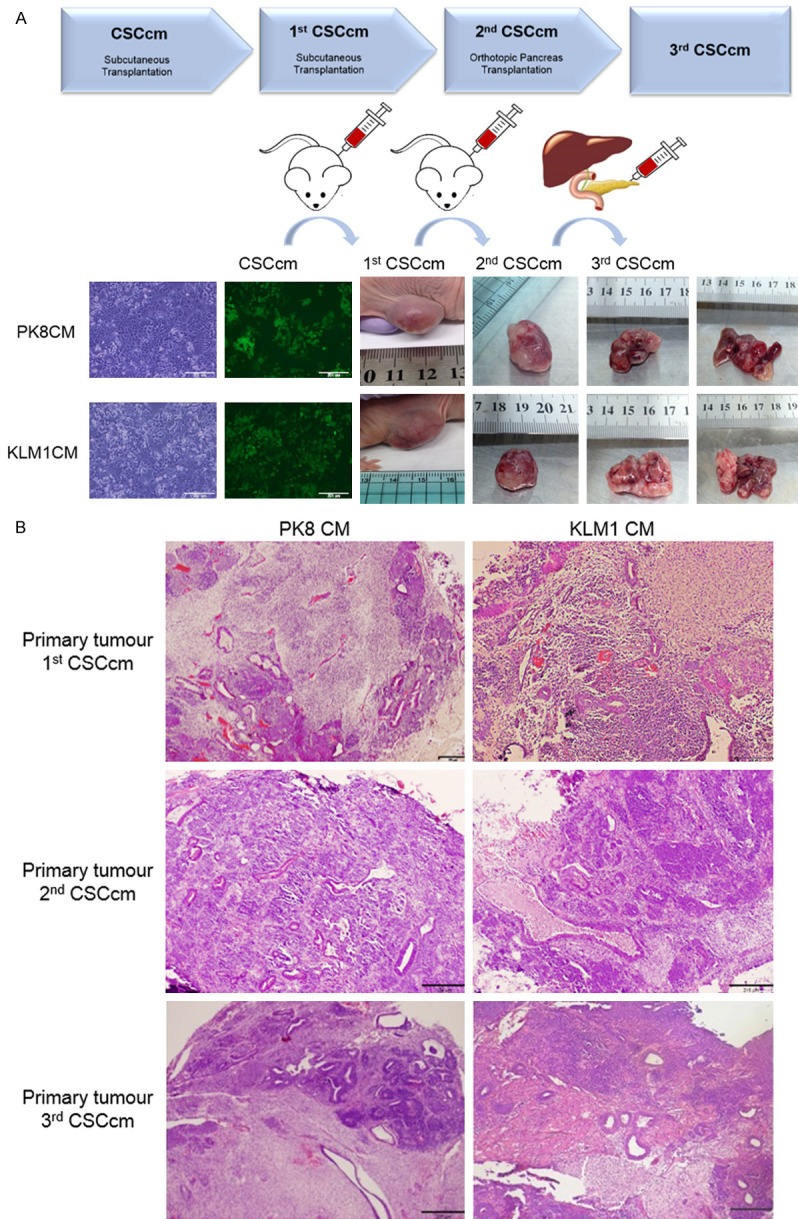
Tumorigenic Potential. A. Serial transplantation scheme and images from sequential excised tumours obtained from subcutaneously transplanted CSCcm and 1st CSCcm primary cultures. Primary tumours and their corresponding metastatic liver nodes generated from orthotopically implanted 2rd CSCcm cells are also shown. Experiments were equally performed for both PK8CM and KLM1CM lines. B. Micrographs from serial primary tumours showing the histopathological adenocarcinoma-like morphology by H&E staining. Original magnification 4×.
Table 1.
Primary CSCcm tumours from PK8CM cell lines and KLM-1CM cell lines obtained after the serial transplantation
| Primary Tumours | Time (days) | Volume (mm3) | Metastasis |
|---|---|---|---|
| 1st CSCcm PK8 | 30 | 2300 | No |
| 2nd CSCcm PK8 | 25 | 2600 | No |
| 3rd CSCcm PK8 | 20 | 3000 | Liver |
| 1st CSCcm KLM1 | 30 | 2500 | No |
| 2nd CSCcm KLM1 | 28 | 2100 | No |
| 3rd CSCcm KLM1 | 18 | 3000 | Liver |
Interestingly, subcutaneously transplanted tumours displayed indistinguishable histopathological morphologies whereas orthotopic primary tumours were richer in stroma mostly located in the inner mass and less epithelial ductal-like structures were found at the edges of the tumour. This may likely be due to difference in differentiation time whereby the myofibroblast-like phenotype is acquired earlier the epithelial-like phenotype requires a longer period of time (Figure 3B).
When the liver metastatic nodes were examined a remarkable difference in the histopathological features was observed. Unlike primary tumours, liver metastatic nodes displayed a teratocarcinoma phenotype containing very few structures corresponding to PDAC. To determine that CSCcm had the ability to metastasise we searched evidences of GFP protein. Its expression was strongly localised in areas with undifferentiated embryonic-like cells and no evidences were seen in more differentiated regions (Figure 4A). This difference made us question the integrity of CSCcm and whether a possible reversion towards the iPSCs phenotype was occurring. Nonetheless, complementary lines of investigation by our group reported tumours harbouring adenocarcinoma phenotype when transplantations were held into mammary gland and confirmed the teratocarcinoma phenotype when transplanted directly into the liver most likely because of the strong endocrine potential that liver microenvironment owns (data not shown). Thus, determining the major influence that the microenvironment exerts on the CSCcm fate.
Figure 4.
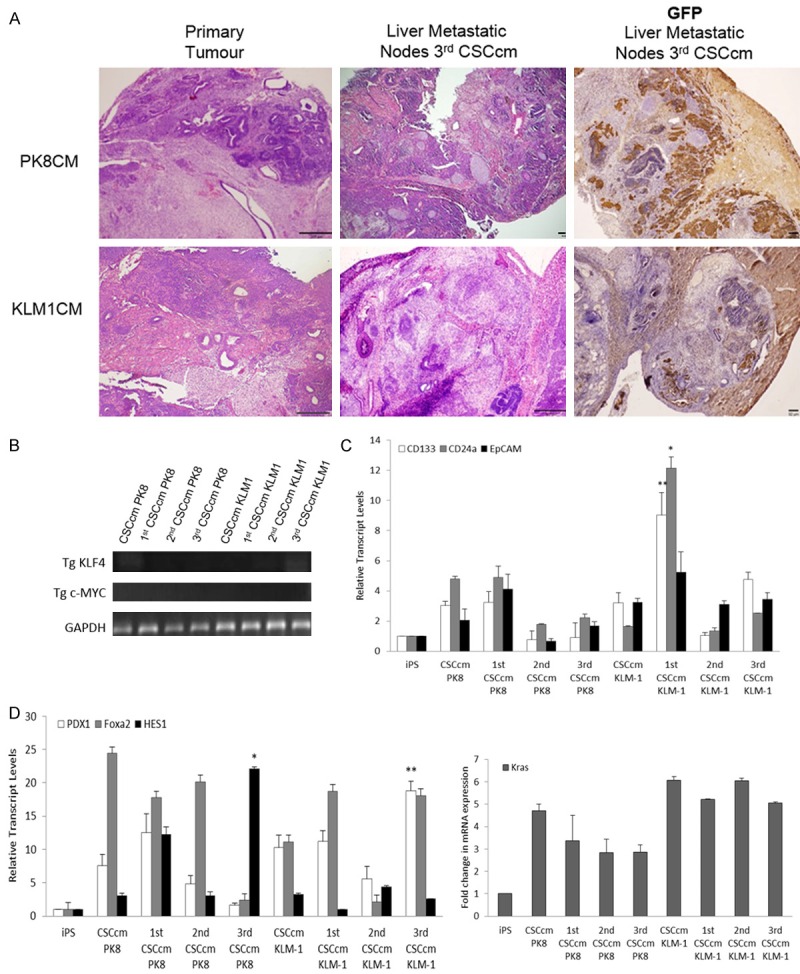
Established PDAC phenotype. A. Comparison of the histopathological features between 3rd CSCcm primary tumours and 3rd CSCcm liver nodes. 3rd CSCcm primary tumours displayed adenocarcinoma-like phenotype while a clear teratocarcinoma structure was found in liver nodes. GFP protein expression was shown to be located in undifferentiated cells areas within the liver metastatic nodes. Original magnification 10×. B. Agarose gel images from RT-qPCR products for the detection of Klf4 and cMyc transgenes. GAPDH was taken as a housekeeping control gene. C. RT-qPCR analysis of the preferentially expressed CSCs markers in a panel of serial transplantation samples. D. Pancreatic progenitor markers Pdx1, Foxa2 and Hes1 transcript levels and Kras mRNA expression levels were analysed through RT-qPCR analysis in a panel of all serial transplantation samples.
Given that the reprogramming of iPSCs was performed with the proto-oncogenes Sox2, Oct3/4, Klf4 and c-Myc, and that the implication specifically of c-Myc and Klf4 have been linked to PDAC we explored the expression of the transgenes in order to determine that there was no residual activity (Figure 4B). Hence, strongly supported that the malignant transformation arose by means of the intrinsically activated mechanisms of the cells.
Even though the resulting CSCcm populations from the serial transplantation gave rise to more aggressive phenotype, with regard to the assessment of pancreatic CSC markers intriguingly a decreased expression was observed in 2nd CSCcm and 3rd CSCcm suggesting a possible establishment of the lineage that results into more differentiated pancreatic cancer cells (Figure 4C). This is later discussed by RNA-seq DEG analysis that demonstrated an up-regulation of the pancreatic cancer cell hallmarks (Figure 7C).
Figure 7.
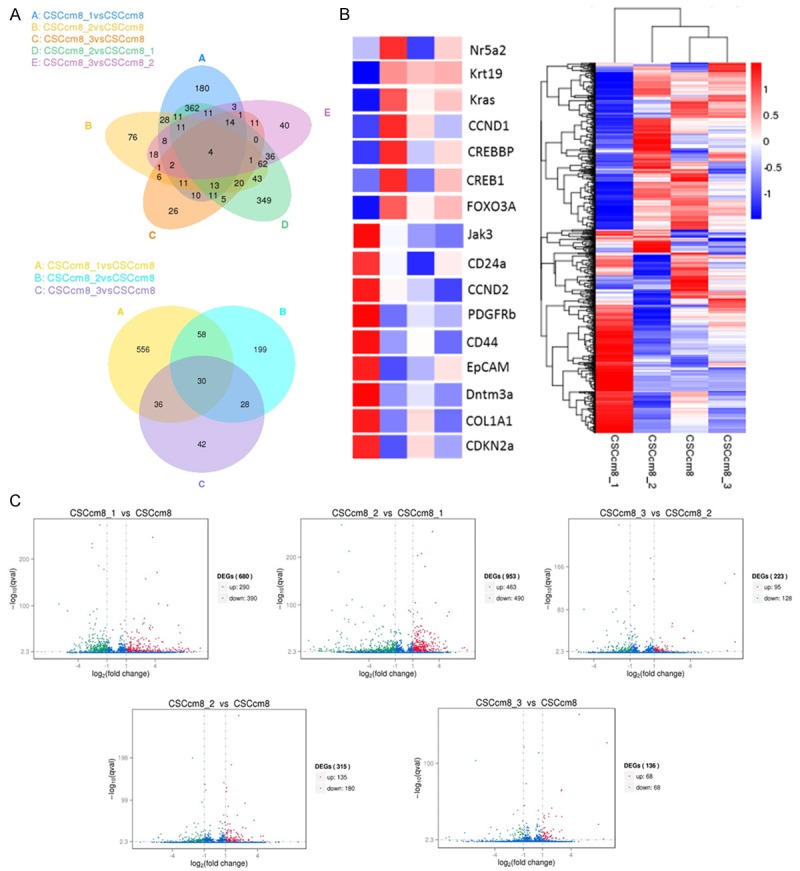
Molecular characterization. A. Venn diagram shows the differentially expressed genes (DEG) of the 5 possible combinations among the CSCcm PK8 lines (upper), and the combination of 1st CSCcm, 2nd CSCcm and 3rd CSCcm versus the CSCcm (lower). B. Representative cluster analysis among the four CSCcm PK8 lines (right). The colour range represents the log10 (FPKM+1) value from large (red) to small (blue). Main oncogenes and CSCs markers implicated in PDAC carcinogenesis were enlarged (left). C. Volcano plots for the differentially expressed genes screening (DESeq) distributed in significantly up-regulated genes (red), donw-regulated (green) and not differentially expressed (blue). Threshold set as: |log2(FoldChange)| > 1 and qvalue < 0.005.
Characterization of CSCcm lines and primary tumours by PDAC hallmarks
Bailey P. et al. (March 2016) presented a new definition for 4 pancreatic cancer subtypes based on an integrated genomic analysis. Among these 4 different subtypes is found the pancreatic progenitor class wherein the main transcriptional networks are comprised by transcription factors involved during the embryonic pancreatic differentiation [10]. In order to assess PDAC characteristics in the serial CSCcm lines, we hypothesise that CSCcm populations most likely may share similar molecular pattern as found in the pancreatic progenitor subtypes. Within the main transcription factors described by Bailey P. et al., PDX1, FOXA2 and HES1 were cited to be essentially expressed in the progenitor subtype. Thus, their transcript levels were examined along with the Kras which is well-known to be involved in PDAC. Despite the variation found between the populations most likely due to the uncontrollable fate of the lineage, the up-regulation was apparent for all pancreatic progenitor markers as well as it was for Kras (Figure 4D). Likewise to the described pancreatic progenitor class, the adenocarcinoma markers MUC1 and MUC5aC were co-expressed in all CSCcm tumours samples (Figure 5) specifically located at the membrane of epithelial ductal cells. Interestingly few structures that presented ductal ectasia displayed strong expression in the infiltrating immune cells. Therefore these evidences infer in the achievement of CSCcm lines to recapitulate the PDAC phenotype.
Figure 5.
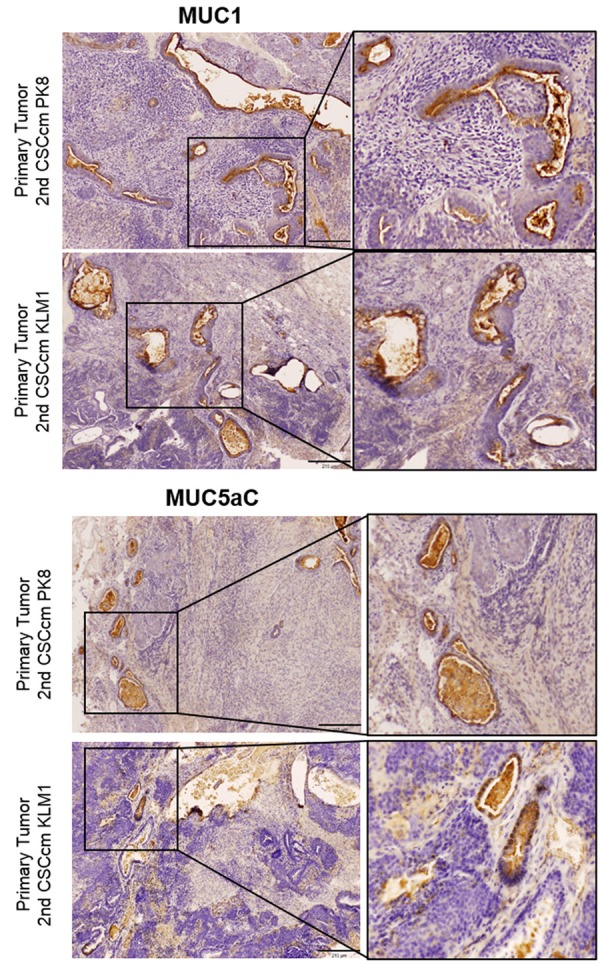
PDAC hallmarks. The adenocarcinoma phenotype was evaluated through the co-expression of the apomucinous of MUC1 and MUC5aC which were predominantly located in the membrane of ductal structures. Infiltrating ductal immune cells also displayed high expression was also observed in mucinous ductal ectasia (MDE). Original magnification 10×.
A possible model for lineage tracing ADM events
The acinoductal metaplasia (ADM) describes the process whereby the islet neogenesis is accompanied by the transdifferentiation of the normal exocrine tissue into ductal complexes. This pancreatic metaplasia may turn into a premalignant state by means of progressive changes in the ductal epithelium that gives rise to PanIN lesions and may eventually lead to the progression of PDAC [11,12]. Thus, proposed linear progression models for PDAC where ductal cells evolve into hyperplastic and later into dysplastic epithelium resulting in an invasive carcinoma had to be reconsidered since there are no formal evidences of these sequential events. Instead, in vitro and genetically engineered mouse models of PDAC have shown that tumours can arise from acinar cells [7].
When orthotopically transplanted 3rd CSCcm tumours were examined in detail large regions composed of highly desmoplastic stroma could be seen surrounding clusters of cells that resembled the acini morphology. Moreover, multiple ducts were also found to co-localize with the acinar-like areas and few of them showed evidences of ADM transition (Figure 6A, 6B). With the purpose to determine the acinar phenotype the presence of the protein Ptf1a was assessed. The resulting Ptf1a expression was specifically located in the cell clusters demonstrating a primary level of acinar differentiation. In addition, few PanIN lesions together with well-differentiated ductal structures were found nearby the acinar-like cells and the intermediate stages of ADM. The expression of GFP was found to co-localized in the Ptf1a positive cell clusters validating the acinar cells as the lineage of CSCcm (Figure 6C). Supporting these evidences we detected the overexpression of Akt which through the PI3K/Akt pathway have been reported to be involved in the acinar dedifferentiation along with the accumulation of the β-catenin in the cytoplasm that has also been associated (Figure 6D) [13-16]. Since it is important to elucidate whether the process of dedifferentiation plays an important role in the appearance of early events in pancreatic cancer our CSCcm model may be suitable for lineage tracing how acinar cells undergo into dedifferentiation losing its mature features and discern whether eventually develop a malignant transformation.
Figure 6.
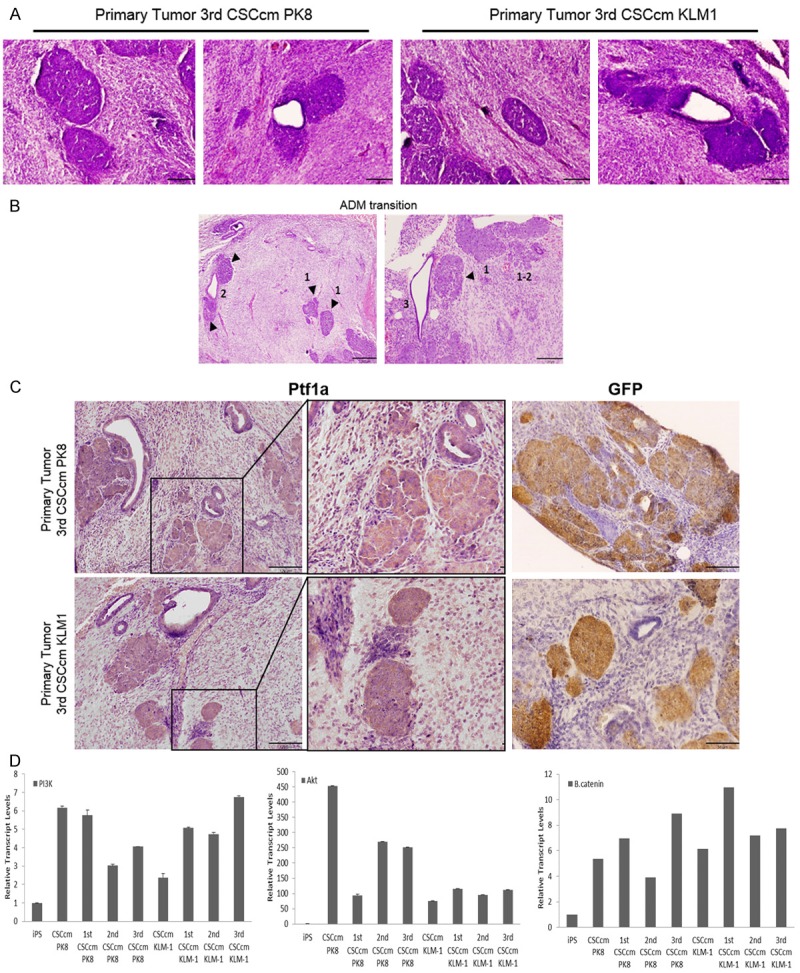
Acino-ductal metaplasia. A. H&E staining of cluster cells areas with acinar morphology and ADM structures found in 3rd CSCcm primary tumours. B. Images showing evidences of ADM transition originated from acinar structures (arrowheads) indicated as 1, acino-ductal interphase as 2 and final ductal-like acquired phenotype 3. C. Cell clusters and few ductal cells were positive for the acinar marker Ptf1a. GFP was equally predominantly located in cell clusters and few cells from ductal epithelial cells. D. RT-qPCR of the relative transcript levels for PI3K, AKT and β-catenin (from left to right) in a panel of the samples obtained from serial transplantation. Micrographs original magnification 10× and 20×.
Molecular characterization confirms the acquisition of CSC features and a subsequent established PDAC pattern
To further validate the molecular nature of the CSCcm from PK8CM lines a transcriptome analysis by RNA-sequencing was performed. The expression quantification of 35,276 total described genes was estimated by Fragments Per Kilobase Million (FPKM) [log10(FPKM+1) value] and members from the transcriptional pancreatic progenitor network Pdx1, Hes1, Foxa2, Hnf1a, Hnf4a, Pax6, Nr5a2, Rbpj, Rbpgl, MafA and MafB were found to be expressed together with PDAC related hallmarks such as Kras, Krt19, Col8a1, Col1a1, Cxcr4, Muc1, Muc5aC, Mmp2 or Malat1 as well as the most representative pancreatic CSC markers CD133, CD24a, EpCAM and CD44.
Analysis for the differentially expressed genes (DEG) of single tumours were applied based on the following comparisons: i) 1st CSCcm (CSCcm8_1), 2nd CSCcm (CSCcm8_2) and 3rd CSCcm (CSCcm8_3) to CSCcm (CSCcm8), ii) 1st CSCcm to 2nd CSCcm and iii) 2nd CSCcm versus 3rd CSCcm. Venn diagrams indicated 1st CSCcm to bear a particular transcriptome programme among the 4 groups (Figure 7A). Thus, to gain insights of the similar expression patterns DEG data were arranged in a cluster analysis using the log10(FPKM+1) value which revealed CSCcm and 3rd CSCcm to be closer in expression whereas the 2nd CSCcm slightly differed and finally corroborated 1st CSCcm as the most differentially expressed group (Figure 6B). The gene identification and their corresponding localization within the heat map suggested possible new roles for already described genes in PDAC. Pdgfrb and Cdkn2a which are known to participate in the development and progression of PDAC [17,18] co-localise with the CSC markers EpCAM and CD44 which leads to question their level of implication in the acquisition of the CSC-like genotype pattern (Figure 7B).
In order to elucidate potential candidates involved in the malignant transformation the distribution of differentially expressed genes (DESeq) was analysed for each of the aforementioned comparisons and represented in volcano plots |(log2 (FoldChange)| > 1&qvalue < 0.005) (Figure 7C). The transcript levels of pancreatic CSC markers CD24a, EpCAM and CD44 were remarkably enhanced in 1st CSCcm whilst decreased in the subsequent generations. Instead, the popular hallmarks of PDAC Kras, Krt19 and Myc were notably activated in 2nd CSCcm and persisted in 3rd CSCcm. Suggesting that the fate of the 1st CSCcm lineage may probably be established. The activation of Nr5a2 which its heterozygosity has been recently proposed to likely contribute to the occurrence of the PDAC [38], was sustained in 2nd CSCcm whereas in 1st CSCcm was dysregulated. Interestingly, the expression of GATA6 which have been found to be spontaneously lost in mouse model of Kras(G12V)-driven PDAC was not found in any of the groups [39]. On the other hand, the original converted cell line CSCcm only differed from 2nd CSCcm in the increased expression of Krt19 and the activation of PI3K in 3rd CSCcm. Since tumours from all CSCcm lines were rich in desmoplastic stroma we sought for corresponding PSCs markers. Col8a1, Col1a1 and Col1a2 including TIMPs and Mmp2 which have been linked to tumour progression and invasion [19-21] were observed in 1st CSCcm. In addition, family members from CXC/CC chemokines Ccl2, Cxcl1 and Cxcl5 that are reported to favour the occurrence, maintenance and progression of the tumour [22] were also expressed in a correlated manner with PSCs markers. In overall, DESeq analysis consistently supported the gain of CSCs features and the established PDAC molecular pattern in the subsequent generations of CSCcm. Nonetheless a full transcriptome comparison between other RNA-seq datasets from other tumour types would be required.
“Mutation or not mutation-that is the question”
There is a wide range of genetic abnormalities in PDAC most of them derived from somatic single nucleotide variants (SNVs). In particular, the activation of the point mutation in Kras codon 12 has an 85-90% of prevalence in PDAC cases. Several mouse models have demonstrated that endogenous expression of oncogenic Kras G12D induces phenotypic changes at molecular and cellular levels that eventually recapitulate PDAC [7,23-25].
The variants obtained from the screening of repeated reads showed no evidences of single point mutation in Kras codon 12 nor 13 for any of the groups. In fact, the resulting variants were found to be located in the 3’-UTR region of the oncogene and were present in all CSCcm lines. To discern whether the Kras variants had any biological relevance in the malignant transformation of the CSCcm, and due to iPSCs have a high SNPs variability, we decided to screen the mouse strains sources from where the original fibroblast was obtained previously to the iPSCs reprogramming [26]. Through the database platforms http://www.informatics.jax.org and http://www.sanger.ac.uk, DBA mouse strains which are supposed to compose the 50% of the fibroblast genome were found to harbour the exact Kras SNPs as CSCcm lines. Likewise, the presence of INDELs were not localised within the oncogene. Thereby, there were no evidences of correlation between the SNPs found in Kras and the malignant transformation.
Discussion
PDAC also known as pancreatic ductal adenocarcinoma is the most representative form of pancreatic cancer. PDAC is characterised by heterogeneous population of tumour cells structured in CSCs, differentiated cancer cells, tumour-associated PSCs and immune cells [1,4,6]. Similarly to iPSCs, CSCs are considered as cells bearing stem cell properties that give rise to a diverse lineage of cancer cells. Driven by this convergence between CSCs and iPSCs, we opened a new avenue to generate new cell lines endowed of CSCs properties and enable to recapitulate the PDAC tumour phenotype.
Based on the observations that the xenograft tumours generated from CSCcm lines recapitulate the ductal adenocarcinoma phenotype, and that puromycin-enriched primary cultures ensured to start over from the top of the hierarchy where CSCs are residing, we can postulate that CSCcm indeed give rise heterogeneous progenies composed by more differentiated cancer cells and PSCs in a hierarchical manner. Nevertheless, it is important to note that our results have pointed out 1st CSCcm lines as the potential candidates to represent the pancreatic CSCs. Thus, even though the conditioned medium from cancer cell lines provides an appropriate microenvironment able to initiate a malignant transformation, it is not until CSCcm get in touch with an in vivo system that the CSCs features become robust at the level of transcriptome. Supporting this, clear experimental evidences were seen when enriched primary cultures from 1st CSCcm generated a particular cell distribution wherein specific myofibroblast-like cells arose favouring the maintenance and stability of the CSCcm colonies. Therefore, the intervention of the organisms is still essential. Certainly, this reliance on in vivo systems is an unresolved matter that must eventually be overcome. Recent studies in the tumour microenvironment (TME) have provided new insights about how the cancerous niche plays a significant role in disease progression sustaining cell proliferation, activating invasion and metastasis [27]. It has been appreciated for some time the influence of the TME, but the precise function of each constituent remains unknown. We expect that future analysis on the components of the conditioned medium from cancer cell lines will help to elucidate new mechanisms to optimize the conversion of CSCcm and end up being independent of the murine organisms.
Important early steps in pancreatic tumour initiation and progression are the genome reprogramming and dedifferentiation. Thus, the acino-ductal metaplasia has a relevant implication in the development of the pancreatic cancer. We observed that CSCcm tumours also generated ADM. In order to confirm the acinar phenotype the expression protein Ptf1a was sought in tumour samples and strong expression was found in the cell cluster structures. The expression of Hes1 together with Ptf1a are attributed to the characterization of centroacinar cells (CACs) which have been ascribed to bear stem-like features [7,28]. Since the RNA-seq expression quantification analysis indicate Hes1 as one of the most highly expressed genes, prompted us to question whether Ptf1a positive cells found in CSCcm tumours are either acinar or CACs. Further experiments will be required in order to clarify the real identity of the cells responsible for the ADM transition.
Pdgfrb and Cdkn2a were found to be highly expressed in 1st CSCcm and strongly decreased in the rest of the groups. The overexpression of Pdgfrb has been linked to a poor disease-free survival promoting metastasis in a cell-autonomous manner in mutant p53 mice and glioma stem cells have been reported to preferentially express PDGFRb which its activation promotes self-renewal and its depletion completely abrogated their tumorigenicity [17,29]. Intriguingly, CDKN2a acts as a tumour suppressor and is usually aberrant in 95% of pancreatic cancer. This alteration may be due to either an abnormal methylation or several reported germline mutations which leads to its inactivation [18,35]. When SNPs in Cdkn2a were screened in CSCcm lines there were no evidences of single point mutations, but a single INDEL C < CAA located in the 3’-UTR region in Cdkn2a was present only in 1st CSCcm group. Nevertheless the same INDEL was found in the murine genome but neither relevant clinical nor functional information has been described so far. Thus, the sudden enhanced expression of Pdgfrb may be related to the CSCs phenotype of 1st CSCcm. On the other hand, from the results obtained for Cdkn2a we could not discern properly its implication, although its sudden downregulation in 2nd CSCcm and maintained in 3rd CSCcm may be linked to the establishment of the PDAC phenotype.
As it has been previously aforementioned, the frequency of single point mutations in Kras occur at 85-90% in pancreatic cancer [25,30,31]. The activation of the oncogenic Kras together with its downstream effector Myc have been implicated in self-renewal and tumour plasticity events such as dedifferentiation [24]. In our study we demonstrated that the tumorigenicity of CSCcm and consequent recapitulation of PDAC phenotype is not given by variants in the oncogene Kras since no formal evidences were found in SNPs analysis. However, this premature idea should be developed in extended eQTLs analysis which will provide information at the level of genotype-gene relation in addition to karyotype and genomic profiling analysis that would enable clear discrimination. MYC is generally overexpressed in PDAC. In accordance to this DESeq analysis showed increased expression of Myc in the 2nd CSCcm generation, however it is important to note that it was remarkably downregulated in 1st CSCcm. Recent findings on genome-wide DNA methylation profiling in pancreatic CSCs determine CpG sites annotated to Myc to be more methylated [5]. This may be correlated with the overexpression of Dnmt3a and Dntm3b observed in 1st CSCcm and the subsequent downregulation in 2nd CSCcm. The predominant modification of DNMTs is the catalysis of the DNA methylation at 5-position cytosine. Dntm3a and Dnmt3b function as de novo methyltransferases and are highly expressed in embryonic cells and down-regulated in adult tissues. They have also been involved in self-renewal and maintenance of colon cancer stem cells, and pancreatic cancer patients with higher levels have been significantly attributed to have an overall lower survival [41]. Thereby, the activation of Kras and Myc are required but not sufficient to originate a PDAC tumour and their turnover between 1st CSCcm and more differentiated 2nd CSCcm may be tightly related to epigenetic alterations.
In the context of inflammation, the combination of Kras and the activation of NFkB have been reported to induce the conversion of non-stem into stem cell properties of intestinal epithelial cells (IEC) through the stabilization of β-catenin [32]. Despite there are not strong evidences in pancreatic cancer models that implicate NFkB in PDAC progression [25], it was noteworthy that in 2nd CSCcm the levels of NFkB arose as Kras and Myc did. Experimental evidences are required to evaluate whether the acquisition of stem-like features may arise in a stochastic way since our model is based on iPSCs, however this fact may involve NFkB pathway in the prevalence of the CSCcm in pancreatic cancer cells progeny. On the other hand, inflammatory cells constitute an important part of the stromal tissue in pancreatic cancer and the existence of a consistent feedback between PSCs and CXC/CC chemokines family members makes to consider them as potential candidates to be effectors in the occurrence and progression of the tumour [19-22]. Hence, these facts were corroborated with our DESeq data which indeed showed that the PSCs markers Col81a, Col1a1, Col1a2 were correlated with the expression of Ccl2, Cxcl1 and Cxcl5 chemokines interestingly with the stimulation of the CSCs markers overexpression. In addition, IL-33 is known to activate mast cells and stimulate pro-inflammatory cytokine production and has been found to be expressed in the nuclei of activated PSCs [36,37]. Intrapancreatic mast cells also express the cytokine stem cell factor (SCF) which plays a constitutively important role in proliferation and survival of pluripotent progenitor cells together with its receptor proto-oncogene tyrosine-kinase KIT (cKIT) [33,34]. Although pancreatic cancer cells express cKIT, its role in CSCs still needs to be elucidated. Interestingly, in the expression quantification analysis it appears to be overexpressed and DESeq results highlighted a differential expression of cKit in 1st CSCcm indicating that it could be a potential target to abrogate the acquisition of CSCs properties. Thus, the aforementioned results notably remark the involvement of the inflammatory system as the main effector for the acquisition of the CSCs properties.
Pancreatic CSCcm is a feasible model for pancreatic CSCs which recapitulates PDAC phenotype. It is important to note that the conversion process has not been achieved under any genetic manipulation and the present study has demonstrated a preliminary analysis where the expected single point mutations were not found. Therefore, our model may provide new insights about the actual occurrence of the pancreatic cancer leading to develop different approaches for the early detection and find new effectors in order to target CSCs and abrogate the progression of this fatidic disease.
Acknowledgements
The authors thank with much appreciation Francisco X Real for his assistance and guidance. Francisco X Real for his guidance and valuable discussion, and Akifumi Mizutani for his assistance. This research was supported by the Grant-in-Aid for Scientific Research (A) No. 25242045 (MS); Grant-in-Aid for Scientific Research (C) No. 16K07116 (YI); the Grant-in-Aid for Challenging Ex-ploratory Research No. 26640079 (MS) and the Japan Science and Technology Agency, Matching Planner Program-Tansaku Shiken-Grant (TK).
Disclosure of conflict of interest
None.
References
- 1.Zhan HX, Xu JW, Wu D, Zhang TP, Hu SY. Pancreatic cancer stem cells: New insight into a stubborn disease. Cancer Lett. 2014;357:429–37. doi: 10.1016/j.canlet.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 2.Shakya R, Gonda T, Quante M, Salas M, Kim S, Brooks J, Hirsch S, Davies J, Cullo A, Olive K, Wang TC, Szabolcs M, Tycko B, Ludwig T. Hypomethylating therapy in an aggressive stroma-rich model of pancreatic carcinoma. Cancer Res. 2012;73:885–896. doi: 10.1158/0008-5472.CAN-12-1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sainz B Jr, Martín B, Tatari M, Heeschen C, Guerra S. ISG15 is a critical microenvironmental factor for pancreatic cancer stem cells. Cancer Res. 2014;74:7309–20. doi: 10.1158/0008-5472.CAN-14-1354. [DOI] [PubMed] [Google Scholar]
- 4.Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313–23. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 5.Zagorac S, Alcala S, Fernandez Bayon G, Bou Kheir T, Schoenhals M, González-Neira A, Fernandez Fraga M, Aicher A, Heeschen C, Sainz B Jr. DNMT1 inhibition reprograms pancreatic cancer stem cells via upregulation of the miR-17-92 cluster. Cancer Res. 2016;76:4546–58. doi: 10.1158/0008-5472.CAN-15-3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sancho P, Alcala S, Usachov V, Hermann PC, Sainz B Jr. The ever-changing landscape of pancreatic cancer stem cells. Pancreatology. 2016;16:489–96. doi: 10.1016/j.pan.2016.04.004. [DOI] [PubMed] [Google Scholar]
- 7.Rooman I, Real FX. Pancreatic ductal adenocarcinoma and acinar cells: a matter of differentiation and development? Gut. 2012;61:449–458. doi: 10.1136/gut.2010.235804. [DOI] [PubMed] [Google Scholar]
- 8.Kasai T, Chen L, Mizutani A, Kudoh T, Murakami H, Fu L, Seno M. Cancer stem cells converted from pluripotent stem cells and the cancerous niche. J Stem Cells Regen Med. 2014;10:2–7. doi: 10.46582/jsrm.1001002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen L, Kasai T, Li Y, Sugii Y, Jin G, Okada M, Vaidyanath A, Mizutani A, Satoh A, Kudoh T, Hendrix MJ, Salomon DS, Fu L, Seno M. A model of cancer stem cells derived from mouse induced pluripotent stem cells. PLoS One. 2012;7:e33544. doi: 10.1371/journal.pone.0033544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, Nourse C, Murtaugh LC, Harliwong I, Idrisoglu S, Manning S, Nourbakhsh E, Wani S, Fink L, Holmes O, Chin V, Anderson MJ, Kazakoff S, Leonard C, Newell F, Waddell N, Wood S, Xu Q, Wilson PJ, Cloonan N, Kassahn KS, Taylor D, Quek K, Robertson A, Pantano L, Mincarelli L, Sanchez LN, Evers L, Wu J, Pinese M, Cowley MJ, Jones MD, Colvin EK, Nagrial AM, Humphrey ES, Chantrill LA, Mawson A, Humphris J, Chou A, Pajic M, Scarlett CJ, Pinho AV, Giry-Laterriere M, Rooman I, Samra JS, Kench JG, Lovell JA, Merrett ND, Toon CW, Epari K, Nguyen NQ, Barbour A, Zeps N, Moran-Jones K, Jamieson NB, Graham JS, Duthie F, Oien K, Hair J, Grutzmann R, Maitra A, Iacobuzio-Donahue CA, Wolfgang CL, Morgan RA, Lawlor RT, Corbo V, Bassi C, Rusev B, Capelli P, Salvia R, Tortora G, Mukhopadhyay D, Petersen GM Australian Pancreatic Cancer Genome Initiative. Munzy DM, Fisher WE, Karim SA, Eshleman JR, Hruban RH, Pilarsky C, Morton JP, Sansom OJ, Scarpa A, Musgrove EA, Bailey UM, Hofmann O, Sutherland RL, Wheeler DA, Gill AJ, Gibbs RA, Pearson JV, Waddell N, Biankin AV, Grimmond SM. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52. doi: 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- 11.Schmid RM. Acinar-to-ductal metaplasia in pancreatic cancer development. J Clin Invest. 2002;109:1403–1404. doi: 10.1172/JCI15889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Means AL, Meszoely IM, Suzuki K, Miyamoto Y, Rustgi AK, Coffey RJ Jr, Wright CV, Stoffers DA, Leach SD. Pancreatic epithelial plasticity mediated by acinar cell transdifferentiation and generation of nestin-positive intermediates. Development. 2005;132:3767–3776. doi: 10.1242/dev.01925. [DOI] [PubMed] [Google Scholar]
- 13.Elghazi L, Weiss AJ, Barker DJ, Callaghan J, Staloch L, Sandgren EP, Gannon M, Adsay VN, Bernal-Mizrachi E. Regulation of pancreas plasticity and malignant transformation by Akt signaling. Gastroenterology. 2009;136:1091–103. doi: 10.1053/j.gastro.2008.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jensen JN, Cameron E, Garay MV, Starkey TW, Gianani R, Jensen J. Recapitulation of elements of embryonic development in adult mouse pancreatic regeneration. Gastroenterology. 2005;128:728–41. doi: 10.1053/j.gastro.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 15.Minami K, Okano H, Okumachi A, Seino S. Role of cadherin mediated cell-cell adhesion in pancreatic exocrine-to-endocrine transdifferentiation. J Biol Chem. 2008;283:13753–61. doi: 10.1074/jbc.M710034200. [DOI] [PubMed] [Google Scholar]
- 16.Morris JP 4th, Cano DA, Sekine S, Wang SC, Hebrok M. Beta-catenin blocks Kras dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J Clin Invest. 2010;120:508–20. doi: 10.1172/JCI40045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weissmueller S, Manchado E, Saborowski M, Morris JP 4th, Wagenblast E, Davis CA, Moon SH, Pfister NT, Tschaharganeh DF, Kitzing T, Aust D, Markert EK, Wu J, Grimmond SM, Pilarsky C, Prives C, Biankin AV, Lowe SW. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor β signaling. Cell. 2014;157:382–94. doi: 10.1016/j.cell.2014.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rachakonda PS, Bauer AS, Xie H, Campa D, Rizzato C, Canzian F, Beghelli S, Greenhalf W, Costello E, Schanne M, Heller A, Scarpa A, Neoptolemos JP, Werner J, Buchler M, Hoheisel JD, Hemminki K, Giese N, Kumar R. Somatic mutations in exocrine pancreatic tumors: association with patient survival. PLoS One. 2013;8:e60870. doi: 10.1371/journal.pone.0060870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sato N, Maehara N, Goggins M. Gene expression profiling of tumor-stromal interactions between pancreatic cancer cells and stromal fibroblasts. Cancer Res. 2004;64:6950–6956. doi: 10.1158/0008-5472.CAN-04-0677. [DOI] [PubMed] [Google Scholar]
- 20.Li X, Ma Q, Xu Q, Duan W, Lei J, Wu E. Targeting the cancer-stroma interaction: a potential approach for pancreatic cancer treatment. Curr Pharm Des. 2012;18:2404–2415. doi: 10.2174/13816128112092404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seton-Rogers S. Pancreatic cancer: Fibroblast co-conspirators. Nat Rev Cancer. 2011;11:758. doi: 10.1038/nrc3157. [DOI] [PubMed] [Google Scholar]
- 22.Wang S, Wu Y, Hou Y, Guan X, Castelvetere MP, Oblak JJ, Banerjee S, Filtz TM, Sarkar FH, Chen X, Jena BP, Li C. CXCR2 macromolecular complex in pancreatic cancer: a potential therapeutic target in tumor growth. Transl Oncol. 2013;6:216–225. doi: 10.1593/tlo.13133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ischenko I, Petrenko O, Hayman MJ. Analysis of the tumor-initiating and metastatic capacity of PDX1-positive cells from the adult pancreas. Proc Natl Acad Sci U S A. 2014;111:3466–71. doi: 10.1073/pnas.1319911111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ischenko I, Zhi J, Moll UM, Nemajerova A, Petrenko O. Direct reprogramming by oncogenic Ras and Myc. Proc Natl Acad Sci U S A. 2013;110:3937–42. doi: 10.1073/pnas.1219592110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, Dubus P, Sandgren EP, Barbacid M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 26.Fujishiro SH, Nakano K, Mizukami Y, Azami T, Arai Y, Matsunari H, Ishino R, Nishimura T, Watanabe M, Abe T, Furukawa Y, Umeyama K, Yamanaka S, Ema M, Nagashima H, Hanazono Y. Generation of naive-like porcine-induced pluripotent stem cells capable of contributing to embryonic and fetal development. Stem Cells Dev. 2013;22:473–82. doi: 10.1089/scd.2012.0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen F, Zhuang X, Lin L, Yu P, Wang Y, Shi Y, Hu G, Sun Y. New horizons in tumor microenvironment biology: challenges and opportunities. BMC Med. 2015;13:45. doi: 10.1186/s12916-015-0278-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rovira M, Scott SG, Liss AS, Jensen J, Thayer SP, Leach SD. Isolation and characterization of centroacinar/terminal ductal progenitor cells in adult mouse pancreas. Proc Natl Acad Sci U S A. 2010;107:75–80. doi: 10.1073/pnas.0912589107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang Y, Boije M, Westermark B, Uhrbom L. PDGF-B can sustain self-renewal and tumorigenicity of experimental glioma-derived cancer-initiating cells by preventing oligodendrocyte differentiation. Neoplasia. 2011;13:492–503. doi: 10.1593/neo.11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, Weinberg RA. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gidekel Friedlander SY, Chu GC, Snyder EL, Girnius N, Dibelius G, Crowley D, Vasile E, DePinho RA, Jacks T. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell. 2009;16:379–389. doi: 10.1016/j.ccr.2009.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Göktuna SI, Ziegler PK, Canli O, Heijmans J, Huels DJ, Moreaux G, Rupec RA, Gerhard M, Schmid R, Barker N, Clevers H, Lang R, Neumann J, Kirchner T, Taketo MM, van den Brink GR, Sansom OJ, Arkan MC, Greten FR. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell. 2013;152:25–38. doi: 10.1016/j.cell.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 33.Esposito I, Kleeff J, Bischoff SC, Fischer L, Collecchi P, Iorio M, Bevilacqua G, Büchler MW, Friess H. The stem cell factor-c-kit system and mast cells in human pancreatic cancer. Lab Invest. 2002;82:1481–92. doi: 10.1097/01.lab.0000036875.21209.f9. [DOI] [PubMed] [Google Scholar]
- 34.Yasuda A, Sawai H, Takahashi H, Ochi N, Matsuo Y, Funahashi H, Sato M, Okada Y, Takeyama H, Manabe T. The stem cell factor/c-kit receptor pathway enhances proliferation and invasion of pancreatic cancer cells. Mol Cancer. 2006;5:46. doi: 10.1186/1476-4598-5-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao R, Choi BY, Lee MH, Bode AM, Donga Z. Implications of genetic and epigenetic alterations of CDKN2A (p16INK4a) in cancer. EBioMedicine. 2016;8:30–39. doi: 10.1016/j.ebiom.2016.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu D, Jiang HR, Kewin P, Li Y, Mu R, Fraser AR, Pitman N, Kurowska-Stolarska M, McKenzie AN, McInnes IB, Liew FY. IL-33 exacerbates antigen-induced arthritis by activating mast cells. Proc Natl Acad Sci U S A. 2008;105:10913–10918. doi: 10.1073/pnas.0801898105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Masamune A, Watanabe T, Kikuta K, Satoh K, Kanno A, Shimosegawa T. Nuclear expression of interleukin-33 in pancreatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2010;299:G821–G832. doi: 10.1152/ajpgi.00178.2010. [DOI] [PubMed] [Google Scholar]
- 38.Flandez M, Cendrowski J, Cañamero M, Salas A, del Pozo N, Schoonjans K, Real FX. Nr5a2 heterozygosity sensitises to, and cooperates with, inflammation in KRas(G12V)-driven pancreatic tumourigenesis. Gut. 2014;63:647–55. doi: 10.1136/gutjnl-2012-304381. [DOI] [PubMed] [Google Scholar]
- 39.Martinelli P, Madriles F, Cañamero M, Pau EC, Pozo ND, Guerra C, Real FX. The acinar regulator Gata6 suppresses KrasG12V-driven pancreatic tumorigenesis in mice. Gut. 2016;65:476–86. doi: 10.1136/gutjnl-2014-308042. [DOI] [PubMed] [Google Scholar]
- 40.Bruns CJ, Harbison MT, Kuniyasu H, Eue I, Fidler IJ. In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice. Neoplasia. 1999;1:50–62. doi: 10.1038/sj.neo.7900005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Subramaniam D, Thombre R, Dhar A, Anant S. DNA methyltransferases: a novel target for prevention and therapy. Front Oncol. 2014;4:80. doi: 10.3389/fonc.2014.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–7. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]