
Figure 1.
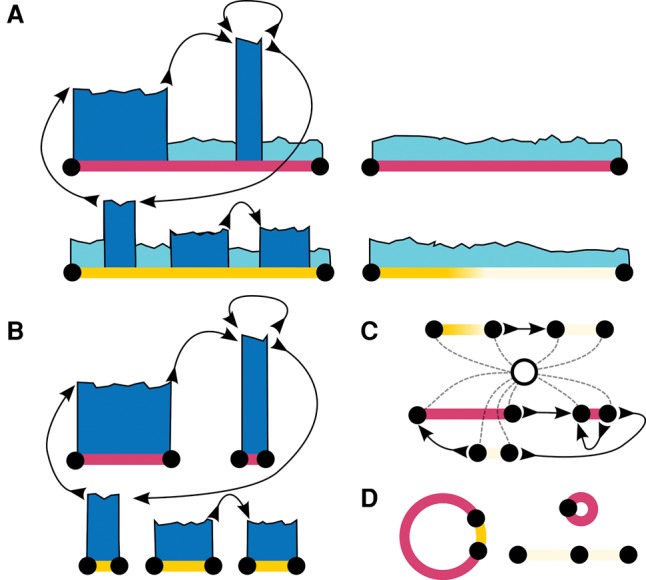
Overview of CouGaR algorithm. Tumor and normal samples are processed through a five-step algorithm. (A) We identify regions that are potentially amplified (dark blue) across two different chromosomes (red and yellow lines) in the tumor samples (left two contigs) compared to normal samples (right two contigs). We compute depth of coverage (DOC) information and cluster discordant read pairs to represent novel (with respect to hg19) adjacencies in the genome. (B) We identify continuous regions of amplification in the tumor genome using an HMM and DOC information from both tumor and normal samples. (C) We add a single super-source/-sink node, and using a min-cost circulation algorithm, we solve for the copy count of each region in the tumor genome. (D) Finally, a minimal set of circular and linear contigs that explain the coverage is found by formulating an integer programming problem that puts a penalty term on the number of unique contigs used.