

ABSTRACT
Group A Streptococcus (GAS) acquires mutations of the virulence regulator CovRS in human and mouse infections, and these mutations result in the upregulation of virulence genes and the downregulation of the protease SpeB. To identify in vivo mutants with novel phenotypes, GAS isolates from infected mice were screened by enzymatic assays for SpeB and the platelet-activating factor acetylhydrolase Sse, and a new type of variant that had enhanced Sse expression and normal levels of SpeB production was identified (the variants had a phenotype referred to as enhanced Sse activity [SseA+] and normal SpeB activity [SpeBA+]). SseA+ SpeBA+ variants had transcript levels of CovRS-controlled virulence genes comparable to those of a covS mutant but had no covRS mutations. Genome resequencing of an SseA+ SpeBA+ isolate identified a C605A nonsense mutation in orphan kinase gene rocA, and 6 other SseA+ SpeBA+ isolates also had nonsense mutations or small indels in rocA. RocA and CovS mutants had similar levels of enhancement of the expression of CovRS-controlled virulence genes at the exponential growth phase; however, mutations of RocA but not mutations of CovS did not result in the downregulation of speB transcription at stationary growth phase or in subcutaneous infection of mice. GAS with RocA and CovS mutations caused greater enhancement of the expression of hasA than spyCEP in mouse skin infection than wild-type GAS did. RocA mutants ranked between wild-type GAS and CovS mutants in skin invasion, inhibition of neutrophil recruitment, and virulence in subcutaneous infection of mice. Thus, GAS RocA mutants can be selected in subcutaneous infections in mice and exhibit gene expression patterns and virulences distinct from those of CovS mutants. The findings provide novel information for understanding GAS fitness mutations in vivo, virulence gene regulation, in vivo gene expression, and virulence.
KEYWORDS: CovRS, PAF acetylhydrolase, RocA, SpeB, Streptococcus pyogenes, group A Streptococcus, in vivo expression, mutation, virulence, virulence regulation
INTRODUCTION
The human pathogen group A Streptococcus (GAS) commonly causes pharyngitis and superficial skin infections. GAS also causes severe invasive infections, such as necrotizing fasciitis and sepsis. The most recently available data indicate that from 2005 to 2012 severe invasive infections in the United States were most frequently associated with GAS of the M protein serotypes M1, M12, M28, M89, and M3 (1), and the currently circulating M1 GAS belongs to the pandemic M1T1 clone (2). GAS produces an abundance of extracellular virulence factors to mediate its pathogenesis (3, 4). These virulence factors include the M protein (5), C5a peptidase ScpA (6), the hyaluronic acid capsule synthesized by HasABC (7), CXC chemokine peptidase SpyCEP (8), platelet-activating factor (PAF) acetylhydrolase Sse (9), streptolysin O (Slo) (10), NADase Nga (11), streptokinase (12), protease SpeB (13), and opsonophagocytosis-inhibiting protein Mac (14). Most of these virulence factors are involved in innate immune evasion.
While the M protein gene, emm, and scpA are regulated by the transcription activator Mga (15), most of the virulence genes are negatively regulated by the two-component regulatory system CovRS (also known as CsrRS) (16–19). Mutation of the histidine kinase covS causes the downregulation of speB and enhancement of expression of many CovRS-controlled virulence genes, resulting in hypervirulence (19–22). The rocA gene, an orphan kinase, regulates CovRS expression and capsule production (23) and functions through CovR (24). Natural mutations and deletion of rocA lead to considerable increases in the levels of transcription of multiple CovRS-controlled virulence genes, at least in M3 and M1 GAS isolates (24).
Invasive GAS isolates frequently carry CovRS mutations that are associated with their hypervirulence (25, 26). CovRS mutations appear to arise during human infections with GAS isolates carrying wild-type CovRS and are not transmissible (27). RocA mutations are also present in clinical isolates. In particular, all M3 GAS isolates isolated from diverse geographic locations since the 1930s have a truncation mutation that enhances the levels of the hyaluronic acid capsule (28), and a nonsense mutation in rocA is present in serotype M18 GAS isolates and contributes to the hyperencapsulation of serotype M18 strains (29). Serotype M89 strains have polymorphisms in the region upstream of the rocA gene that alter rocA expression (30). RocA mutations are detected in clinical M1 GAS isolates (31). Mutations that lead to the loss of RocA expression or function enhance virulence (24, 30, 31). Whether null CovS and RocA mutations have any distinct effects on gene expression is not known.
The emergence of CovRS mutants of invasive M1T1 GAS isolates has readily been demonstrated during experimental mouse infections (20, 22, 32–36), and M12 GAS isolates have recently been demonstrated to arise in subcutaneous infections in mice as well (36). In these studies, the SpeB casein hydrolysis assay used to identify GAS variants in skin infections in mice inevitably identifies CovRS mutants for which SpeB activity is lacking in the culture supernatant and may miss mutants that have enhanced expression of virulence genes but have normal SpeB production. The objective of this study was to determine whether such M1T1 GAS mutants can be selected in infected mice and, if so, to characterize such mutants. We screened GAS isolates recovered from mice infected with M1T1 GAS strain MGAS2221 by enzymatic assays for the expression of the protease SpeB and CovRS-controlled PAF acetylhydrolase Sse. We indeed identified MGAS2221 variants that produced high levels of Sse but that had normal levels of SpeB expression, a phenotype referred to as enhanced Sse activity (SseA+) and normal SpeB activity (SpeBA+). This new phenotype was caused by RocA mutations. Although null RocA and CovS mutants showed similar enhancements in the levels of transcription of CovRS-controlled virulence genes at the exponential growth phase, CovS mutations but not RocA mutations downregulated speB transcription and enhanced hasA and sse transcription at the stationary phase and in skin infections in mice. RocA mutants ranked between CovS mutants and MGAS2221 in lesion sizes, GAS loads in the spleen, inhibition of neutrophil recruitment, and virulence in subcutaneous infections in mice. Thus, we demonstrated the in vivo selection of M1T1 GAS RocA mutants and the distinct effects of CovS and RocA mutations on in vitro and in vivo gene expression and virulence.
RESULTS
SseA+ SpeBA+ variants of MGAS2221 in subcutaneous mouse infection.
To identify the novel GAS mutants selected in vivo, we screened 48 GAS isolates that were recovered from skin infection sites from each of 4 mice 4 days after inoculation using two assays, one for the activity of the Sse PAF acetylhydrolase and the other for SpeB protease activity in their culture supernatants. Some isolates retained the phenotype of MGAS2221 without significant levels of Sse activity in the culture supernatant at the exponential growth phase (Fig. 1A, black curves) and with detectable SpeB activity in the supernatant of overnight cultures (the SpeB assay results are not shown). This wild-type phenotype is referred to here as SseA− SpeBA+. A second type of isolate had no detectable SpeB activity but had high levels of Sse PAF acetylhydrolase activity (Fig. 1A, blue curves), and this phenotype, referred to here as SseA+ SpeBA−, is similar to that of the previously characterized SpeBA− variants (22). Several isolates had SpeB activity at levels comparable to those of MGAS2221 but had levels of Sse activity similar to those of the SseA+ SpeBA− isolates (Fig. 1A, red curves), and this new phenotype is referred to as SseA+ SpeBA+. The remaining isolates were SpeBA+ and had enhanced Sse activity, but their Sse activity was not as high as that of the SseA+ SpeBA− and SseA+ SpeBA+ variants (Fig. 1A, green curves), and these isolates were not further characterized. GAS isolates with the SseA+ SpeBA− and SseA+ SpeBA+ phenotypes were present in all four mice (Fig. 1B). The percentages of SseA+ SpeBA+ variants in mice were less variable than the percentages of SseA+ SepBA− variants (average ± standard deviation, 14.58% ± 3.82% for SseA+ SpeBA+ variants versus 25.55% ± 23.10% for SseA+ SpeBA− variants; P = 0.7715). The possible implication for this difference is discussed in Discussion.
FIG 1.
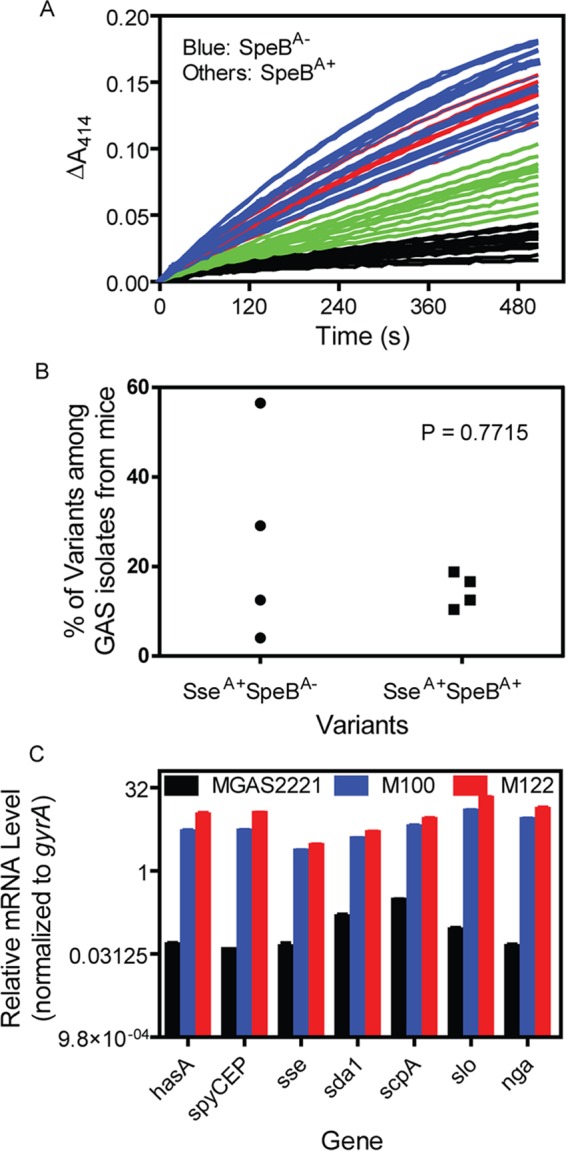
Identification of MGAS2221 variants selected in mouse skin infections that had enhanced virulence gene expression and normal levels of SpeB production. Four mice were inoculated subcutaneously with 108 CFU MGAS2221. GAS bacteria were recovered from the skin infection sites, and the Sse and SpeB activities in the culture supernatant of 48 colonies from each mouse were determined. (A) The time courses of changes of the A414 due to the hydrolysis of 2-thio-PAF catalyzed by Sse in culture supernatants of 48 isolates from the same mouse. Isolates without detectable SpeB activity (SpeBA−) had high levels of Sse activity (SseA+) (blue curves) and are referred to as SseA+ SpeBA− variants. Red lines, SseA+ SpeBA+ variants; black lines, isolates with the wild-type SseA− SpeBA+ phenotype. (B) Percentages of SseA+ SpeBA− and SseA+ SpeBA+ variants among the analyzed isolates from each of four mice. (C) Relative mRNA levels of hasA, spyCEP, sda1, scpA, slo, nga, and sse in isolates M100 (SseA+ SpeBA−), M122 (SseA+ SpeBA+), M177 (SseA− SpeBA+), and parent strain MGAS2221 (SseA− SpeBA+) at the exponential growth phase. The transcript data from triplicate analyses of three independent samples of each strain were first normalized to the data for the internal gyrA control and then to the data for MGAS2221.
Next we randomly chose one isolate from each of the three phenotypic categories, M100 (SseA+ SpeBA−), M122 (SseA+ SpeBA+), and M177 (SseA− SpeBA+), for further analyses. These isolates were compared with the parent strain for the transcript levels of the CovRS-controlled virulence genes hasA, spyCEP, sda1, scpA, slo, nga, and sse at the exponential growth phase. The M100 and M122 isolates had similar high levels of mRNA for these virulence genes, which were ≥21-fold higher than those in MGAS2221 and M177 (Fig. 1C), even though M122, but not M100, had normal levels of SpeB production (Fig. 2A). Thus, in addition to known SseA+ SpeBA− variants, we identified a new kind of in vivo-selected variant of M1T1 GAS that has enhanced expression of CovRS-controlled virulence genes but unaltered SpeB expression in comparison with the gene expression of wild-type M1T1 GAS.
FIG 2.
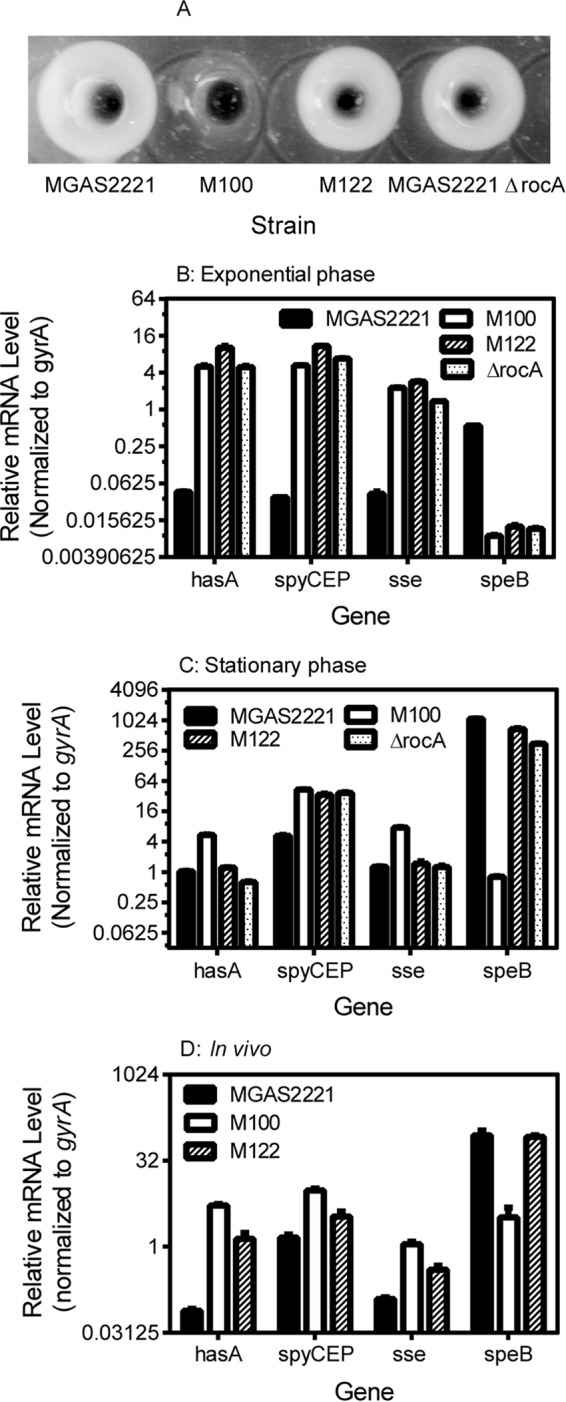
Comparison of the levels of SpeB production and the hasA, sse, speB, and spyCEP transcription levels in vitro and in vivo in isolates MGAS2221, M100 (covSΔ133TTCTT137), M122, and MGAS2221 ΔrocA. (A) The overnight culture supernatant for MGAS2221 and its rocA mutants but not its covS mutant had SpeB protease activity, as determined by the casein hydrolysis assay. (B to D) Relative hasA, sse, and spyCEP transcript levels in the indicated strains at the exponential growth phase (B) and stationary phase (C) in THY and in infected mouse skin at 8 h after inoculation (D). The real-time RT-PCR data from triplicate analyses of three independent in vitro and in vivo samples of each strain were normalized to the data for the internal gyrA control.
Genetic variations for the SseA+ SpeBA+ phenotype.
The transcript levels of the CovRS-controlled virulence genes in the SseA+ SpeBA+ M122 isolate were similar to the transcript levels of these genes in MGAS2221 covS-null mutants reported previously (20, 21). One possibility for the basis of the SseA− SpeBA+ phenotype is that certain CovRS mutations enhance the expression of these virulence genes but have no detrimental effect on SpeB expression. To examine this possibility, the covRS genetic locus in isolates M100, M122, and M177 was sequenced. M100 had a 5-bp deletion of TTCTT from bases 133 to 137 of the covS gene (covSΔ133TTCTT137) (Table 1), consistent with the previous finding that SpeBA− variants from skin infections in mice usually have covS mutations (22, 36). As expected, M177 had the wild-type covRS genes. Unexpectedly, M122 also had the wild-type covRS genes. Thus, the SseA+ SpeBA+ phenotype of isolate M122 was caused by a variation other than covRS mutations.
TABLE 1.
Mutations of virulence regulators in SseA− SpeBA+, SseA+ SpeBA+, and SseA+ SpeBA− isolates from mice with subcutaneous MGAS2221 infection
| Isolate | Mouse no. | Activity in culture supernatant |
Mutation |
||
|---|---|---|---|---|---|
| Sse | SpeB | rocA | covRS | ||
| M27 | 1 | + | + | C605A | wta |
| M76 | 2 | + | + | G insertion at 205 | wt |
| M108 | 3 | + | + | C605A | wt |
| M116 | 3 | + | + | C605A | wt |
| M122 | 3 | + | + | C605A | wt |
| M138 | 3 | + | + | 204TGATGTT insertion | wt |
| M153 | 4 | + | + | 1228A deletion | wt |
| M100 | 4 | + | − | wt | covSΔ133TTCTT137 |
| M177 | 4 | − | + | wt | wt |
wt, wild type.
To identify the genetic alteration that was responsible for the SseA+ SpeBA+ phenotype, M122 was subjected to Illumina MiSeq sequencing analysis, and 99.49% of 14,199,275 reads that passed quality control were aligned to the genome of M1T1 isolate MGAS5005 (NCBI accession number NC_007297.2). The analysis identified 21 single nucleotide polymorphisms (SNPs), two 1-bp inserts, and a 1-bp deletion. Excluding the known SNPs between MGAS5005 and MGAS2221 and the 1-bp deletion in MGAS5005 covS (20), there were three alterations in two loci: (i) M5005_spy0556, encoding phosphopyruvate hydrolase, had two alterations, a 1-bp addition, and a 1-bp deletion, in a 7-bp segment from positions 547753 to 547759, and (ii) the C605A mutation was found in the rocA gene. Sanger sequencing found that the sequence of the M5005_spy0556 gene in MGAS5005, MGAS2221 (the parent strain of M122), M122, and M100 was identical to the M5005_spy0556 gene sequence in the M122 genome determined by Illumina MiSeq sequencing analysis and the M5005_spy0556 gene sequence in many GAS genome sequences in the GenBank database. Apparently, the M5005_spy0556 polymorphism between the M122 sequence and the MGAS5005 reference sequence was due to either an error in the MGAS5005 genome sequence or the acquisition of the mutations by the sequenced bacterium of MGAS5005. Thus, we detected the rocA C605A mutation in M122, and this mutation resulted in a stop codon and, thus, a truncated RocA comprising just the first 201 amino acids of the 451 amino acid residues in the wild type.
We next sequenced by Sanger sequencing the rocA and covRS genes in M122, six other SseA+ SpeBA+ isolates from four mice, one SseA− SpeBA+ isolate (M177), and one SseA+ SpeBA− isolate (M100). None of the SseA+ SpeBA+ isolates had mutations in covRS genes, but they did have either the C605A replacement or an open reading frame-shifting insertion or deletion in the rocA gene, whereas M177 and M100 had the wild-type rocA sequence (Table 1). Thus, all 7 SseA+ SpeBA+ isolates analyzed had rocA mutations, indicating that rocA-null mutations result in the SseA+ SpeBA+ phenotype. To further confirm that the rocA-null mutations result in the SseA+ SpeBA+ phenotype, we deleted the rocA gene in MGAS2221. The MGAS2221 ΔrocA mutant had SpeB activity in its overnight culture supernatant at levels similar to those of the parent strain (Fig. 2A). Consistent with the recent finding that the rocA deletion in MGAS2221 enhances the expression of CovRS-controlled virulence genes (24), at the exponential growth phase the MGAS2221 ΔrocA mutant had levels of hasA, spyCEP, and sse transcripts that were similar to those of the rocAC605A and covSΔ133TTCTT137 mutants and >40-fold higher than those of the parent strain (Fig. 2B and Table 2). Thus, rocA mutations are selected in mice and enhance the expression of multiple virulence genes but do not affect SpeB production.
TABLE 2.
Relative mRNA levels of virulence genes in strains MGAS2221, M100 (covSΔ133TTCTT137), and M122 (rocAC605A) under in vitro and in vivo conditions
| Condition | Gene | Relative mRNA level or fold change in expressiona |
||
|---|---|---|---|---|
| MGAS2221 | M122 | M100 | ||
| Exponential growth phase | hasA | 1 | 197 | 111 |
| spyCEP | 1 | 252 | 141 | |
| sse | 1 | 55 | 54 | |
| speB | 1 | −9 | −63 | |
| sda1 | 1 | 33 | 26 | |
| scpA | 1 | 29 | 21 | |
| slo | 1 | 241 | 142 | |
| nga | 1 | 301 | 198 | |
| sagA | 1 | 1.8 | 0.86 | |
| Stationary phase | hasA | 1 | 1 | 5 |
| spyCEP | 1 | 6 | 8 | |
| sse | 1 | 1 | 6 | |
| speB | 1 | 0.50 | −1,366 | |
| sda1 | 1 | 5.8 | 4 | |
| scpA | 1 | 2 | 6 | |
| slo | 1 | 6 | 18 | |
| nga | 1 | 5 | 15 | |
| sagA | 1 | 6.4 | 1.4 | |
| In vivo | hasA | 1 | 24 | 64 |
| spyCEP | 1 | 3 | 6 | |
| sse | 1 | 4 | 8 | |
| speB | 1 | 1 | −76 | |
| sda1 | 1 | 1.8 | 2.1 | |
| scpA | 1 | 2 | 3.9 | |
| slo | 1 | 1.5 | 1.6 | |
| nga | 1 | 1.9 | 1.9 | |
| sagA | 1 | 3.5 | −2.6 | |
| In vivo and exponential phaseb | hasA | 2 | 0.22 | 0.97 |
| spyCEP | 40 | 0.46 | 1.84 | |
| sse | 3 | 0.22 | 0.49 | |
| speB | 155 | 1,458 | 122 | |
| sda1 | 3.2 | 0.20 | 0.22 | |
| scpA | 1.8 | 0.13 | 0.33 | |
| slo | 16 | 0.25 | 0.36 | |
| nga | 15 | 0.09 | 0.15 | |
| sagA | 66 | 125 | 30 | |
The real-time RT-PCR data in Fig. 2 and for additional genes were first normalized to those for the internal gyrA control and then to those for MGAS2221.
The normalized in vivo data for each gene of each strain were divided by those for the same strain at the exponential growth phase.
Distinct effects of rocA and covS mutations on virulence gene expression at the stationary phase.
Because SpeB is produced in the stationary growth phase by RocA mutants but not by CovS mutants (Fig. 2A and B), we hypothesized that null mutations of covS but not rocA alter speB transcription in the stationary phase. Thus, we compared the transcript levels of hasA, spyCEP, sse, and speB in MGAS2221, M100 (covSΔ133TTCTT137), M122 (rocAC605A), and MGAS2221 ΔrocA at the exponential growth phase and stationary phase. On the basis of the threshold cycle numbers for the speB and gyrA transcripts, the levels of speB mRNA were about 50% of those of gyrA transcripts in MGAS2221 at the exponential growth phase, and SpeB activity was not detectable by the casein assay at this level of speB mRNA expression. The levels of the speB transcript in MGAS2221 at the stationary phase were 310-fold greater than those at the exponential growth phase in the same amount of RNA. At the exponential growth phase, all the RocA and/or CovS mutants had an enhancement in the levels of mRNA for hasA, spyCEP, sda1, scpA, slo, nga, and sse and suppression of speB transcription (Fig. 1C and 2B and Table 2). At the stationary phase, the levels of the speB transcript decreased by about 50% compared with those for the parent strain but were still at very high levels in M122 (rocAC605A) and MGAS2221 ΔrocA, and the level of speB expression decreased by 130,000% in M100 (covSΔ133TTCTT137) compared with that in the parent strain (Fig. 2C and Table 2). M122 (rocAC605A) had a 6.4-fold higher level of sagA expression at stationary phase than MGAS2221, and the covSΔ133TTCTT137 mutation in M100 had a negligible effect on sagA expression at exponential growth and stationary phases (Table 2). M122 (rocAC605A) and MGAS2221 had similar levels of hasA, scpA, and sse transcripts, while M100 (covSΔ133TTCTT137) had ≥5-fold more hasA and sse mRNA at the stationary phase, though all the rocA and covS mutants had ≥6-fold more spyCEP mRNA than MGAS2221 (Fig. 2C and Table 2). The order of the levels of expression for the other CovRS-controlled virulence genes at stationary phase was M100 (covSΔ133TTCTT137) > M122 (rocAC605A) > MGAS2221 (Table 2). Thus, the RocA- and CovS-mediated regulation of virulence genes, especially speB and sagA, can be distinguished at the stationary phase.
In vivo expression of virulence genes in MGAS2221, M122 (rocAC605A), and M100 (covSΔ133TTCTT137).
The in vitro expression data described above raise an important question: are the distinct effects of RocA and CovS mutations on SpeB expression at the stationary phase relevant in infections? To address this question, we compared by quantitative PCR (qPCR) analysis the hasA, spyCEP, sse, sda1, scpA, slo, nga, sagA, and speB transcript levels in MGAS2221, M100 (covSΔ133TTCTT137), and M122 (rocAC605A) in subcutaneous infections in mice at 8 h after inoculation. As shown in Fig. 2D, M122 (rocAC605A) and M100 (covSΔ133TTCTT137) had 24- and 64-fold higher levels of hasA transcripts than MGAS2221, respectively, and the levels of the spyCEP and sse transcripts in M100 (covSΔ133TTCTT137) and M122 (rocAC605A) were ≤8-fold higher than those in MGAS2221. M100 (covSΔ133TTCTT137) had 2-fold higher spyCEP and sse transcript levels than M122 (rocAC605A). As summarized in Table 2, the levels of expression of sda1, scpA, slo, and nga in M100 (covSΔ133TTCTT137) and M122 (rocAC605A) were 1.4 to 3.9 and 1.5 to 2.0 times higher than those in MGAS2221, respectively. For speB expression, M122 (rocAC605A) and MGAS2221 had similar levels of speB transcription, and M100 (covSΔ133TTCTT137) had a 76-fold decrease in the levels of speB transcription in comparison with that in MGAS2221 and M122 (rocAC605A) (Fig. 2D and Table 2). Similar to the speB expression patterns, sagA expression was higher in M122 (rocAC605A) than M100 (covSΔ133TTCTT137). Thus, the effects of RocA and CovS mutations on speB and sagA expression in vivo are closer to those in the stationary phase than those in the exponential growth phase in vitro.
The comparison of the levels of gene expression in vivo and at exponential phase also revealed some distinct expression patterns among the virulence genes. The levels of hasA mRNA in MGAS2221 in vivo were very low, just 8% of the levels of gyrA mRNA, and increased just 2-fold compared with the levels at the exponential growth phase in vitro (Table 2); however, the levels of mRNA for spyCEP, sagA, and speB in MGAS2221 increased ≥40-fold in vivo compared with the levels at the exponential growth phase in vitro. Apparently, different virulence genes exhibit different sensitivities to the in vivo signal-mediated relief of CovR repression of virulence genes.
Comparison of MGAS2221, M122 (rocAC605A), M100 (covSΔ133TTCTT137), and M177 (rocAwt covSwt) in skin invasion and neutrophil infiltration.
Because of the distinct effects of rocA and covS mutations on virulence gene expression, we were interested in whether the mutants had different phenotypes for skin infection. Thus, we compared parent strain MGAS2221 with M100 (covSΔ133TTCTT137), M122 (rocAC605A), and M177 (which expressed wild-type [wt] rocA [rocAwt] and covS [covSwt]) in subcutaneous infections in mice. M122 (rocAC605A) caused a mean lesion size ± standard error of the mean (SEM) of 256 ± 40 mm2, which was significantly smaller than the lesions caused by M100 (covSΔ133TTCTT137) (406 ± 69 mm2) but larger than the lesions caused by M177 (rocAwt covSwt) (128 ± 6 mm2) and MGAS2221 (131 ± 12 mm2) (Fig. 3A). The order of magnitude of the burdens of these GAS isolates in the spleen was similar to that for the lesion size data (Fig. 3B). The mean bacterial number ± SEM in the spleen in M122 (rocAC605A) infection was (2.2 ± 1.8) × 106 CFU/g, which was less than that in M100 (covSΔ133TTCTT137) infection [(6.6 ± 5.6) × 107 CFU/g] but greater than that in M177 (rocAwt covSwt) infection (753 ± 695 CFU/g) and in MGAS2221 infection (97 ± 96 CFU/g) (P values, 0.0097 for MGAS2221 versus M100, 0.0178 for MGAS2221 versus M122, 0.3465 for MGAS2221 versus M177, and 0.2222 for M100 versus M122) (Fig. 3B). The lesion and spleen GAS burden data were inversely related to the neutrophil levels at infection sites. The mean neutrophil number ± SEM at M122 (rocAC605A) infection sites was (3.7 ± 1.0) × 104 neutrophils/mm2, which was greater than that at M100 (covSΔ133TTCTT137) infection sites [(1.9 ± 0.3) × 104 neutrophils/mm2] but less than that at M177 (rocAwt covSwt) [(1.2 ± 0.2) × 105 neutrophils/g] and MGAS2221 [(1.1 ± 0.1) × 105 neutrophils/g] infection sites (Fig. 3C). Thus, the rocAC605A mutant ranked between the covS mutant and wild-type strains in lesion size, bacterial burdens in the spleen, and inhibition of neutrophil infiltration.
FIG 3.

The RocA-null mutant ranks between MGAS2221 and the covS mutant in skin invasion, inhibition of neutrophil infiltration, GAS loads in the spleen, and virulence in subcutaneous infection of mice. Groups of 15 or 10 mice were subcutaneously inoculated with 108 CFU of MGAS2221, M100 (covSΔ133TTCTT137), M122 (rocAC605A), M177 (wild-type covS and rocA), or MGAS2221 ΔrocA. (A to C) Five mice from each group were euthanized at 24 h after GAS inoculation to measure lesion sizes (A), GAS loads in spleen (B), and neutrophil (polymorphonuclear leukocyte [PMN]) infiltration at skin infection sites (C). (D) Ten mice were monitored for up to 14 days after inoculation to determine survival rates (P values, 0.0001 for M122 versus MGAS2221, 0.0003 for the ΔrocA mutant versus MGAS2221, 0.0291 for M122 versus M100, and 0.8706 for the ΔrocA mutant versus M122).
rocA-null mutants rank between MGAS2221 and the covS mutants in virulence in mice.
Consistent with the lesion size, GAS load, and neutrophil infiltration data described above, M122 (rocAC605A) and MGAS2221 ΔrocA were significantly more virulent in subcutaneous infections in mice than MGAS2221 and less virulent than M100 (covSΔ133TTCTT137) (P values, 0.0001 for M122 versus MGAS2221, 0.0003 for the ΔrocA mutant versus MGAS2221, and 0.0291 for M122 versus M100) (Fig. 3D). In addition, MGAS2221 ΔrocA and M122 (rocAC605A) had similar virulences (P = 0.8706). Thus, while rocA mutations increased the virulence of M1T1 GAS, the rocA mutants were less virulent than the covS mutants.
DISCUSSION
Using the assays for SpeB protease and Sse PAF acetylhydrolase activities in the culture supernatant, we identified a new kind of MGAS2221 mutant that is selected in mouse infection and that has enhanced expression of CovRS-controlled virulence genes but normal SpeB expression. This novel phenotype is caused by rocA-null mutations. MGAS2221 RocA and CovS mutants had similar enhancements in the levels of expression of CovRS-controlled virulence genes at the exponential growth phase; however, mutations of RocA but not CovS did not have a dramatic effect on the level of speB expression at stationary phase and in subcutaneous infection in mice. The levels of expression of speB and spyCEP in wild-type M1T1 GAS strains in skin infections in mice were similar to those at stationary phase and were considerably higher than those at exponential growth phase in vitro; however, hasA expression in vivo was repressed nearly as much as it was at exponential growth phase. RocA mutants rank between wild-type MGAS2221 and its CovS mutants in the lesion sizes that they cause, the GAS loads in the spleen that they cause, their levels of inhibition of neutrophil recruitment, and their virulence in subcutaneous infection in mice. As discussed below, the findings provide novel insight into the understanding of GAS fitness mutations in vivo, virulence gene regulation, in vivo gene expression, and virulence.
Clinical GAS isolates frequently have mutations in the virulence regulators CovRS and RocA (24–26, 28–31, 37). GAS isolates with wild-type CovRS and CovRS mutants are present in the same patients (38). Mice with skin infection have been a valuable model with which to demonstrate the arising of CovRS mutants of the contemporary invasive M1T1 serotype. The capsule synthesis gene hasA and the M protein gene are required for the selection of M1T1 GAS CovRS mutants in skin infection (34). The DNase Sda1 is reported to be the trigger for the in vivo selection of the CovRS mutant (33), but this finding could not be reproduced (35). The arising of CovRS mutants of M1T1 GAS requires neutrophils, and CovS mutants survive better than wild-type GAS against neutrophils in mice (22). This is not surprising because many of the CovRS-regulated genes are involved in evasion of the neutrophil responses. Thus, CovRS mutations are apparently fitness mutations that are selected for to obtain a survival advantage over wild-type GAS against killing by neutrophils. This study shows that RocA mutants of M1T1 GAS can be selected during subcutaneous infections in mice. RocA mutants are more virulent than wild-type GAS and are most likely fitness mutants in vivo. During infections, GAS is in a hostile environment and faces elimination by various immune system killing mechanisms. Fitness mutants that survive killing by the innate immune of the host better than parent strains would therefore arise.
RocA mutants have the SseA+ SpeBA+ phenotype, while CovS mutations cause the SseA+ SepBA− phenotype. These data are consistent with the findings that no RocA mutations are detected but CovS mutations are frequent in clinical SpeBA− isolates (39). As shown in Fig. 1B, the percentage of SseA+ SpeBA+ isolates in different mice was less variable than the percentage of SsEA+ SepBA− isolates. We have shown that the levels of neutrophils vary at skin MGAS2221 infection sites (9). Since neutrophils are required for the selection of CovRS mutations, the high degree of variation of the percentage of SseA+ SepBA− isolates among different mice may reflect the variation in the levels of neutrophil infiltration at skin infection sites. Several genes involved in evasion of the neutrophil response are upregulated in RocA mutants (24). Neutrophils most likely play a role in the selection of RocA mutants. However, the selection of RocA mutants not only may be dependent on neutrophils, but an additional selection force may also be present. Otherwise, the variation in the percentage of SseA+ SpeBA+ isolates among mice should be similar to that for SseA+ SepBA− isolates. Invasive M1T1 GAS can evade autophagic killing inside epithelial cells, and SpeB is required for evasion of autophagic killing (40). The SseA+ SpeBA+ phenotype may provide a fitness advantage not only against killing by neutrophils but also against killing by other mechanisms, such as autophagic killing by epithelial cells.
The relative virulence of MGAS2221 and its RocA and CovS mutants in subcutaneous infections in mice appears to be correlated with the relative level of virulence gene expression in vivo. MGAS2221 RocA-null mutants are more virulent than wild-type MGAS2221 in subcutaneous infections in mice, confirming the recent findings that RocA mutations or rocA-downregulating mutations enhance the virulence of GAS in different serotype backgrounds (24, 29–31). The rocAC605A mutant had higher levels of expression of hasA, spyCEP, scpaA, sda1, slo, nga, sagA, and sse than MGAS2221 in vivo, while the two strains had similar levels of speB expression in skin infections in mice. The higher level of virulence of the rocA mutant than of MGAS2221 is apparently due to the enhanced expression of virulence genes. The other new information regarding the virulence of RocA mutants from this study is that RocA mutants have a significantly lower capacity to cause local tissue damage, disseminate to organs, and inhibit neutrophil infiltration and are significantly less virulent than CovS mutants in subcutaneous infections in mice. These results are consistent with the attenuation of the virulence of hypervirulent, covS and rocA mutation-carrying serotype M3 strain MGAS315 by the replacement of the covS mutant gene with the wild-type covS gene (41). CovS and RocA mutations cause similar levels of enhancement of the expression of the same set of the virulence genes in vitro (20, 24, 42), and M122 (rocAC605A) and M100 (covSΔ133TTCTT137) had similar high levels of expression of these virulence genes in vitro (Fig. 1C). These in vitro expression data would support the idea that the repression of speB expression in M100 (covSΔ133TTCTT137) but not in M122 (rocAC605A) is critical for the difference in virulence between them. However, M100 (covSΔ133TTCTT137) has higher levels of in vivo expression of virulence genes than M122 (rocAC605A). Thus, the higher levels of in vivo expression of the virulence genes in CovS mutants than RocA mutants likely contribute at least partially to the enhanced virulence of the CovS mutants in comparison with that of the RocA mutants.
The covS mutant had a 76-fold decrease in the level of in vivo speB expression in comparison with that of MGAS2221 and the RocA mutant. Whether this speB downregulation critically contributes to the difference in virulence between the CovS and RocA mutants is not known. It has been proposed that the downregulation of SpeB in CovS mutants preserves virulence factors so that they may contribute to the hypervirulence of CovRS mutants (43, 44). The results of a number of studies also suggest that high levels of SpeB expression may enhance localized infection in soft tissue infections. Deletions of covR and covS similarly enhance the expression of many virulence genes (19); however, covR deletion enhances SpeB expression (17), whereas covS deletion downregulates SpeB expression (19, 21). In addition, SpeB critically contributes to the dermal ulceration caused by an M1 GAS covR deletion mutant (45). Interestingly, the covR deletion enhanced the skin lesion but reduced the level of virulence and the GAS loads in the spleen of SpeBA+ M3 GAS strain AM3 (46). These studies suggest that high levels of SpeB expression reduce systemic dissemination and virulence in skin infections. On the other hand, speB is critical for the virulence of the M3 AM3 strain in intraperitoneal infections in mice (13). In addition, the majority (84.6%) of invasive M1 GAS isolates are SpeBA+, and no MGAS2221 CovRS mutants were detected among isolates recovered from muscle infections in nonhuman primates (39). However, it should be noted that the percentage of SpeB isolates among clinical invasive emm1 GAS isolates in that study (39) was similar to the percentage of SpeBA− variants among GAS isolates from infected mice obtained at day 4 after inoculation in this study. The GAS isolates were recovered at day 1 after inoculation in that nonhuman primate study, and a lack of CovRS mutants in that study could have been due to the short duration of infection (39). Furthermore, the downregulation of SpeB expression and speB deletion may have different consequences. Thus, whether the downregulation of SpeB expression critically contributes to the hypervirulence of CovRS mutants is still an outstanding question.
A covR deletion mutant of M1T1 GAS strain MGAS5005 has higher speB transcript levels than MGAS5005 after subcutaneous infection in mice (47). Because MGAS5005 has a natural covS mutation (21), the data indicate the downregulation of in vivo speB expression by the covS mutation but do not indicate the in vivo speB expression of wild-type M1T1 GAS. The level of in vivo speB expression of M23 GAS strain HSC5 at days 1 and 2 after inoculation was similar to that at the stationary phase (48). The level of speB expression of MGAS2221 in skin infection at 8 h after inoculation was also similar to that at the stationary phase. The levels of expression of spyCEP and sse were also lower at the exponential growth phase than at the stationary phase and in MGAS2221 skin infection. The levels of expression of virulence genes in vivo were more similar to those at stationary phase in vitro. An exception was the level of hasA expression in MGAS2221, which was very low in skin infection and at exponential growth phase. The basis for this exception is not known.
A large body of data support a model in which CovR directly and negatively regulates virulence genes by binding to the promoter region of its target genes and in which CovR phosphorylation is regulated by CovS and RocA. In vitro CovR phosphorylation induces dimerization and enhances CovR binding to the promoter regions of hasA, sagA, covRS, speMF, and speB (49–52). In bacteria, CovS senses Mg2+ to phosphorylate CovR (53). Mutation or deletion of covS reduces the level of CovR phosphorylation by about 50% in M3 GAS strain MGAS1870 with a natural rocA-null mutation and about 70% in M1 strain MGAS2221 with wild-type rocA. Natural GAS RocA mutations enhance the production of the capsule synthesized by HasA (28, 29). A natural rocA mutation of M3 GAS and the rocA deletion in M1T1 GAS reduce the level of CovR phosphorylation and enhance the expression of CovRS-regulated genes at exponential growth phase in vitro (24). Based on these findings, Sumby and colleagues proposed that RocA and CovS form a heterodimer to regulate CovR activity (24).
RocA mutations repress speB expression at the exponential growth phase, which is consistent with the model of Sumby and colleagues (24). However, the model cannot explain the difference in the effects of RocA and CovS mutations on the expression of speB and sagA at the stationary phase in vitro and in vivo. RocA and CovS mutations cause similar levels of downregulation of RopB expression (24, 53), and thus, RopB appears not to play a role in the differential effects of RocA and CovS mutations on speB expression. There are also other unique features of speB and sagA expression described in the literature. Both speB and sagA are expressed at higher levels at stationary phase than the mid-exponential growth phase, and speB and sagA expression is downregulated by covS mutations, whereas the majority of CovRS-controlled genes are upregulated in covS mutants (19). In addition, the promoter region of sagA has two CovR-binding sites, the upstream promoter region and the downstream untranslated leader region of sagA (51), and CovR also protects the region covering the upstream and downstream regions of the speB promoter (49). In contrast, CovR binds to the region from position −35 to the downstream promoter region (49). We purely speculate that CovR can function as both an activator and a repressor of speB and sagA expression. In this case, speB cannot be expressed at high levels when the levels of CovR phosphorylation and ropB expression are low in covS mutants and the level of CovR phosphorylation in RocA mutants is still enough to cause a high level of speB expression and a higher level of sagA expression. We further speculate that RocA may sense a signal unique to stationary phase and the environment in vivo. SpeB expression is enhanced by acidic conditions that do not alter RopB levels (48). Thus, RocA may sense an acidic pH to phosphorylate CovR. More studies are needed to understand the mechanisms of the complicated regulation of the virulence genes by CovRS and RocA. The CovRS system but not RocA is present in group B Streptococcus (23). The RocA regulation of virulence genes that has evolved in GAS appears to provide an additional variety of means for virulence gene regulation to achieve fitness in different environments.
CovRS mutants appear to arise de novo during infections, but the rocA mutations of M3 and M18 GAS are preserved. In addition, CovS mutations are significantly more associated with invasive isolates, but the RocA mutations of M3 and M18 GAS are present in both invasive and noninvasive isolates (24, 28, 29). It has been proposed that RocA mutants are less attenuated in the ability to cause pharyngeal infections than covRS mutants, which is a possible reason for these observations (24). An alternative mechanism may be the normal production of SpeB in RocA mutants. Higher percentages of pharyngeal M1 isolates than severe invasive isolates possess the SpeBA+ phenotype (36). Normal SpeB production may provide an advantage in GAS transmission and fitness in pharyngeal infections.
MATERIALS AND METHODS
Declaration of ethical approval.
All animal experimental procedures were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Research Council (54). The protocols for the mouse experiments were approved by the Institutional Animal Care and Use Committee at MSU (permit numbers 2011-57 and 2014-45).
Bacterial strains and growth.
Serotype M1 strain MGAS2221 has been described previously (20). The strain and its derivative strains from mouse infection and in vitro manipulation were grown in Todd-Hewitt broth supplemented with 0.2% yeast extract (THY).
In vivo selection of MGAS2221 variants with alterations in expression of Sse and SpeB.
Details about the infection and care of animals for GAS variant selection have recently been described (36). Briefly, MGAS2221 was grown in THY to an optical density at 600 nm (OD600) of 0.35 and harvested by centrifugation. The bacteria were washed with pyrogen-free Dulbecco's phosphate-buffered saline (DPBS) twice and resuspended in DPBS to an OD600 of 0.9. The GAS suspension, 0.2 ml, was subcutaneously inoculated into each of four 5-week-old female C57BL/6J mice. CovS mutants of M1T1 GAS can be selected in both male and female C57BL/6J mice (22), but only female mice were used to minimize the number of mice used in the experiment. At day 4 after inoculation, skin samples containing infection sites were collected, homogenized in DPBS using a Kontes pestle, and plated at appropriate dilutions. Forty-eight colonies were randomly picked from each mouse and stored frozen. These isolates were analyzed by the SpeB casein hydrolysis and Sse PAF acetylhydrolase activity assays. The details of the SpeB casein hydrolysis assay have recently been described (36). Briefly, isolates recovered from mice were inoculated in 200 μl THY in 96-well plates and cultured in 5% CO2 at 37°C overnight. The cultures in the plates were centrifuged at 3,500 rpm for 10 min, and the culture supernatants were transferred into new plates. Three microliters of 10% β-mercaptoethanol was added into each well, the contents of the wells were mixed, and the plates were incubated for 20 min. The supernatant samples, 15 μl each, were then loaded into wells on casein gel plates, and the plates were incubated at 37°C for 3 h. The formation of a cloudy ring around the wells due to casein hydrolysis was an indication of the presence of SpeB activity in the culture supernatant (SpeBA+), and the lack of a cloudy ring indicated the lack of SpeB activity in the samples (SpeBA−).
The Sse PAF acetylhydrolase activity assay was performed with the same isolates as previously described (55). The isolates were grown in THY to an OD600 of 0.38 ± 0.02 and centrifuged to collect the culture supernatants. Each culture supernatant, 100 μl, was mixed with 100 μl of reactant solution containing 0.9 mM 2-thio-PAF and 1.3 mM 5,5′-dithiobis-(2-nitrobenzoic acid) at room temperature in the wells of a 96-well plate. The change in the absorbance at 414 nm (ΔA414), which was used as a measure of Sse-catalyzed 2-thio-PAF hydrolysis, was recorded with time using a SpectraMax 384 Plus spectrophotometer (Molecular Devices).
Whole-genome sequencing.
Illumina whole-genome sequencing of isolate M122 with the SseA+ SpeBA+ phenotype was performed by Otogenetics (Atlanta, GA). The sequencing data sets were subjected to analysis with the cutAdapt program to remove the adapters from the Illumina library preparation kit and then quality filtered. The sequences of the parsed data sets were mapped against the MGAS5005 genome sequence (NCBI accession number NC_007297.2) using the BWA-MEM algorithm (56). Polymorphisms were called with the GATK (v2.39) program.
Targeted DNA sequencing.
The covRS, rocA, ropB, and M5005_spy0556 genes of the test strains were sequenced by Sanger sequencing using a BigDye Terminator (v3.1) cycle sequencing kit and an Applied Biosystems 3130 genetic analyzer. The covRS genes were amplified from the test strains using a Phusion high-fidelity PCR kit from New England BioLabs and the primer pair 5′-TCGCTAGAAGACTATTTGAC-3′ and 5′-TTCATGTCATCCATCATTGC-3′. The primers used for covRS sequencing were 5′-TCGCTAGAAGACTATTTGAC-3′, 5′-TTCATGTCATCCATCATTGC-3′, 5′-AACGGCTTCATCATATTTCC-3′, 5′-AAATCCACAAAACCGTTCAG-3′, 5′-TGATACACACGACCGATAG-3′, 5′-TTGATGACAGAAAGGGCAG-3′, 5′-TACGCGAACCATGTCTAAC-3′, and 5′-GTTGGGGTAAAGATGACAG-3′. The rocA gene was amplified using primers 5′-CGAAATGAAAAGAAAATCGAG-3′ (rocAseq1) and 5′-CTATTGTCTCAGACTCTTAAG-3′ (rocAseq2) and sequenced with primers rocAseq1, rocAseq2, 5′-TACTGATTTACAGCATTACTTG-3′, and 5′-TATCTCTATTGTGAGACTTAC-3′. The ropB gene was amplified and sequenced using primers 5′-GTAACAATAACCACATAGTAGGCG-3′ and 5′-TCGTCATTGCTTTTTATGATTTGTC-3′. Part of the M5005_spy0556 gene was amplified from isolates MGAS5005, MGAS2221, M100, and M122 using primers 5′-GAAGCTTTTCATGAGATTGC-3′ (M5005_spy0556p1) and 5′-CAAAACCGATCATAATGCCG-3′ (M5005_spy0556p2) and sequenced with primer M5005_spy0556p1. Sequence data were analyzed using the software Sequencer (v5.1) from Gene Codes Corporation.
Generation of MGAS2221 ΔrocA.
Suicide plasmid p740-ΔrocA was used for the deletion of bases 152 to 1346 of rocA and was constructed as follows. 5′ and 3′ flanking fragments, each of which was ∼1,000 bp long, were amplified from MGAS2221 DNA using primer pairs 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTGTGTATGATTGAGTGGCAATAAC-3′ (primer 1)/5′-CATACCTTTAACATCAGTCAGGCTTGAAACAAAACCATACTAAGTG-3′ (primer 2) and 5′-CACTTAGTATGGTTTTGTTTCAAGCCTGACTGATGTTAAAGGTATG-3′ (primer 3)/5′-GGGGACCACTTTGTACAAGAAAGCTGGGTATAACCAAATGACTAATCATCGC-3′ (primer 4). The two PCR fragments were fused together in a subsequent overlay PCR via the 21-bp complementary sequences that are underlined in primers 2 and 3 using primers 1 and 4. The fused PCR product was first cloned into the donor vector pDONR221 in the BP Clonase reaction and then into p740-RFA (57) in the LR Clonase reaction using a Gateway Cloning kit, yielding p740-ΔrocA.
MGAS2221 ΔrocA was then generated as described previously (58). Briefly, the suicide plasmid p740-ΔrocA was introduced into MGAS2221 via electroporation. Chloramphenicol-resistant merodiploid transconjugants were selected on THY agar with 10 μg/ml chloramphenicol, and the sequences were confirmed by PCR. One transconjugant was grown on THY agar without chloramphenicol selection (one passage), and passaging was repeated ≥12 times. GAS colonies from the last passage were spotted in parallel on THY agar plates with and without chloramphenicol to identify chloramphenicol-sensitive colonies derived from a second homologous crossover. Chloramphenicol-sensitive strains were then analyzed by PCR using primers 5′-TTTAACTGTTAGAATGACAGAAC-3′ and 5′-GACTTTAGCCTAAGCCTGCCA-3′ to identify MGAS2221 ΔrocA, which had a shorter PCR product from the target gene than MGAS2221. The MGAS2221 ΔrocA sequence was confirmed by DNA sequencing.
Quantitative reverse transcription-PCR (RT-PCR) analyses.
For in vitro gene expression measurements, GAS was grown in THY at 37°C in 5% CO2 to an OD600 of 0.25 (exponential growth phase) or for 30 min after GAS growth reached a plateau (stationary phase). For in vivo gene expression measurements, 200 μl of a GAS suspension in DPBS with an OD600 of 1.0 was subcutaneously injected into each of three 6-week-old female C57BL/6 mice. At 8 h after inoculation, skin samples containing the infection site were collected, and each sample was ground in 1 ml of RNApro solution (catalog no. 6055-050; MP Biomedicals) using a conical tissue grinder (catalog no. 47732-446; VWR International). Large pieces of the remaining skin tissue were discarded, and there were about 4 × 108 CFU bacteria in each sample according to the plating results. The bacteria from the in vitro cultures and murine skin were immediately processed to isolate total RNA, as described previously (41). RNA was then converted into cDNA using an All-in-One first-strand cDNA synthesis kit from GeneCopoeia (Rockville, MD). Quantitative real-time PCR assays were performed using an All-in-One SYBR qPCR mix from GeneCopoeia. The following primers were used: for hasA, 5′-GGAGTTCAAACACAGATGCAATAC-3′ and 5′-CAAGGGAACGGTGAACGATAA-3′; for sse, 5′-TGCAGCTAGTTCTCGTTCTTG-3′ and 5′-GGAAGCTTGGGTCATCTTGT-3′; for spyCEP, 5′-CCGCTTGAACAGTCCTTGTA-3′ and 5′-CCTTCGATACGGTAGCCTTTAG-3′; for scpA, 5′-ACAGCCGATCAGCAAGATAAA-3′ and 5′-CATCCTCTTTCATCCCACGATTA-3′; for slo, 5′-ACCCTACCTATGCCAATGTTTC-3′ and 5′-GTTCTGGCAGGAAGCGTATTA-3′; for nga, 5′-GCGTCACGTGCTGAGTATTA-3′ and 5′-AAGCTCCGCTTTCTTTGTAGA-3′; for sagA, 5′-CTGCTGTTGCTGCTGTACTA-3′ and 5′-ATAACTTCCGCTACCACCTTG-3′; for sda1, 5′-CGTAAAGGTGGGATGCAGTAT-3′ and 5′-GGTAGCAATTCGGATCCTTGA-3′; and for gyrA, 5′-GCCGTTGGGATGGCAACTAACATT-3′ and 5′-TAACAAGGGCACCAGTCGGAAAGT-3′. All RNA samples were assayed in triplicate, and the levels of mRNA for each target gene were normalized to the level of mRNA for the gyrA gene.
Mouse infections.
C57BL/6J mice were bred at the Animal Resource Center (ARC) at Montana State University using breeding pairs of mice from The Jackson Laboratory (Bar Harbor, ME). Five-week-old female C57BL/6J mice were used to compare the test strains for virulence, neutrophil recruitment, skin invasion, and systemic GAS dissemination. Groups of 15 mice were subcutaneously inoculated with 0.2 ml of DPBS containing GAS at an OD600 of 1.0 (about 108 CFU). Five mice from each group were euthanized at day 1 after inoculation to collect skin samples for measurement of lesion size and neutrophil recruitment, as described below, and the spleen was also harvested to measure the numbers of viable GAS. Ten other mice from each group were monitored four times a day in the first 5 days after subcutaneous inoculation and twice a day (8:00 a.m. and 4:30 p.m.) after day 5 for 14 days to determine survival rates.
Quantification of neutrophil infiltration.
At day 1 after inoculation as described above, the skin around the infection sites was peeled off and the skin lesion was recognized by the boundary of the inflammation area. The size of the skin lesions was measured by analyzing the lesion pictures using the area measurement tool of the Adobe Acrobat (v9) software program. The skin containing the area of infection was excised for neutrophil measurement. The numbers of neutrophils recruited to the infected skin samples were determined by the myeloperoxidase assay, as described previously (9).
Statistical analysis.
The GraphPad Prism (v7) software program (GraphPad Software, Inc.) was used for statistical analyses. The data in Fig. 1B and 3A to C were analyzed using the two-tailed Mann-Whitney t test. The survival data in Fig. 3D were analyzed using the log-rank (Mantel-Cox) test.
Accession number(s).
The sequence data were deposited in the Sequence Read Archive (SRA) at the National Center for Biotechnology Information under accession number SRP091565.
ACKNOWLEDGMENTS
This work was supported in part by grants AI095704, AI097703, and GM110732 from the National Institutes of Health, Montana University System Research Initiative 51040-MUSRI2015-03, the Hatch Fund from the National Institute of Food and Agriculture, and the Montana State Agricultural Experiment Station.
REFERENCES
- 1.Nelson GE, Pondo T, Toews KA, Farley MM, Lindegren ML, Lynfield R, Aragon D, Zansky SM, Watt JP, Cieslak PR, Angeles K, Harrison LH, Petit S, Beall B, Van Beneden CA. 2016. Epidemiology of invasive group A streptococcal infections in the United States, 2005-2012. Clin Infect Dis 63:478–486. doi: 10.1093/cid/ciw248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nasser W, Beres SB, Olsen RJ, Dean MA, Rice KA, Long SW, Kristinsson KG, Gottfredsson M, Vuopio J, Raisanen K, Caugant DA, Steinbakk M, Low DE, McGeer A, Darenberg J, Henriques-Normark B, Van Beneden CA, Hoffmann S, Musser JM. 2014. Evolutionary pathway to increased virulence and epidemic group A Streptococcus disease derived from 3,615 genome sequences. Proc Natl Acad Sci U S A 111:E1768–E1776. doi: 10.1073/pnas.1403138111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lei B, Mackie S, Lukomski S, Musser JM. 2000. Identification and immunogenicity of group A Streptococcus culture supernatant proteins. Infect Immun 68:6807–6818. doi: 10.1128/IAI.68.12.6807-6818.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cunningham MW. 2000. Pathogenesis of group A streptococcal infections. Clin Microbiol Rev 13:470–511. doi: 10.1128/CMR.13.3.470-511.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fischetti VA. 1989. Streptococcal M protein: molecular design and biological behavior. Clin Microbiol Rev 2:285–314. doi: 10.1128/CMR.2.3.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cleary P, Prabu U, Dale J, Wexler D, Handley J. 1992. Streptococcal C5a peptidase is a highly specific endopeptidase. Infect Immun 60:5219–5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wessels MR, Goldberg JB, Moses AE, DiCesare TJ. 1994. Effects on virulence of mutations in a locus essential for hyaluronic acid capsule expression in group A streptococci. Infect Immun 62:433–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Edwards RJ, Taylor GW, Ferguson M, Murray S, Rendell N, Wrigley A, Bai Z, Boyle J, Finney SJ, Jones A, Russell HH, Turner C, Cohen J, Faulkner L, Sriskandan S. 2005. Specific C-terminal cleavage and inactivation of interleukin-8 by invasive disease isolates of Streptococcus pyogenes. J Infect Dis 192:783–790. doi: 10.1086/432485. [DOI] [PubMed] [Google Scholar]
- 9.Liu M, Zhu H, Li J, Garcia CC, Feng W, Kirpotina LN, Hilmer J, Tavares LP, Layton AW, Quinn MT, Bothner B, Teixeira MM, Lei B. 2012. Group A Streptococcus secreted esterase hydrolyzes platelet-activating factor to impede neutrophil recruitment and facilitate innate immune evasion. PLoS Pathog 8:e1002624. doi: 10.1371/journal.ppat.1002624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Seaghdha M, Wessels MR. 2013. Streptolysin O and its co-toxin NAD-glycohydrolase protect group A Streptococcus from xenophagic killing. PLoS Pathog 9:e1003394. doi: 10.1371/journal.ppat.1003394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bricker AL, Carey VJ, Wessels MR. 2005. Role of NADase in virulence in experimental invasive group A streptococcal infection. Infect Immun 73:6562–6566. doi: 10.1128/IAI.73.10.6562-6566.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun H, Ringdahl U, Homeister JW, Fay WP, Engleberg NC, Yang AY, Rozek LS, Wang X, Sjöbring U, Ginsburg D. 2004. Plasminogen is a critical host pathogenicity factor for group A streptococcal infection. Science 305:1283–1286. doi: 10.1126/science.1101245. [DOI] [PubMed] [Google Scholar]
- 13.Lukomski S, Sreevatsan S, Amberg C, Reichardt W, Woischnik M, Podbielski A, Musser JM. 1997. Inactivation of Streptococcus pyogenes extracellular cysteine protease significantly decreases mouse lethality of serotype M3 and M49 strains. J Clin Invest 99:2574–2580. doi: 10.1172/JCI119445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lei B, DeLeo FR, Hoe NP, Graham MR, Mackie SM, Cole RL, Liu M, Hill HR, Low DE, Federle MJ, Scott JR, Musser JM. 2001. Evasion of human innate and acquired immunity by a bacterial homolog of CD11b that inhibits opsonophagocytosis. Nat Med 7:1298–1305. doi: 10.1038/nm1201-1298. [DOI] [PubMed] [Google Scholar]
- 15.Hondorp ER, McIver KS. 2007. The Mga virulence regulon: infection where the grass is greener. Mol Microbiol 66:1056–1065. doi: 10.1111/j.1365-2958.2007.06006.x. [DOI] [PubMed] [Google Scholar]
- 16.Levin JC, Wessels MR. 1998. Identification of csrR/csrS, a genetic locus that regulates hyaluronic acid capsule synthesis in group A Streptococcus. Mol Microbiol 30:209–219. doi: 10.1046/j.1365-2958.1998.01057.x. [DOI] [PubMed] [Google Scholar]
- 17.Heath A, DiRita VJ, Barg NL, Engleberg NC. 1999. A two-component regulatory system, CsrR-CsrS, represses expression of three Streptococcus pyogenes virulence factors, hyaluronic acid capsule, streptolysin S, and pyrogenic exotoxin B. Infect Immun 67:5298–5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Federle MJ, McIver KS, Scott JR. 1999. A response regulator that represses transcription of several virulence operons in the group A Streptococcus. J Bacteriol 181:3649–3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Treviño J, Perez N, Ramirez-Peña E, Liu Z, Shelburne SA III, Musser JM, Sumby P. 2009. CovS simultaneously activates and inhibits the CovR-mediated repression of distinct subsets of group A Streptococcus virulence factor-encoding genes. Infect Immun 77:3141–3149. doi: 10.1128/IAI.01560-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sumby P, Whitney AR, Graviss EA, DeLeo FR, Musser JM. 2006. Genome-wide analysis of group A streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog 2:e5. doi: 10.1371/journal.ppat.0020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, Zhu H, Feng W, Liu M, Song Y, Zhang X, Zhou Y, Bei W, Lei B. 2013. Regulation of inhibition of neutrophil infiltration by the two-component regulatory system CovRS in subcutaneous murine infection with group A Streptococcus. Infect Immun 81:974–983. doi: 10.1128/IAI.01218-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li J, Liu G, Feng W, Zhou Y, Liu M, Wiley JA, Lei B. 2014. Neutrophils select hypervirulent CovRS mutants of M1T1 group A Streptococcus during subcutaneous infection of mice. Infect Immun 82:1579–1590. doi: 10.1128/IAI.01458-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biswas I, Scott JR. 2003. Identification of rocA, a positive regulator of covR expression in the group A Streptococcus. J Bacteriol 185:3081–3090. doi: 10.1128/JB.185.10.3081-3090.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller EW, Danger JL, Ramalinga AB, Horstmann N, Shelburne SA, Sumby P. 2015. Regulatory rewiring confers serotype-specific hyper-virulence in the human pathogen group A Streptococcus. Mol Microbiol 98:473–489. doi: 10.1111/mmi.13136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ikebe T, Ato M, Matsumura T, Hasegawa H, Sata T, Kobayashi K, Watanabe H. 2010. Highly frequent mutations in negative regulators of multiple virulence genes in group A streptococcal toxic shock syndrome isolates. PLoS Pathog 6:e1000832. doi: 10.1371/journal.ppat.1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shea PR, Beres SB, Flores AR, Ewbank AL, Gonzalez-Lugo JH, Martagon-Rosado AJ, Martinez-Gutierrez JC, Rehman HA, Serrano-Gonzalez M, Fittipaldi N, Ayers SD, Webb P, Willey BM, Low DE, Musser JM. 2011. Distinct signatures of diversifying selection revealed by genome analysis of respiratory tract and invasive bacterial populations. Proc Natl Acad Sci U S A 108:5039–5044. doi: 10.1073/pnas.1016282108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cole JN, Barnett TC, Nizet V, Walker MJ. 2011. Molecular insight into invasive group A streptococcal disease. Nat Rev Microbiol 9:724–736. doi: 10.1038/nrmicro2648. [DOI] [PubMed] [Google Scholar]
- 28.Lynskey NN, Turner CE, Heng LS, Sriskandan S. 2015. A truncation in the regulator RocA underlies heightened capsule expression in serotype M3 group A streptococci. Infect Immun 83:1732–1733. doi: 10.1128/IAI.02892-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lynskey NN, Goulding D, Gierula M, Turner CE, Dougan G, Edwards RJ, Sriskandan S. 2013. RocA truncation underpins hyper-encapsulation, carriage longevity and transmissibility of serotype M18 group A streptococci. PLoS Pathog 9:e1003842. doi: 10.1371/journal.ppat.1003842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu L, Olsen RJ, Horstmann N, Shelburne SA, Fan J, Hu Y, Musser JM. 2016. Intergenic variable-number tandem-repeat polymorphism upstream of rocA alters toxin production and enhances virulence in Streptococcus pyogenes. Infect Immun 84:2086–2093. doi: 10.1128/IAI.00258-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ikebe T, Matsumura T, Nihonmatsu H, Ohya H, Okuno R, Mitsui C, Kawahara R, Kameyama M, Sasaki M, Shimada N, Ato M, Ohnishi M. 2016. Spontaneous mutations in Streptococcus pyogenes isolates from streptococcal toxic shock syndrome patients play roles in virulence. Sci Rep 6:28761. doi: 10.1038/srep28761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Engleberg NC, Heath A, Miller A, Rivera C, DiRita VJ. 2001. Spontaneous mutations in the CsrRS two-component regulatory system of Streptococcus pyogenes result in enhanced virulence in a murine model of skin and soft tissue infection. J Infect Dis 183:1043–1054. doi: 10.1086/319291. [DOI] [PubMed] [Google Scholar]
- 33.Walker MJ, Hollands A, Sanderson-Smith ML, Cole JN, Kirk JK, Henningham A, McArthur JD, Dinkla K, Aziz RK, Kansal RG, Simpson AJ, Buchanan JT, Chhatwal GS, Kotb M, Nizet V. 2007. DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat Med 13:981–985. doi: 10.1038/nm1612. [DOI] [PubMed] [Google Scholar]
- 34.Cole JN, Pence MA, von Köckritz-Blickwede M, Hollands A, Gallo RL, Walker MJ, Nizet V. 2010. M protein and hyaluronic acid capsule are essential for in vivo selection of covRS mutations characteristic of invasive serotype M1T1 group A Streptococcus. mBio 1:e00191-10. doi: 10.1128/mBio.00191-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu G, Feng W, Li D, Liu M, Nelson DC, Lei B. 2015. The Mga regulon but not deoxyribonuclease Sda1 of invasive M1T1 group A Streptococcus contributes to in vivo selection of CovRS mutations and resistance to innate immune killing mechanisms. Infect Immun 83:4293–4303. doi: 10.1128/IAI.00857-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feng W, Liu M, Chen DG, Yiu R, Fang FC, Lei B. 2016. Contemporary pharyngeal and invasive emm1 and invasive emm12 group A Streptococcus isolates exhibit similar in vivo selection for CovRS mutants in mice. PLoS One 11:e0162742. doi: 10.1371/journal.pone.0162742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoshida H, Ishigaki Y, Takizawa A, Moro K, Kishi Y, Takahashi T, Matsui H. 2015. Comparative genomics of the mucoid and nonmucoid strains of Streptococcus pyogenes, isolated from the same patient with streptococcal meningitis. Genome Announc 3(2):e00221-15. doi: 10.1128/genomeA.00221-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Flores AR, Sahasrabhojane P, Saldaña M, Galloway-Peña J, Olsen RJ, Musser JM, Shelburne SA. 2014. Molecular characterization of an invasive phenotype of group A Streptococcus arising during human infection using whole genome sequencing of multiple isolates from the same patient. J Infect Dis 209:1520–1523. doi: 10.1093/infdis/jit674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olsen RJ, Raghuram A, Cantu C, Hartman MH, Jimenez FE, Lee S, Ngo A, Rice KA, Saddington D, Spillman H, Valson C, Flores AR, Beres SB, Long SW, Nasser W, Musser JM. 2015. The majority of 9,729 group A Streptococcus strains causing disease secrete SpeB cysteine protease: pathogenesis implications. Infect Immun 83:4750–4758. doi: 10.1128/IAI.00989-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barnett TC, Liebl D, Seymour LM, Gillen CM, Lim JY, Larock CN, Davies MR, Schulz BL, Nizet V, Teasdale RD, Walker MJ. 2013. The globally disseminated M1T1 clone of group A Streptococcus evades autophagy for intracellular replication. Cell Host Microbe 14:675–682. doi: 10.1016/j.chom.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stetzner ZW, Li D, Feng W, Liu M, Liu G, Wiley J, Lei B. 2015. Serotype M3 and M28 group A streptococci have distinct capacities to evade neutrophil and TNF-α responses and to invade soft tissues. PLoS One 10:e0129417. doi: 10.1371/journal.pone.0129417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bao YJ, Liang Z, Mayfield JA, Lee SW, Ploplis VA, Castellino FJ. 2015. CovRS-regulated transcriptome analysis of a hypervirulent M23 strain of group A Streptococcus pyogenes provides new insights into virulence determinants. J Bacteriol 197:3191–3205. doi: 10.1128/JB.00511-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aziz RK, Pabst MJ, Jeng A, Kansal R, Low DE, Nizet V, Kotb M. 2004. Invasive M1T1 group A Streptococcus undergoes a phase-shift in vivo to prevent proteolytic degradation of multiple virulence factors by SpeB. Mol Microbiol 51:123–134. [DOI] [PubMed] [Google Scholar]
- 44.Cole JN, McArthur JD, McKay FC, Sanderson-Smith ML, Cork AJ, Ranson M, Rohde M, Itzek A, Sun H, Ginsburg D, Kotb M, Nizet V, Chhatwal GS, Walker MJ. 2006. Trigger for group A streptococcal M1T1 invasive disease. FASEB J 20:1745–1747. doi: 10.1096/fj.06-5804fje. [DOI] [PubMed] [Google Scholar]
- 45.Engleberg NC, Heath A, Vardaman K, DiRita VJ. 2004. Contribution of CsrR-regulated virulence factors to the progress and outcome of murine skin infections by Streptococcus pyogenes. Infect Immun 72:623–628. doi: 10.1128/IAI.72.2.623-628.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dalton TL, Hobb RI, Scott JR. 2006. Analysis of the role of CovR and CovS in the dissemination of Streptococcus pyogenes in invasive skin disease. Microb Pathog 40:221–227. doi: 10.1016/j.micpath.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 47.Graham MR, Smoot LM, Migliaccio CA, Virtaneva K, Sturdevant DE, Porcella SF, Federle MJ, Adams GJ, Scott JR, Musser JM. 2002. Virulence control in group A Streptococcus by a two-component gene regulatory system: global expression profiling and in vivo infection modeling. Proc Natl Acad Sci U S A 99:13855–13860. doi: 10.1073/pnas.202353699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Loughman JA, Caparon M. 2006. Regulation of SpeB in Streptococcus pyogenes by pH and NaCl: a model for in vivo gene expression. J Bacteriol 188:399–408. doi: 10.1128/JB.188.2.399-408.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miller AA, Engleberg NC, DiRita VJ. 2001. Repression of virulence genes by phosphorylation-dependent oligomerization of CsrR at target promoters in S. pyogenes. Mol Microbiol 40:976–990. doi: 10.1046/j.1365-2958.2001.02441.x. [DOI] [PubMed] [Google Scholar]
- 50.Federle MJ, Scott JR. 2002. Identification of binding sites for the group A streptococcal global regulator CovR. Mol Microbiol 43:1161–1172. doi: 10.1046/j.1365-2958.2002.02810.x. [DOI] [PubMed] [Google Scholar]
- 51.Gao J, Gusa AA, Scott JR, Churchward G. 2005. Binding of the global response regulator protein CovR to the sag promoter of Streptococcus pyogenes reveals a new mode of CovR-DNA interaction. J Biol Chem 280:38948–38956. doi: 10.1074/jbc.M506121200. [DOI] [PubMed] [Google Scholar]
- 52.Gusa AA, Gao J, Stringer V, Churchward G, Scott JR. 2006. Phosphorylation of the group A streptococcal CovR response regulator causes dimerization and promoter-specific recruitment by RNA polymerase. J Bacteriol 188:4620–4626. doi: 10.1128/JB.00198-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Horstmann N, Sahasrabhojane P, Saldaña M, Ajami NJ, Flores AR, Sumby P, Liu CG, Yao H, Su X, Thompson E, Shelburne SA. 2015. Characterization of the effect of the histidine kinase CovS on response regulator phosphorylation in group A Streptococcus. Infect Immun 83:1068–1077. doi: 10.1128/IAI.02659-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 55.Liu M, Feng W, Zhu H, Lei B. 2015. A neutralizing monoclonal IgG1 antibody of platelet-activating factor acetylhydrolase Sse protects mice against lethal subcutaneous group A Streptococcus infection. Infect Immun 83:2796–2805. doi: 10.1128/IAI.00073-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997v2. https://arxiv.org/abs/1303.3997v2. [Google Scholar]
- 57.Zhou Y, Hanks TS, Feng W, Li J, Liu G, Liu M, Lei B. 2013. The sagA/pel locus does not regulate the expression of the M protein of the M1T1 lineage of group A Streptococcus. Virulence 4:698–706. doi: 10.4161/viru.26413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu H, Liu M, Sumby P, Lei B. 2009. The secreted esterase of group A Streptococcus is important for invasive skin infection and dissemination in mice. Infect Immun 77:5225–5232. doi: 10.1128/IAI.00636-09. [DOI] [PMC free article] [PubMed] [Google Scholar]