

Abstract
Objective
Superstimulatory treatment of one-month-old lambs can achieve synchronous development of numerous growing follicles. However, these growing follicles cannot complete maturation and ovulation. Oocyte maturation and competence are acquired during follicular development, in which granulosa cells play an essential role.
Methods
In this study, we applied RNA sequencing to analyze and compare gene expression between prepubertal and adult superstimulated follicle granulosa cells in sheep.
Results
There were more than 300 genes that significantly differed in expression. Among these differently expressed genes, many extracellular matrix genes (EGF containing Fibulin Like Extracellular Matrix Protein 1, pentraxin 3, adrenomedullin, and osteopontin) were significantly down-regulated in the superstimulated follicles. Ingenuity pathway and gene ontology analyses revealed that processes of axonal guidance, cell proliferation and DNA replication were expressed at higher levels in the prepubertal follicles. Epidermal growth factor, T-Box protein 2 and beta-estradiol upstream regulator were predicted to be active in prepubertal follicles. By comparison, tumor protein P53 and let-7 were most active in adult follicles.
Conclusion
These results may contribute to a better understanding of the mechanisms governing the development of granulosa cells in the growing follicle in prepubertal sheep.
Keywords: Lamb, Granulosa Cells, Sheep, RNA Seq, Superstimulation
INTRODUCTION
Ovarian superstimulation protocols are commonly used components of assisted reproductive technology in monovular species in order to improve reproductive performance and genetic characteristics. Superstimulation involves the application of follicle-stimulating hormone (FSH) to stimulate the growth of a cohort of follicles to a pre-ovulatory stage. The continued growth of medium and large follicles depends on sufficient levels of gonadotrophins, and therefore exogenous gonadotrophins are used to reduce the incidence of atresia in these follicles [1]. Antral follicles become visible in the ovary of the ovine fetus at approximately day 135 of gestation and a relatively large number is present at birth. This number further increases until reaching a maximum between 4 and 8 weeks of age, before declining to relatively stable numbers as puberty approaches [2,3]. It has been shown that the number of antral follicles present postnatally is not affected by the season or by weight at birth and follicular atresia is minimal at this point in time, at which no evidence for the presence of follicle waves could be detected [4,5]. Hormonal regimes that usually combine gonadotrophins are available for ovarian stimulation [6]. Subsequently, embryos can be generated from prepubertal lambs by follicle aspiration, in vitro maturation and in vitro fertilization [6,7]. Offspring have been successfully produced from 4- to 8-week-old lambs by embryo transfer [7,8]. This approach shortens the generation interval and accelerates the genetic gain. Therefore, lambs are a reliable source of oocytes that offer a potential means of rapidly multiplying/reproducing animals of economic importance.
In the adult sheep, the superovulated oocyte competence is similar to natural ovulation [9]. However, the superstimulation treatment of lamb follicles leads to their synchronously growing to 3 to 4 mm in diameter and infrequent ovulation, following which the granulosa cells (GCs) cease dividing and stop differentiate into the specialized cells of the corpus luteum [6–8]. Furthermore, in vitro- adenosine triphosphate produced embryos derived from prepubertal oocytes show lower developmental rates [10]. The lower developmental competence maybe associated with deficiencies in the mRNA storage, incomplete nuclear maturation and adenosine triphosphate (ATP) content during oocyte growth [11–13]. It is well known that the oocytes and the GCs are in constant communication and the development of one influences the other’s compartment. During follicle development, GCs replicate, secrete hormones and support the growth of the oocyte [14]. Previous studies suggested that oocytes obtained after superstimulation may lag behind in maturation, differentiation and angiogenesis as well as activation of genes involved in the oxidative stress response and endoplasmic reticulum stress response in GCs in cows [15].
Transcriptome analysis of GCs may facilitate a better understanding of the molecular basis of follicular development in superstimulated lamb by evaluating the expression levels of thousands of genes at the same time [16].
The objective of this study was to determine the effect of ovarian superstimulation on major molecular and cellular pathways in ovine follicular GCs as a function of age. According to our hypothesis, molecular pathways affecting cell differentiation may be altered following superstimulation and this effect may differ between lambs and adult ewes. Therefore we analyzed the transcriptome of lamb GCs and compared it to that obtained from adult sheep follicles that underwent an equivalent/identical growing phase. An improved efficiency of juvenile in vitro embryo transfer due to adjustments of the hormone stimulation regimes and a better understanding of the differences existing between oocytes from adult and prepubertal animals may result in considerable economic benefits.
MATERIALS AND METHODS
Animals and treatments
Lambs used in experiments originated from the natural mating of mature Merino ewes kept in the Sheep Research Centre, Urmuqi, Xinjiang Uygur Autonomous Region. Ovarian superstimulation was induced three times (two individuals each time) in a total of 6 donor Merino lambs aged 7 weeks following a previously reported treatment schedule [6]. Briefly, lambs were stimulated with 4×40 mg FSH (Folltropin-V, Bioniche, Belleville, ON, Canada) that was administered in 12 h intervals. Sixty hours after the first FSH injection, the lambs underwent ovariectomy. To minimize genetic noise, the six lambs’ mother ewes formed the control group for gene expression analysis after superstimulation. The adult ewes were implanted with controlled internal drug release devices (CIDR, 0.3 g progesterone, Pharmacia and Upjohn Limited, Auckland, New Zealand) for oestrus synchronization on the first day. On the twelfth day, the ewes were given 4 progressively decreasing doses of FSH (120, 100, 80, and 60 mg) in 12 h intervals (Figure 1). Sixty hours after the first injection, the ewes were ovariectomized and the CIDR was removed.
Figure 1.

Experimental protocol used to test the effect of ovarian superstimulation on gene expression in granulosa cells. FSH, follicle-stimulating hormone; CIDR, controlled internal drug release.
All experiments were performed in accordance with relevant guidelines and regulations set by the Ministry of Agriculture of the People’s Republic of China.
Surgery and tissue collection
Ovariectomies were performed using an abdominal surgery approach. Briefly, anesthesia was induced by intramuscular injection of xylidinothiazoline (2 mg/kg body weight). The lower abdomen was shaved, washed and disinfected with an iodine-based detergent solution. A small incision was made in the midline of the abdomen, close to the mammary gland. A hole was punctured in the peritoneum with a surgical knife and then enlarged manually, allowing direct access to and palpation of the reproductive tract. Local anesthesia was applied to the ovarian pedicle using gauze soaked with procaine and a plastic clip was then applied on the pedicle to minimize hemorrhage. Intestinal suture was used to ligate the ovarian pedicle, the ligature was slowly tightened and then the ovary was removed. The ovaries were placed in polyethylene bags, kept on ice, and were transported to the laboratory within 5 min after collection. In order to harvest antral and mural GCs, the growing antral follicles (3 to 4 mm in diameter) were stripped away from the ovaries using ophthalmic tweezers. The follicular fluid was collected by aspiration of the follicular antrum using a 4.5-gauge needle and a syringe. Follicles were flushed three times with Dulbecco’s phosphate buffered saline (D-PBS). The collapsed follicles were cut using a scalpel blade and small tweezers in D-PBS to expose the inner follicular wall; the inner follicular wall was subsequently scraped with a microbiology culture loop to harvest the mural layer of GCs. The mural GCs were collected by centrifugation at 1,500 rpm for 5 min. The cell pellet was resuspended in Sample Protector (TaKaRa, Biotechnology (Dalian) Co., LTD, Dalian, China) and stored at −80°C until RNA extraction.
RNA extraction and amplification
Each individual sample total RNA was extracted using QIAzol (miRNeasy Mini Kit, QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. Quality of the extracted RNA was assessed by gel electrophoresis and quantity was measured by NanoDrop 2000 spectrophotometer. Purity of extracted RNA was considered good when the ratio of optical density between 260 nm and 280 nm fell between 1.8 and 2.0, while the ratio of values measured at 260 nm and 230 nm was ≥2.0. The RNA integrity value required for RNA sequencing was >8.0. After total RNA extraction (three lambs or three ewes pooled in each group) and DNase treatment, construction of cDNA libraries and sequencing were performed as described previously [17].
Sequence reads mapping and assembly
After Illumina HiSeq2500 sequencing, raw data were processed using Perl and Python scripts provided by Novogene (Novogene Bioinformatics Technology Co. Ltd, Beijing, China). In this step, the clean data (clean reads) were obtained by removing reads containing adapter, reads containing poly-N and low quality reads from raw data. At the same time, quality parameters of clean data including Q20, Q30, GC-content and sequence duplication level were used for data filtering. All the succeeding analyses were carried out using high quality clean data.
Reference genome and gene model annotation files were downloaded from the sheep genome website at ftp://ftp.ensembl.org/pub/release-76/fasta/ovis_aries/dna/. Paired-end clean reads were aligned to the reference genome using TopHat2 [18]. TopHat was chosen as the mapping tool because it can generate a database of splice junctions based on the gene model annotation file, and thus give a better mapping result than other non-splice mapping tools.
Expression profiling
The parameter FPKM (fragments per kilobase of exon per million fragments mapped) was used to evaluate the gene expression levels. HTSeq v0.5.3 (http://www-huber.embl.de/users/anders/HTSeq) was used to count the reads numbers mapped to each transcript. FPKM was calculated based on the mapped transcript fragments, transcript length and sequencing depth. Currently, this is the most commonly used method for estimating transcript expression [19].
Differential expression analysis of two groups was performed using the DESeq R package (1.12.0). DESeq provides statistical routines to determine differential expression in digital gene expression data using a model based on the negative binomial distribution. Absolute value of log2≥1 and q-value<0.005 were considered to reflect significant differences in gene expression [20].
Gene ontology analysis
Gene ontology (GO) enrichment analysis of differentially expressed transcripts was implemented using GOseq software package. GO terms with corrected P-value less than 0.05 were considered significantly enriched by differentially expressed transcripts.
A differentially expressed gene (DEG) data set was imported into ingenuity pathway analysis (IPA, http://www.ingenuity.com) to examine functional and molecular pathway enrichment for DEGs based on Fisher’s exact test. Analisis of canonical pathways and regulator effects as well as network analysis were performed as previously described [21]. The activation score (z-score) was used to infer the activation state of transcriptional regulators (z-score<−2 and >2). The expression and/or differential expression of transcriptional regulators in the data set provided evidence for biological mechanisms. Combining lamb and ewe GCs regulator analyses provided clues to the putative regulation involved in the lamb GCs on the follicle development stage.
Real-time quantitative polymerase chain reaction
In order to validate our RNA sequencing data, SYBR green real-time quantitative polymerase chain reaction were performed with the TransStart Green qPCR SuperMix (Transgen, Beijing, China) on a Roche HOLD CYCLE LightCycler 480 II. Three lambs and three ewes that have not been used for the RNA sequencing experiments were selected. Total RNA (1 μg) was used to synthesize first-strand cDNA with the TransScript-Uni One-Step gDNA Removal and cDNA Synthesis SuperMix (Transgen, China) with oligo (dT) primer. All qPCR experiments were run in duplicates and included no-RT and no-template controls. The qPCR reactions comprised a denaturation step of 3 min at 95°C followed by 40 cycles of product amplification (5 s at 95°C, 15 s at 60°C, and 15 s at 72°C). Quantities of mRNA corresponding to individual genes were normalized to glyceraldehyde-3phosphate dehydrogenase (GAPDH) expression.
Relative gene expression was calculated with the comparative Ct (ΔCt) method as follows: Ct target gene −Ct GAPDH. ΔCt-values for each target gene were then compared across samples (ΔΔCt) in the following way: ΔCt (sample 1) minus ΔCt (sample 2). Results of real-time quantitative polymerase chain reaction analysis were expressed as fold changes of RNA transcripts and were calculated by the 2−ΔΔCt method [22].
Unless otherwise stated, all chemicals were obtained from Sigma Chemical Company (St. Louis, MO, USA).
RESULTS
RNA seq data of granulosa cells
We sequenced the RNA from two groups, superstimulated lamb follicles vs superstimulated (adult) ewe follicles, and performed sequencing quality assessment. In this study, 29627505 and 32774030 raw reads were generated for each sample. After quality control, 3.23 G and 3.56 G clean bases were obtained from the two groups (Supplementary Table S1). Approximately 85% of the total reads were mapped to sheep chromosomes and about 83% of the reads were uniquely mapped to the sheep genome (Supplementary Table S2). Regarding coding characteristics of the mapped reads, 54.1% (ewe) and 59.0% (lamb) corresponded to exon sequences, 8% (ewe) and 15.4% (lamb) to introns, and 26.0% (ewe) and 25.6% (lamb) were matched to intergenic genome regions (Supplementary Figure S1). Read density in chromosomal distrbution and number of mapped reads in chromosomes were shown in Supplementary Figure S2. The correlation of transcripts expression between samples is the most important indicator for reliability of experimental results and rationality of sampling. Generally, the correlation value should be up to 0.92 (r2≥0.92). In our study, the two samples correlation value was up to 0.966. This analysis validates the quality of the library and its utility for further analysis.
Differential gene expression profile
To explore the biological mechanism of follicle development following ovarian stimulation in lamb, we sought to identify the DEGs between lamb and ewe GCs. Comparing the two samples, we identified 311 DEGs with the fold change ≥2 and q-value<0.005 used as cutoff values (Supplementary Table S3). Of these, 175 genes (42 novel) were down-regulated and 136 genes (5 novel) were up-regulated in lamb versus ewe GCs (Figure 2). Among them we identified 33 DEGs with more than four-fold change in expression, including 20 downregulated and 13 upregulated in lamb versus ewe GCs (Table 1).
Figure 2.
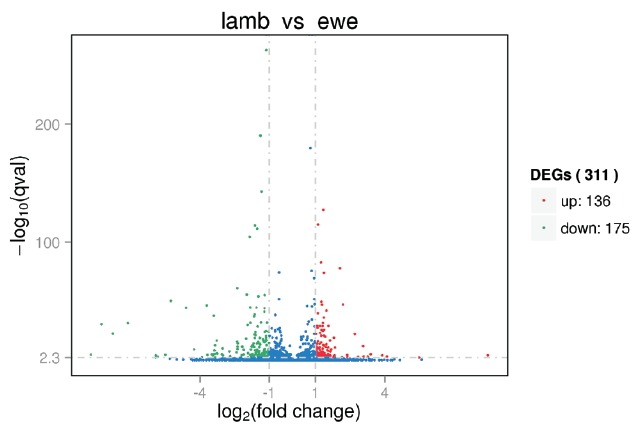
Volcano plot depicting the fold differences in gene expression levels between the lamb granulosa cells and ewe granulosa cells. Colored points refer to transcripts with expression changes of greater than 2-fold with q<0.005, up-regulated (red) and down-regulated (green). DEGs, differentially expressed genes.
Table 1.
Greater than four-fold upregulated and downregulated genes in granulosa cells of lamb after ovarian superstimulation treatment compared with ewe controls
| Genes | Description | log2Fold change | q-value |
|---|---|---|---|
| Upregulated | |||
| FGL2 | Fibrinogen-like 2 | 4.09 | 4.99×10−4 |
| COL6A6 | Collagen, type VI, alpha 6 | 3.88 | 5.47×10−5 |
| CDCP1 | CUB domain containing protein 1 | 3.39 | 8.33×10−6 |
| HTRA3 | HtrA serine peptidase 3 | 3.26 | 6.58×10−4 |
| GRIK2 | Glutamate receptor, ionotropic, kainate 2 | 3.08 | 6.02×10−4 |
| APOA2 | Apolipoprotein A-II | 3.06 | 1.01×10−12 |
| CAPN6 | Calpain 6 | 2.84 | 1.19×10−3 |
| REG4 | Regenerating islet-derived family, member 4 | 2.70 | 5.27×10−23 |
| LPL | Ovis aries lipoprotein lipase (LPL) | 2.38 | 1.98×10−3 |
| SERPINA14 | Ovis aries serpin peptidase inhibitor, member 14 (SERPINA14) | 2.37 | 3.43×10−5 |
| COBLL1 | Cordon-bleu WH2 repeat protein-like 1 | 2.19 | 5.36×10−48 |
| CEMIP | Cell migration inducing protein, hyaluronan binding | 2.14 | 2.70×10−3 |
| CDO1 | Cysteine dioxygenase type 1 | 2.06 | 1.25×10−78 |
| Downregulated | |||
| IL1RL1 | Interleukin 1 receptor-like 1 | −7.12 | 3.31×10−32 |
| SV2C | Synaptic vesicle glycoprotein 2C | −5.91 | 7.06×10−5 |
| CA5A | Carbonic anhydrase VA, mitochondrial | −5.49 | 2.28×10−5 |
| OXT | Oxytocin-neurophysin 1 precursor | −5.25 | 6.04×10−51 |
| EFEMP1 | EGF containing fibulin-like extracellular matrix protein 1 | −4.60 | 4.29×10−45 |
| TMEM52B | Transmembrane protein 52B | −4.26 | 4.80×10−10 |
| PIWIL2 | Uncharacterized protein | −3.92 | 2.36×10−3 |
| RGS4 | Ovis aries regulator of G-protein signaling 4 | −3.48 | 1.18×10−5 |
| CLEC1B | C-type lectin domain family 1, member B | −3.44 | 6.46×10−5 |
| STC1 | Stanniocalcin 1 | −3.37 | 4.89×10−6 |
| CXorf57 | Chromosome X open reading frame 57 | −3.35 | 1.84×10−13 |
| SPP1 | Osteopontin precursor | −3.27 | 1.45×10−17 |
| NOV | Nephroblastoma overexpressed | −3.05 | 9.67×10−6 |
| IL6R | Interleukin 6 receptor | −2.42 | 1.99×10−7 |
| KCNK12 | Potassium channel, subfamily K, member 12 | −2.39 | 1.16×10−16 |
| NRP1 | Neuropilin 1 | −2.39 | 6.87×10−62 |
| MT1A | Metallothionein-1A | −2.38 | 2.04×10−12 |
| SEMA3A | Sema domain, immunoglobulin domain (Ig), short basic domain, secreted, (semaphorin) 3A | −2.19 | 6.94×10−5 |
| ADAMTS9 | ADAM metallopeptidase with thrombospondin type 1 motif, 9 | −2.10 | 1.09×10−9 |
| ARID5B | AT rich interactive domain 5B (MRF1-like) | −2.00 | 5.54×10−19 |
Gene ontology analysis
We hypothesized that the DEGs we identified were important for follicle development. The 311 identified DEGs were classified into three major functional categories: cellular component, molecular function, and biological process. The top five functional groups included binding, catalytic activity, cellular process, metabolic process, and organic substance metabolic process (Figure 3). Further enrichment analysis was related to cellular functions and subcellular locations, and the detected DEGs were enriched in different terms related to follicle development. For example, the DEGs cytochrome P450, family 19 (CYP19A1), steroidogenic acute regulatory protein (STAR), neuropilin 1 (NRP1), EGF containing fibulin-like extracellular matrix protein 1 (EFEMP1) and proliferating cell nuclear antigen (PCNA) were enriched in binding, lipid binding, protein binding, cellular process, catalatic activity and metabolic process, respectively (Supplementary Table S4).
Figure 3.
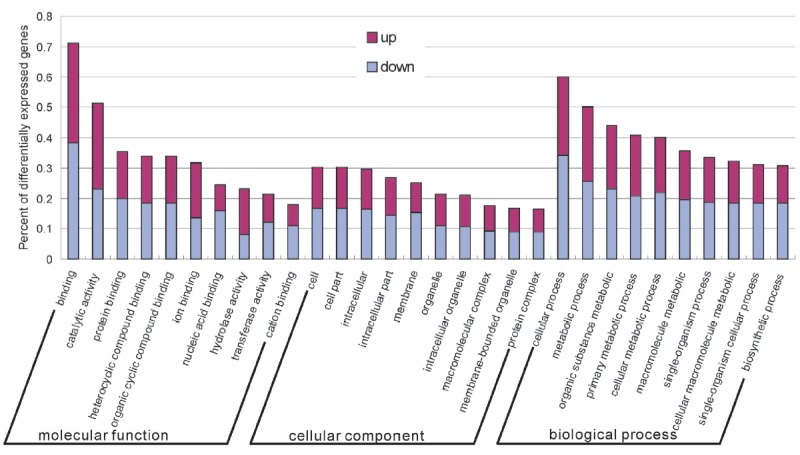
Functional categorization of differentially expressed genes based on known genes in the Uniprot database. The x-axis shows the 2nd level term of gene ontology; y-axis shows percent of genes in differentially expressed genes.
Pathway, upstream regulator and network analyses
The DEG list was subjected to IPA and pathway, network and upstream regulator analyses of lamb versus ewe were performed. Among 311 DEGs, the 234 genes (119 up-regulated and 115 down-regulated) have been annotated (Supplementary Table S3) and subjected to pathway network and upstream regulator analyses in IPA.
Forty-two IPA canonical pathways were identified in lamb compared to ewe with threshold −log (p-value) ≥ 1.3 (Supplementary Table S5). The fourteen pathways were with values greater than 2 (Figure 4). Among these pathways, the top three IPA canonical pathways were base excision repair (BER) pathway (ligase I [LIG1, DNA, ATP-dependent], PCNA, polymerase [DNA directed], epsilon, catalytic subunit [POLE], [flap structure-specific endonuclease 1]), cell cycle control of chromosomal replication (MCM3, MCM4, MCM5, MCM6 (minichromosome maintenance complex component 3, 4, 5, 6), CDC6 [cell division cycle 6]) and GADD45 (growth arrest and DNA-damage-inducible) signaling (PCNA, GADD45G, BRCA1 [breast cancer 1], CDK1 [cyclin-dependent kinase 1]) (Figure 3). The top three pathways by the number of included DEGs were axonal guidance signaling SEMA3A (semaphorin 3A), SEMA6D (semaphorin 6D), PAPPA (pregnancy-associated plasma protein A), NFAT5 (nuclear factor of activated T-cells 5), PTCH1 (patched 1), NTRK1 (neurotrophic tyrosine kinase, receptor, type 1), GNB2 (guanine nucleotide binding protein [G protein], beta polypeptide 2), TUBA1C (tubulin, alpha 1c), ADAMTS9 (ADAM metallopeptidase with thrombospondin type 1 motif, 9), ADAMTS4 (ADAM metallopeptidase with thrombospondin type 1 motif, 4), NRP1, SEMA7A (semaphorin 7A), protein ubiquitination pathway CDC20 (cell division cycle 20), USP27X (ubiquitin specific peptidase 27, X-linked), USP53 (ubiquitin specific peptidase 53), DNAJB11 (DnaJ [Hsp40] homolog, subfamily B, member 11), DNAJC27 (DnaJ [Hsp40] homolog, subfamily C, member 27), UBE2S (ubiquitin-conjugating enzyme E2S), NEDD4L (neural precursor cell expressed, developmentally down-regulated 4-like, E3 ubiquitin protein ligase), BRCA1, HSPA5 (heat shock 70 kDa protein 5), UBE2C (ubiquitin-conjugating enzyme E2C), and LPS (lipopolysaccharide)/IL-1 (Interleukin 1) mediated inhibition of retinoid × receptor (RXR) function MGST1 (microsomal glutathione S-transferase 1), ALDH1L2 (aldehyde dehydrogenase 1 family, member L2), IL1RL1 (interleukin 1 receptor-like 1), SREBF1 (sterol regulatory element binding transcription factor 1), CHST11 (carbohydrate [chondroitin 4] sulfotransferase 11), ALAS1 (aminolevulinate, delta-, synthase 1), CHST15 (carbohydrate [N-acetylgalactosamine 4-sulfate 6-O] sulfotransferase 15. In contrast, there were six pathways with only one DEG each, namely andamide degradation (FAAH), taurine biosynthesis (CDO1 [cysteine dioxygenase type 1]), glutathione biosynthesis (GCLC), arginine degradation I (arginase pathway) (OAT, ornithine aminotransferase), L-cysteine degradation I (CDO1), and myo-inositol biosynthesis (ISYNA1) (Supplementary Table S5).
Figure 4.

Top ranked canonical pathways generated in ingenuity pathway analysis. The bar chart represents the −log(p-value) of genes from the data set that map to each canonical pathway.
On the IPA function analysis, these DEGs have been involved in 500 function cases (Supplementary Table S6). There were 17 functional groups with activation z-score greater than 2 and predicted activation state activated (Table 2). The most significantly affected functions in lamb follicles were found to be proliferation of cells, DNA replication, cellular movement, organ development and tissue development (Table 2). IPA upstream regulator analysis was used to identify upstream transcriptional regulators. We had identified 32 activated upstream regulators, including transcription regulators (MYC [v-myc avian myelocytomatosis viral oncogene homolog], E2F1 [E2F transcription factor 1], E2F2 [E2F transcription factor 2], TBX2 [T-box protein 2], CCND1 [cyclin D1], SREBF1, GATA4 [GATA binding protein 4], STAT6 [signal transducer and activator of transcription 6], MYOD1 [myoblast determination protein 1], XBP1 [X-box binding protein 1]), growth factors (IGF1 [insulin-like growth factor 1], HGF [hepatocyte growth factor], EGF, VEGFA [vascular endothelial growth factor A], INHA [inhibin, alpha]), enzymes and kinases (HRAS [Harvey rat Sarcoma viral oncogene homolog], CAT [catalase], KRAS [Kirsten rat Sarcoma viral oncogene homolog], ERBB2 [erb-b2 receptor tyrosine kinase 2]), and others (Table 3). The top activated upstream regulator was the growth factor EGF. The EGF receptor upstream regulator target molecules in the dataset included ACTB (actin, beta), ANGPT2 (angiopoietin 2), BIRC5 (baculoviral IAP repeat containing 5), CAV1 (caveolin 1), CDCP1 (CUB domain containing protein 1), CDK1, CLK1 (CDC-like kinase 1), CTGF (connective tissue growth factor), CYP19A1, FAP (fibroblast activation protein), FOSL2 (FOS-like antigen 2), HK2 (hexokinase 2), MYBL2 (v-myb avian myeloblastosis viral oncogene homolog-like 2), NOTCH1 (notch 1), PCNA, POSTN (periostin), and THBS1 (thrombospondin 1). They interacted within a mechanistic network which included Akt (serine/threonine-specific protein kinase), CTNNB1 (catenin [cadherin-associated protein], beta 1), EGFR (epidermal growth factor receptor), ERK1/2 (extracellular regulated protein kinases), ESR1 (estrogen receptor 1), FOXO1 (forkhead box O1), HIF1A (hypoxia inducible factor 1, alpha subunit), HRAS, JUN (Jun Proto-Oncogene), MYC, NFkB (complex), P38 MAPK (mitogen-activated protein kinase), PI3K (phosphatidylinositol-4,5-bisphosphate 3-kinase, complex), PPARG (peroxisome proliferator-activated receptor gamma), RELA (v-rel avian reticuloendotheliosis viral oncogene homolog A), RUNX2 (runt-related transcription factor 2), SMAD3 (SMAD family member 3), SP1 (specificity protein 1), STAT3 (signal transducer and activator of transcription 3), and TP53 (tumor protein P53). In addition, 23 upstream regulators were predicted to be inhibited, including transcription regulators (RB1 [retinoblastoma 1], RBL1 [retinoblastoma-like 1], TP53, CDKN2A [cyclin-dependent kinase inhibitor 2A], E2F6 [E2F transcription factor 6], SMARCB1 [SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily B, member 1], HDAC2 [histone deacetylase 2], SIM1 [single-minded family BHLH transcription factor 1], ARNT2 [aryl-hydrocarbon receptor nuclear translocator 2]), kinases (CDKN1A [cyclin-dependent kinase inhibitor 1A], DYRK1A [dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1A], TGFBR2 [transforming growth factor, beta receptor II]), a phosphatase (PTEN), micro-RNAs (miR-24-3p, let-7), and others (Table 4). Upstream regulator let-7 predicted target molecules including cell division cycle family members (CDC20, CDC6, CDCA3 [cell division cycle associated 3]), minichromosome maintenance complex components (MCM3, MCM4, MCM5, MCM6), ribonucleotide reductase (RRM1 [ribonucleotide reductase M1], RRM2 [ribonucleotide reductase M2]) and others transcripts (BRCA1, CCNA2 [cyclin A2], CDK1, THBS1). The top inhibited upstream regulator was TP53.
Table 2.
Biological functions determined in IPA for genes differentially regulated between lamb and ewe follicles
| Categories | Functions annotation | p-value | Activation z-score | Molecules | No. |
|---|---|---|---|---|---|
| Cellular growth and proliferation | Proliferation of cells | 3.88×10−10 | 3.602 | CTGF, HMMR, GCLC, MKI67, SKAP2, RECQL, PTP4A2, CAV1, JARID2, AMACR, CAPN6, IL4R, PTCH1, RRM2, GRK5, PLD1, NINJ1, OXT, PCNA, NPR3, ZBED6, CTSB, CLSPN, NOTCH1, UBE2C, NRP1, ADM, IL1RL1, MYH11, OGT, HK2, NTRK1, GRIK2, FAP, CITED2, ENPP1, ACTB, CDC6, CDK1, RRM1, MCM3, SEMA3A, NOV, SREBF1, FEN1, NEDD9, SEMA7A, LIG1, CDCP1, FGL2, HSPA5, PBK, LMNB1, GABARAPL1, PIWIL2, POSTN, CHST11, KPNA2, MYBL2, NEDD4L, FOSL2, BRCA1, IFT172, ADAMTS4, PDK4, MCM5, CLEC1B, PTX3, PAPPA, SPP1, IL6R, RGS4, HELLS, MYOF, STC1, LMNB2, SCD, ANGPT2, CDC20, GADD45G, DTYMK, MCM4, CCNA2, NFAT5, CLK1, NFKBIA, CYP19A1, THBS1, CSPG4, MANF, SLC20A1, ARID5B, CKS2, UHRF1, VWF, MT1A, BIRC5, IVNS1ABP, B4GALT1, RNF111, EFEMP1 | 101 |
| Cancer | Digestive system cancer | 2.18×10−12 | 2.804 | SNTB2, MCM6, CTGF, L3MBTL3, SYTL2, PCSK1, TFR2, HMMR, GCLC, MKI67, CHAF1A, RECQL, JARID2, CAV1, TDP2, HSD17B1, AMACR, BCOR, PTCH1, LRRC16A, GRAMD1B, RRM2, GRK5, UBE2S, PLD1, PCNA, NPR3, ST3GAL1, CTSB, ARHGEF28, FAM13C, TSPYL4, CEP95, CLSPN, LRP8, NOTCH1, NRP1, UBE2C, CHCHD10, IL1RL1, USP53, ZBED5, HYOU1, MYH11, CRYBG3, REG4, OGT, ISYNA1, AMN1, HK2, PPAP2B, NTRK1, ANTXR2, ABHD2, DSTYK, LNPEP, GRIK2, FAP, VEPH1, CITED2, ADGRB2, STAMBPL1, ENPP1, PMM1, NELL2, ACTB, FAM63A, CDC6, POLE, RRM1, MCM3, SEMA3A, SDF2L1, NOV, LDLR, GPBP1, SREBF1, MAN1A1, FEN1, INADL, NEDD9, KANSL2, WDR19, COBLL1, SEMA7A, LIG1, CDCP1, SERPINA12, KIAA1841, C3orf33, SYNE1, HSPA5, LMNB1, PBK, CHST15, SEMA6D, TMTC2, PIWIL2, LPL, AMPH, POSTN, PDIA4, CHST11, KPNA2, TUBA1C, MYBL2, CDCA3, BRCA1, CCDC64, PDK4, ADAMTS4, IFT172, PTX3, DNAH9, PHYHIPL, MCM5, NBEAL1, ATP6V1C2, PAPPA, SPP1, IL6R, MTUS2, RGS4, HELLS, FAM222B, KCNK12, ADAMTS9, MYOF, RHBDF2, RLF, JAM3, LMNB2, ELL2, C20orf194, GMPPB, ABCD1, SCD, ANGPT2, CDC20, TC2N, RAPGEF4, ALAS1, LRRK1, COL6A6, MCM4, ZNF175, GFPT2, CCNA2, SNX16, CLK1, NFAT5, CSPG4, THBS1, MANF, ASPN, ARID5B, CKS2, KLHL24, FRY, WDR73, UHRF1, NEFM, VWF, BIRC5, ATP1B3, ANGPTL5, IVNS1ABP, CRELD2, B4GALT1, RNF111, DNAJB11, EFEMP1, CEMIP | 173 |
| Cancer, organismal injury and abnormalities | Abdominal neoplasm | 3.33×10−15 | 2.671 | SNTB2, SYTL2, HMMR, PCSK1, GCLC, JARID2, CAV1, AMACR, IL4R, BCOR, RRM2, GRAMD1B, LRRC16A, PLD1, NPR3, ST3GAL1, CTSB, TSPYL4, ARHGEF28, CEP95, CXorf57, UBE2C, HSD17B10, CHCHD10, IL1RL1, ZBED5, CRYBG3, AMN1, ISYNA1, OGT, NTRK1, ANTXR2, LNPEP, FAP, PMM1, MCM3, SEMA3A, SDF2L1, LDLR, NOV, GPBP1, INADL, KANSL2, LIG1, CDCP1, FGL2, HSPA5, TMTC2, PIWIL2, LPL, PDIA4, MYBL2, TUBA1C, CCDC64, IFT172, PHYHIPL, PAPPA, MGST1, ATP6V1C2, NBEAL1, IL6R, MTUS2, HELLS, KCNK12, ADAMTS9, RLF, JAM3, LMNB2, ELL2, ABCD1, SCD, CDC20, TC2N, ALAS1, RAPGEF4, COL6A6, GFPT2, CCNA2, SNX16, NFAT5, CYP19A1, SLC20A1, FRY, BIRC5, IVNS1ABP, ANGPTL5, CRELD2, RNF111, DNAJB11, MCM6, L3MBTL3, CTGF, TFR2, MKI67, RECQL, CHAF1A, TDP2, CRIM1, HSD17B1, CAPN6, OAT, PTCH1, GRK5, UBE2S, NINJ1, PCNA, GNB2, FAM13C, LRP8, CLSPN, NOTCH1, NRP1, ADM, U SP53, HYOU1, MYH11, REG4, HK2, USP27X, PPAP2B, ABHD2, DSTYK, GRIK2, SV2C, VEPH1, ADGRB2, CITED2, ENPP1, STAMBPL1, NELL2, FAM63A, ACTB, POLE, CDC6, CDK1, RRM1, ABHD6, SREBF1, MAN1A1, FEN1, NEDD9, WDR19, SEMA7A, COBLL1, SERPINA12, KIAA1841, APOA2, C3orf33, SYNE1, PBK, LMNB1, CHST15, SEMA6D, PDE7B, AMPH, POSTN, CHST11, KPNA2, CDCA3, BRCA1, ADAMTS4, PDK4, DNAH9, MCM5, PTX3, SPP1, RGS4, TNNI3, FAM222B, MYOF, STC1, C20orf194, GMPPB, ANGPT2, SLC10A7, LRRK1, MCM4, ZNF175, CLK1, NFKBIA, THBS1, CSPG4, MANF, ASPN, FAAH, ARID5B, CKS2, WDR73, KLHL24, UHRF1, NEFM, VWF, PRKAG3, ATP1B3, B4GALT1, EFEMP1, CEMIP | 197 |
| DNA replication, recombination, and repair | DNA replication | 4.96×10−4 | 2.605 | LIG1, CCNA2, PCNA, CKS2, GADD45G, CDC6, POLE, CAV1, FEN1, BRCA1, CDK1 | 11 |
| DNA replication, recombination, and repair | Metabolism of DNA | 4.67×10−5 | 2.434 | ADM, HSD17B10, LIG1, CKS2, GADD45G, POLE, CDC6, BIRC5, CDK1, CCNA2, PCNA, NFKBIA, CAV1, CYCS, KPNA2, FEN1, BRCA1 | 17 |
| Cancer | Abdominal cancer | 2.65×10−15 | 2.376 | SNTB2, SYTL2, HMMR, PCSK1, GCLC, JARID2, CAV1, AMACR, IL4R, BCOR, RRM2, GRAMD1B, LRRC16A, PLD1, NPR3, ST3GAL1, CTSB, TSPYL4, ARHGEF28, CEP95, CXorf57, UBE2C, CHCHD10, IL1RL1, ZBED5, CRYBG3, AMN1, ISYNA1, OGT, NTRK1, ANTXR2, LNPEP, FAP, PMM1, MCM3, SEMA3A, SDF2L1, LDLR, NOV, GPBP1, INADL, KANSL2, LIG1, CDCP1, FGL2, HSPA5, TMTC2, PIWIL2, LPL, PDIA4, MYBL2, TUBA1C, CCDC64, IFT172, PHYHIPL, PAPPA, MGST1, ATP6V1C2, NBEAL1, IL6R, MTUS2, HELLS, KCNK12, ADAMTS9, RLF, JAM3, LMNB2, ELL2, ABCD1, SCD, CDC20, TC2N, ALAS1, RAPGEF4, COL6A6, GFPT2, CCNA2, SNX16, NFAT5, CYP19A1, SLC20A1, FRY, BIRC5, IVNS1ABP, ANGPTL5, CRELD2, RNF111, DNAJB11, MCM6, L3MBTL3, CTGF, TFR2, MKI67, RECQL, CHAF1A, TDP2, CRIM1, HSD17B1, CAPN6, OAT, PTCH1, GRK5, UBE2S, NINJ1, PCNA, GNB2, FAM13C, LRP8, CLSPN, NOTCH1, NRP1, ADM, USP53, HYOU1, MYH11, REG4, HK2, USP27X, PPAP2B, ABHD2, DSTYK, GRIK2, SV2C, VEPH1, ADGRB2, CITED2, ENPP1, STAMBPL1, NELL2, FAM63A, ACTB, POLE, CDC6, CDK1, RRM1, ABHD6, SREBF1, MAN1A1, FEN1, NEDD9, WDR19, SEMA7A, COBLL1, SERPINA12, KIAA1841, APOA2, C3orf33, SYNE1, PBK, LMNB1, CHST15, SEMA6D, PDE7B, AMPH, POSTN, CHST11, KPNA2, CDCA3, BRCA1, ADAMTS4, PDK4, MCM5, DNAH9, PTX3, SPP1, RGS4, TNNI3, FAM222B, MYOF, STC1, C20orf194, GMPPB, ANGPT2, SLC10A7, LRRK1, MCM4, ZNF175, CLK1, NFKBIA, THBS1, CSPG4, MANF, ASPN, FAAH, ARID5B, CKS2, WDR73, KLHL24, UHRF1, NEFM, VWF, PRKAG3, ATP1B3, B4GALT1, EFEMP1, CEMIP | 196 |
| Cellular development, cellular growth and proliferation | Proliferation of tumor cell lines | 4.72×10−8 | 2.311 | LIG1, CTGF, CDCP1, HMMR, MKI67, HSPA5, PBK, CAV1, POSTN, KPNA2, BRCA1, ADAMTS4, PDK4, AMACR, CAPN6, SPP1, IL6R, PTCH1, RRM2, PLD1, OXT, PCNA, ZBED6, CTSB, NOTCH1, UBE2C, NRP1, ADM, SCD, ANGPT2, GADD45G, CCNA2, OGT, HK2, NFKBIA, CYP19A1, THBS1, CSPG4, NTRK1, UHRF1, ACTB, MT1A, BIRC5, CDK1, RRM1, NOV, RNF111, SREBF1, FEN1, EFEMP1, NEDD9 | 51 |
| Cellular movement | Migration of cells | 5.47×10−10 | 2.276 | CTGF, CDCP1, HMMR, HSPA5, LMNB1, SKAP2, SEMA6D, PTP4A2, CAV1, POSTN, TDP2, KPNA2, BRCA1, CLEC1B, PTX3, PAPPA, IL4R, SPP1, IL6R, LRRC16A, RGS4, PLD1, NINJ1, STC1, RHBDF2, JAM3, CTSB, LMNB2, ARHGEF28, LRP8, NOTCH1, NRP1, ADM, ANGPT2, IL1RL1, HYOU1, MYH11, CCNA2, NFKBIA, NFAT5, CSPG4, CYP19A1, THBS1, PPAP2B, ABHD2, FAP, CITED2, FAAH, ARID5B, ACTB, VWF, CDK1, RRM1, MCM3, SEMA3A, ABHD6, LDLR, NOV, B4GALT1, SREBF1, CEMIP, INADL, NEDD9, ST3GAL4 | 64 |
| Cancer | Growth of pancreatic tumor | 4.19×10−5 | 2.219 | CTGF, THBS1, HMMR, CTSB, NRP1 | 5 |
| Cardiovascular system development and function | Proliferation of endothelial cell lines | 1.11×10−4 | 2.219 | ADM, PTX3, THBS1, RGS4, MYH11, PLD1, CDK1 | 7 |
| Skeletal and muscular system development and function | Thickness of bone | 1.26×10−4 | 2.219 | STC1, CTGF, SPP1, CAV1, BRCA1, LRRK1, PDK4 | 7 |
| Cellular assembly and organization | Association of chromosome components | 1.35×10−5 | 2.2 | CDC6, HELLS, BRCA1, BIRC5, MCM4 | 5 |
| Cancer | Growth of tumor | 1.98×10−7 | 2.176 | ADM, ANGPT2, CTGF, IL1RL1, HMMR, HYOU1, MKI67, HSPA5, OGT, NFKBIA, CYP19A1, THBS1, CAV1, POSTN, MYBL2, BRCA1, FAP, IL4R, SPP1, IL6R, PTCH1, RRM2, BIRC5, PLD1, NOV, RNF111, JAM3, CTSB, NEDD9, NOTCH1, UBE2C, NRP1 | 32 |
| Cell death and survival | Apoptosis of pancreatic cancer cell lines | 6.32×10−5 | 2.158 | OGT, NFKBIA, SREBF1, ZBED6, HMMR, TDP2, EFEMP1, NOTCH1 | 8 |
| Carbohydrate metabolism | Metabolism of carbohydrate | 1.58×10−5 | 2.011 | GMPPB, SCD, CTGF, ENPP1, SPP1, PMM1, APOA2, RGS4, GCLC, PRKAG3, GRK5, CHST15, PLD1, GFPT2, OXT, HK2, B4GALT1, SREBF1, NTRK1, LPL, CTSB, ASPN, CHST11, CEMIP | 24 |
| Cancer | Gastrointestinal tract cancer | 3.32×10−12 | 2 | SNTB2, L3MBTL3, CTGF, SYTL2, PCSK1, TFR2, HMMR, MKI67, CHAF1A, RECQL, JARID2, CAV1, TDP2, HSD17B1, AMACR, BCOR, PTCH1, RRM2, GRK5, UBE2S, PLD1, PCNA, NPR3, ST3GAL1, CTSB, FAM13C, CEP95, CLSPN, LRP8, NOTCH1, UBE2C, NRP1, IL1RL1, USP53, MYH11, I SYNA1, OGT, HK2, PPAP2B, NTRK1, ANTXR2, ABHD2, DSTYK, LNPEP, GRIK2, VEPH1, CITED2, ADGRB2, ENPP1, STAMBPL1, PMM1, CDC6, POLE, RRM1, MCM3, SEMA3A, SDF2L1, LDLR, NOV, GPBP1, FEN1, I NADL, NEDD9, COBLL1, SEMA7A, LIG1, SERPINA12, CDCP1, KIAA1841, SYNE1, LMNB1, PBK, CHST15, SEMA6D, TMTC2, LPL, AMPH, POSTN, CHST11, KPNA2, MYBL2, TUBA1C, BRCA1, PDK4, ADAMTS4, IFT172, DNAH9, PHYHIPL, MCM5, PTX3, NBEAL1, ATP6V1C2, PAPPA, SPP1, IL6R, MTUS2, RGS4, HELLS, ADAMTS9, MYOF, RHBDF2, JAM3, ELL2, GMPPB, ANGPT2, CDC20, TC2N, ALAS1, LRRK1, COL6A6, MCM4, GFPT2, CCNA2, SNX16, CSPG4, THBS1, ASPN, ARID5B, WDR73, FRY, KLHL24, UHRF1, NEFM, VWF, BIRC5, IVNS1ABP, ANGPTL5, CRELD2, B4GALT1, RNF111, DNAJB11, EFEMP1, CEMIP | 133 |
| Skeletal and muscular system development and function | Thickness of trabecular bone | 6.06×10−4 | 2 | CTGF, SPP1, CAV1, LRRK1, PDK4 | 5 |
The predicted activation state is inferred from the bias-corrected z-score (+, increased; −, decreased). The bias-corrected z-score is computed based on the proportion of target genes present in the dataset which are directionally regulated as expected according to known associations with functions compiled from the literature. The p value of overlap measures the statistical significance of overlap using Fisher’s exact t-test, between genes from the dataset and those known to be associated with a function.
Table 3.
Upstream regulators predicted to be activated in lamb follicles compared with ewe follicles, using the 2-fold differentially-regulated dataset with q<0.005, on the basis of known interactions compiled in the ingenuity pathway analysis upstream regulator analysis
| Upstream regulator | Molecule type | Activation z-score | p-value of overlap | Target molecules in dataset |
|---|---|---|---|---|
| EGF | Growth factor | 3.296 | 8.75×10−10 | B4GALT1, BIRC5, BRCA1, CAV1, CDK1, CKS2, CTGF, HSD17B1, NOTCH1, NRP1, PBK, PCNA, PDK4, PPAP2B, RRM1, RRM2, SCD, SPP1, SREBF1, ST3GAL1, ST3GAL4, STAR, THBS1 |
| TBX2 | Transcription regulator | 3.162 | 4.33×10−9 | CCNA2, CDC6, CDCA3, CDK1, HELLS, LIG1, MCM4, MCM5, MCM6, NCAPG2 |
| E2f | Group | 3.064 | 2.29×10−12 | BRCA1, CAV1, CCNA2, CDC6, CDK1, LIG1, MCM3, MCM4, MCM5, MCM6, MYBL2, PCNA, RECQL, RRM2, UBE2C |
| RARA | Ligand-dependent nuclear receptor | 2.891 | 2.83×10−6 | ABHD2, BIRC5, CCNA2, CDK1, ELL2, FOSL2, HELLS, IL6R, NEDD9, NRP1, NTRK1, OXT, PBK |
| HGF | Growth factor | 2.856 | 5.94×10−12 | ANGPT2, BIRC5, CAV1, CDC20, CDC6, CDK1, CTGF, DTYMK, FEN1, GRK5, HELLS, HK2, HMMR, LDLR, LRP8, MCM5, MKI67, NRP1, PCNA, PPAP2B, SEMA3A, SLC20A1, SPP1, STC1, THBS1, UBE2C |
| EP400 | Other | 2.828 | 6.04×10−12 | CCNA2, CDC20, CDC6, CDCA3, MCM3, MCM4, MYBL2, NCAPG2, PCNA, UHRF1 |
| CSF2 | Cytokine | 2.732 | 3.24×10−8 | BIRC5, CCNA2, CDC20, CDCA3, CDK1, CHAF1A, CHST11, IL1RL1, MCM3, MCM5, MCM6, MKI67, NFKBIA, NRP1, POLE, RRM2, SPP1, THBS1, UBE2C, UHRF1 |
| CCND1 | Transcription regulator | 2.716 | 1.92×10−5 | BIRC5, BRCA1, CCNA2, CDC6, CLSPN, HERC3, KLHL24, MCM4, PCNA, RRM2, SPP1, UHRF1 |
| Vegf | Group | 2.686 | 1.34×10−9 | ANGPT2, BIRC5, CDC20, CDC6, CDK1, DTYMK, GRK5, HELLS, HK2, HMMR, LDLR, LRP8, MCM5, MKI67, NOTCH1, NRP1, PPAP2B, SLC20A1, STC1, UBE2C, VWF |
| E2F1 | Transcription regulator | 2.670 | 2.47×10−12 | ANGPT2, BIRC5, BRCA1, CAV1, CCNA2, CDC20, CDC6, CDK1, CITED2, CTSB, FEN1, HELLS, MCM3, MCM4, MCM5, MCM6, MYBL2, NFKBIA, NRP1, OXT, PCNA, RRM1, RRM2, THBS1, UHRF1 |
| ERBB2 | Kinase | 2.661 | 8.20×10−11 | ACTB, ALAS1, ATP1B3, BIRC5, CCNA2, CDC20, CDK1, CHCHD10, CKS2, CTGF, CTSB, ELL2, FEN1, IL6R, MAN1A1, MKI67, MYBL2, NEDD9, NOTCH1, NRP1, PCNA, PDIA4, PDK4, RRM2, ST3GAL4, THBS1, UBE2C, VWF |
| SREBF1 | Transcription regulator | 2.610 | 2.01×10−5 | APOA2, HK2, HSD17B1, HSPA5, LDLR, LPL, OAT, SCD, SREBF1, STAR |
| MYC | Transcription regulator | 2.529 | 1.92×10−12 | ACTB, ADM, ANGPT2, BCOR, BIRC5, BRCA1, CAV1, CCNA2, CDC20, CDK1, CHST15, CKS2, COBLL1, CSPG4, CTSB, CYCS, EFEMP1, FAP, GADD45G, GCLC, HK2, JARID2, MCM5, MCM6, MKI67, NFAT5, NFKBIA, NRP1, OAT, PCNA, RRM2, SPP1, ST3GAL1, ST3GAL4, THBS1, TNNI3, UBE2C, UBE2S |
| RABL6 | Other | 2.449 | 5.05×10−5 | CCNA2, FEN1, HMMR, MCM5, PBK, UBE2C |
| IGF1 | Growth factor | 2.447 | 2.77×10−12 | ADM, ANGPT2, BIRC5, BRCA1, CCNA2, CDK1, CTGF, CYP19A1, HSPA5, IL4R, LPL, MKI67, MYH11, NFKBIA, NTRK1, OXT, PBK, PCNA, SCD, SLC20A1, SPP1, SREBF1, STAR, THBS1 |
| estrogen | Chemical drug | 2.440 | 8.16×10−14 | APOA2, BRCA1, CTGF, CYP19A1, FEN1, IL1RL1, LDLR, LIG1, LPL, MCM3, MCM4, MCM5, NTRK1, OAT, OXT, PCNA, POLE, PTX3, RRM2, SPP1, UHRF1, WDR73 |
| MYOD1 | Transcription regulator | 2.425 | 1.16×10−2 | BRCA1, CDO1, GADD45G, LMNB2, MKI67, POSTN |
| NR1H2 | Ligand-dependent nuclear receptor | 2.402 | 8.37×10−6 | LDLR, LPL, LRP8, NFKBIA, SCD, SREBF1, STAR |
| NR1H3 | Ligand-dependent nuclear receptor | 2.381 | 2.26×10−4 | LDLR, LPL, NFKBIA, SCD, SREBF1, STAR |
| XBP1 | Transcription regulator | 2.376 | 1.79×10−2 | DNAJB11, HSPA5, HYOU1, PDIA4, SDF2L1, SREBF1 |
| STAT6 | Transcription regulator | 2.375 | 5.03×10−3 | COBLL1, CTSB, GADD45G, IL4R, NEDD9, NFKBIA, PLD1, SLC25A25 |
| GATA4 | Transcription regulator | 2.352 | 2.53×10−4 | CTGF, CYP19A1, FAP, HSPA5, POSTN, SPP1, STAR, TNNI3 |
| beta-estradiol | Chemical - endogenous mammalian | 2.340 | 1.63×10−18 | ACTB, ADAMTS4, ADM, BIRC5, BRCA1, CAV1, CCNA2, CDC20, CDC6, CDK1, CITED2, CLEC1B, COL6A6, CRIM1, CTGF, CTSB, CYP19A1, FAAH, FAM134C, FEN1, FOSL2, GADD45G, GCLC, HELLS, HSD17B1, HSPA5, HTRA3, IL4R, IL6R, KPNA2, LDLR, LPL, LRP8, MAL2, MCM3, MCM5, MCM6, MGST1, MKI67, MYBL2, MYOF, NEDD4L, NEDD9, NELL2, NFKBIA, NPR3, NRP1, NTRK1, OXT, PCNA, PDIA4, PDK4, PPM1K, PTP4A2, PTX3, RRM2, SEMA3A, SLC20A1, STAR, STC1, THBS1 |
| E2F2 | Transcription regulator | 2.236 | 4.18×10−11 | BIRC5, CCNA2, CDC6, CDK1, MCM3, MCM4, MCM5, MCM6, MYBL2, PCNA, RRM2 |
| INHA | Growth factor | 2.213 | 3.99×10−3 | CTGF, CYP19A1, MYH11, PTX3, STAR |
| THBS4 | Other | 2.191 | 6.85×10−6 | CRELD2, HSPA5, HYOU1, MANF, SDF2L1 |
| linoleic acid | Chemical - endogenous mammalian | 2.190 | 4.23×10−5 | HSPA5, LDLR, LPL, NEDD4L, NFKBIA, SCD |
| VEGFA | Growth factor | 2.184 | 2.70×10−6 | ALAS1, ANGPT2, BIRC5, CAV1, CTGF, CTSB, HSD17B1, NOTCH1, PCNA, PPAP2B, STC1, THBS1 |
| KRAS | Enzyme | 2.157 | 8.65×10−3 | AMPH, BIRC5, CAV1, EFEMP1, HSPA5, HYOU1, PLD1, SNX16, THBS1 |
| HRAS | Enzyme | 2.133 | 7.15×10−10 | ACTB, ADM, AMACR, BIRC5, CAV1, CCNA2, CDK1, CHST11, CTGF, CTSB, EFEMP1, ENPP1, FOSL2, GADD45G, IL1RL1, MYH11, NFKBIA, NOTCH1, NRP1, PCNA, POSTN, RRM2, SPP1, SREBF1, THBS1 |
| CAT | Enzyme | 2.000 | 1.52×10−3 | ADM, CAV1, GCLC, THBS1 |
| TRAF3 | Other | 2.000 | 9.66×10−3 | ADM, HSPA5, LRRC16A, NFKBIA |
Table 4.
Upstream regulators predicted to be inhibited in lamb follicles compared with ewe follicles, using the 2-fold differentially-regulated dataset with q<0.005, on the basis of known interactions compiled in the ingenuity pathway analysis upstream regulator analysis
| Upstream regulator | Molecule type | Activation z-score | p-value of overlap | Target molecules in dataset |
|---|---|---|---|---|
| TP53 | Transcription regulator | −3.877 | 8.02×10−12 | ACTB, BIRC5, BRCA1, CAV1, CCNA2, CDC20, CDC6, CDK1, CITED2, CTGF, CTSB, CYP19A1, FEN1, GADD45G, HK2, HMMR, IL4R, KPNA2, MCM3, MCM4, MCM5, MCM6, MKI67, MYBL2, MYOF, NFKBIA, NINJ1, NOTCH1, NRP1, OAT, PBK, PCNA, PMM1, POSTN, PTCH1, RRM1, RRM2, SPP1, SREBF1, TDP2, THBS1, UBE2C, UHRF1, ZNF175 |
| let-7 | Microrna | −3.450 | 1.35×10−9 | BRCA1, CCNA2, CDC20, CDC6, CDCA3, CDK1, MCM3, MCM4, MCM5, MCM6, RRM1, RRM2, THBS1 |
| SMARCB1 | Transcription regulator | −2.795 | 7.79×10−6 | AMPH, CA5A, CCNA2, CDC6, CDK1, MCM3, MCM4, MCM5, MYBL2, POSTN |
| RB1 | Transcription regulator | −2.785 | 2.56×10−14 | ACTB, ANGPT2, BIRC5, BRCA1, CCNA2, CDC6, CDK1, CHAF1A, CITED2, CTGF, FEN1, LIG1, MCM3, MCM4, MCM5, MCM6, MYBL2, PCNA, PTX3, RRM1, RRM2, SNTB2 |
| SPARC | Other | −2.570 | 1.23×10−4 | BRCA1, CHAF1A, LRP8, MCM6, NOTCH1, POLE, RRM1 |
| RBL1 | Transcription regulator | −2.533 | 6.58×10−12 | CCNA2, CDC6, CDK1, MCM3, MCM5, MCM6, MYBL2, NOTCH1, PCNA, RRM1, RRM2, THBS1 |
| PTEN | Phosphatase | −2.464 | 1.77×10−8 | ADM, ANGPT2, BIRC5, CDC20, CITED2, CKS2, CTSB, FAM134C, IL4R, KLHL24, MKI67, MYBL2, NTRK1, PTP4A2, RRM1, SCD, SLC20A1, SPP1, SREBF1, THBS1 |
| DYRK1A | Kinase | −2.438 | 4.81×10−8 | CDC6, CDK1, MCM3, MCM4, MYBL2, UBE2C |
| CDKN2A | Transcription regulator | −2.436 | 2.17×10−9 | BIRC5, CCNA2, CDK1, CHAF1A, CITED2, CTGF, FEN1, GADD45G, MCM4, MCM5, MKI67, MYBL2, PCNA, PTX3, RRM1, RRM2, SNTB2 |
| Rb | Group | −2.425 | 6.69×10−7 | BIRC5, CCNA2, CDC6, CDK1, MCM3, MCM5, MYBL2, PCNA |
| CDKN1A | Kinase | −2.398 | 6.96×10−16 | ACTB, BIRC5, BRCA1, CCNA2, CDC20, CDC6, CDK1, CTGF, CTSB, HSPA5, MCM3, MCM4, MCM6, MKI67, MYBL2, PBK, PCNA, RRM1, UBE2C, UBE2S, UHRF1, ZBED5 |
| dinoprost | Chemical - endogenous mammalian | −2.273 | 4.95×10−9 | ADM, CYP19A1, FOSL2, LPL, MGST1, PDIA4, PTX3, SPP1, STAR, THBS1 |
| miR-24-3p | Mature microrna | −2.236 | 2.95×10−6 | BRCA1, CCNA2, CDK1, FEN1, NOTCH1 |
| Irgm1 | Other | −2.236 | 7.15×10−5 | CCNA2, CDCA3, MKI67, RRM2, UBE2C |
| HDAC2 | Transcription regulator | −2.236 | 1.41×10−3 | BRCA1, GABARAPL1, LIG1, MCM3, MCM5, NFKBIA |
| SIM1 | Transcription regulator | −2.219 | 6.49×10−2 | ANGPT2, ELL2, IL6R, OXT, POSTN |
| TGFBR2 | Kinase | −2.213 | 1.16×10−2 | BIRC5, CTGF, MYOF, NRP1, SPP1, THBS1 |
| PD98059 | Chemical - kinase inhibitor | −2.191 | 1.57×10−9 | ANGPT2, BIRC5, CAV1, CCNA2, CTGF, CTSB, CYCS, DHRS3, HSPA5, HYOU1, LDLR, LPL, MKI67, MYBL2, MYH11, NINJ1, PCNA, POSTN, RRM2, SPP1, SREBF1, ST3GAL4, STAR, THBS1 |
| INSIG1 | Other | −2.180 | 3.82×10−3 | LDLR, LPL, PLD1, SCD, SREBF1 |
| TNF | Cytokine | −2.035 | 1.17×10−11 | ACTB, ADAMTS4, ADM, ANGPT2, B4GALT1, BIRC5, CAV1, CDO1, CHST11, CITED2, CTGF, CTSB, CYP19A1, DHRS3, FOSL2, GADD45G, GCLC, GFPT2, HK2, IL1RL1, IL4R, JARID2, LDLR, LPL, MCM3, MGST1, MT1A, NEDD9, NEFM, NFKBIA, NINJ1, NOTCH1, NOV, NRP1, PAPPA, PDIA4, POSTN, PTX3, RGS4, RRM1, RRM2, SCD, SLC20A1, SPP1, SREBF1, STAR, THBS1 |
| E2F6 | Transcription regulator | −2.000 | 2.72×10−6 | BRCA1, CDC6, LIG1, MCM3, MCM5, RECQL, RRM2 |
| TNFSF13 | Cytokine | −2.000 | 2.58×10−5 | CDC6, MCM4, MCM5, MCM6, PCNA |
| ARNT2 | Transcription regulator | −2.000 | 1.57×10−1 | ANGPT2, ELL2, IL6R, POSTN |
The networks generated by IPA based on the dataset above are shown in Figure 4. The network in Figure 4A shows a role for cellular movement, cell death and survival. There is considerable connectivity associated with Akt, which exerts direct effects on the extracellular matrix molecules and indirectly affects collagen. The other network in Figure 4B shows a role for cardiovascular system development and function indicating significant interaction between the extracellular matrix genes and ADM (adrenomedullin), which appears to mainly signal through the intracellular PI3K pathway. The extracellular matrix genes PTX3 (pentraxin 3), NOV (nephroblastoma overexpressed), EFEMP1, OXT (oxytocin-neurophysin 1 precursor), and ADM and membrane protein gene NRP1 were all downregulated in these networks in lamb.
Real-time quantitative polymerase chain reaction validation
To confirm the reliability of the high-throughput sequencing data, we used qPCR to compare the expression levels. We selected 8 differently expressed genes, two upregulated (CYP19A1, STAR) and 6 downregulated (EFEMP1, SPP1 [osteopontin, secreted phosphoprotein 1], PTX3, NRP1, ADM, OXT), for validating. The results of the qPCR analysis for these 8 genes were in a good agreement with the RNA-seq data. The primers used for each gene and the fold changes are summarized in Table 5.
Table 5.
Primers used for validation of selected RNA-seq based genes by real-time reverse transcription-polymerase chain reaction analysis
| Gene name | Primer sequence | Genebank NO. | RNA seq | qPCR |
|---|---|---|---|---|
| CYP19A1 | TCGTCCTGGTCACCCTTCTG CGGTCTCTGGTCTCGTCTGG |
NM_001123000.1 | 1.34 | 1.08 |
| STAR | TCGCGGCTCTCTCCTAGGTTC ATCACTTTACTCAGCACCTCGT |
NM_001009243.1 | 1.56 | 0.68 |
| EFEMP1 | AACTTCCTCAATCCATCGTCT GACTTCACCAGCACAAGCA |
XM_012130548.1 | −4.60 | −4.44 |
| SPP1 | GAAACCCCTGATGACTCTGACC GCCATCATTTGTGCTTTCCGTA |
NM_001009224.1 | −3.27 | −3.98 |
| PTX3 | CTGTGATTCTGTTTTGTGCTCT TCGGAGTTCTCACGACTGC |
XM_004003220.2 | −1.68 | −1.03 |
| NRP1 | TGTGGAAAGATCGCCCCT ACTTTATTACCCCACTAGGCAT |
XM_012188254.1 | −2.39 | −1.84 |
| ADM | ATGAAGCTGGTTCCCGTC ACTTAGCGCCCATTTATTCCA |
XM_004016162.2 | −1.98 | −1.76 |
| OXT | CCTCCTGGCGTTGACCTCC GAAGCAGCCCAGCTCGTCCC |
NM_001009801.2 | −5.25 | −1.23 |
| GAPDH | TACTGGCAAAGTGGACATCGTT TTGATGACGAGCTTCCCGTTC |
NM_001190390.1 |
DISCUSSION
To our knowledge, this is the first study describing differences in the transcriptomes of follicle GCs from superstimulated lambs and ewes. In the ovary of one-month-old lamb with synchronous developments of numerous small antral follicles, there is no dominant follicle and no follicle wave, and no endocrine negative feedback typical for ewes; also follicular atresia is rare there. Following treatment of the lambs with FSH, the follicles were synchronously developing to middle large follicles but could not complete maturation and ovulation. Considering the unique features of lamb follicle development, the RNA sequencing of developing GCs and the comprehensive, spatiotem-poral gene expression profiles obtained hereby have a major value. In this study, a total of more than 300 differently expressed genes were identified in growing follicle GCs between lamb and ewe. IPA analyses of DEGs revealed that processes of axonal guidance and other IPA canonical pathways were mostly enriched in lamb GCs. Cell proliferation, DNA replication and other functions were activated in lambs. The upstream regulators EGF, TBX2, beta-estradiol and others were predicted to be active, and moreover, TP53, let-7, PTEN and other genes were predicted to be inhibited in lamb follicles. These results should provide important clues on the molecular mechanisms regulating the growth and maturation of dominant follicles in the lamb ovary.
In sheep, high blood concentrations of FSH trigger the emergence and growth of a cohort of antral follicles, which enter terminal development. As the follicle enlarges from 2 to 3.5 mm in diameter, the follicle becomes progressively more mature, GCs become increasingly responsive to FSH (in terms of FSH-induced cyclic adenosine monophosphate [cAMP] production) and they express increasing amounts of CYP19A1, the key enzyme for oestradiol production. The final stage of cell differentiation is reached in the preovulatory follicle when the GCs acquire luteinizing hormone (LH) responsiveness, through a strong and rapid increase in expression of the LH receptor [23]. In our results, three of the steroidogenesis pathway genes (CYP19A1, HSD17B1 [hydroxysteroid (17-beta) dehydrogenase 1] and HSD17B10 [hydroxysteroid (17-beta) dehydrogenase 10]) were all up-regulated in lamb GCs (Table 5). Although the beta-estradiol upstream regulators were predicted activated (including 62 DEGs) (Table 3), the luteinizing hormone receptor (LHR) expression levels were not different between lamb and ewe. In lambs, the gonadotropin stimulated follicles were only growing up to 3–4 mm in diameter and stopped there. In contrast, the ewe follicles could grow up to 5–6 mm in preovulatory stage and whether they would ovulate or undergo atresia depended on their endocrine environment. In this study, we selected the growing follicles (processed for the same period of time and with similar diameters in both lambs and ewes) that did not reach preovulatory stage as a control group. Therefore, the role of beta-estradiol upstream regulator in lamb may not affect the LHR pathway in the growing follicle development function.
In bovine GCs, as healthy follicles enlarge from small to large antral sizes, genes associated with the processes of axonal guidance signaling were upregulated in large versus small follicles [24]. In our results, the five gene transcripts (NRP1 [−2.3896], SEMA3A [−2.1929], ADAMTS9 [−2.102], PTCH1 [−1.6653], NFAT5 [−1.021]) involved in axonal guidance signaling were significantly down-regulated. The inhibition of these genes and subsequent axonal guidance pathways identified in our result indicate that the GCs might be insufficiently prepared for ovulation and luteinisation in lamb.
In mammalian preovulatory follicles, FSH/LH stimulation of GCs and cumulus cells together with the activation of the EGFR-MAPK3/1 pathway and cAMP priming, triggers oocyte nuclear maturation and acquisition of developmental competence as well as cumulus expansion [25–27]. Unlike preovulatory follicles, in primodial follicle EGF stimulates MAPK and PI3K signaling pathways to promote ovarian cell proliferation by maintaining intraovarian primordial follicle viability (but without promoting activation) in prepubertal cats [28]. In our study, EGFR pathway mechanistic network was predicted to be involved in P38 MAPK and PI3K (complex) pathways but not in the MAPK3/1 pathway (Figure 5). In lamb GCs, the EGFR pathway was activated similarly in primodial follicles to promote age-dependent ovarian cell proliferation but not to trigger oocyte nuclear maturation and acquisition of developmental competence.
Figure 5.

Two networks (A, B) generated for molecules mapped to the ingenuity pathway analysis database from a data set containing genes differentially regulated between lamb and ewe follicles. Interactions between molecules are shown as explained in the legend, with focus molecule symbols highlighted in color, based on up- (red) or down- (green) regulation in large follicles and of increasing intensity with degree of fold change.
Tp53 is an activator of apoptotic pathways in response to cell stress and it functions by regulating cell cycle, DNA repair and metabolic genes. The overall processes influenced by transcription factor gene TP53 were predicted to be activated in early antral stages of atretic bovine follicles [29]. In our study, TP53 signaling in lamb GCs was predicted to be inhibited. The inhibited TP53 signaling pathway may explain why follicular atresia rarely happens in one-month-old lambs compared to ewes.
The let-7 microRNA family members were found to be differentially expressed during follicular atresia [30]. Some of them were significantly decreased in early atretic and progressively atretic porcine ovary follicles compared with healthy follicles, and some highly expressed during follicular atresia [30]. In our study, let-7 was predicted to be inhibited, which might affect the cell division cycle as suggested by its target molecules. FSH treatment may change follicle intracellular miRNA diversification and affect target gene transcript and leading to functional changes.
The extracellular matrix plays a prominent role in ovarian function due to its impact on processes such as cell migration, proliferation, growth, and development of follicles [31]. In horses, EFEMP1 and PTX3 transcripts were significantly up-regulated in GCs during early to late health dominance follicle [32]. ADM is possibly involved in the breakdown of the germinal vesicle [33]. SPP1 transcripts have been reported to rise in parallel with follicle size in both horses and cattle [32,34]. These extracellular matrix genes are involved in most pathways (Tables 3 and 4). In our study, these extracellular matrix genes (EFEMP1, PTX3, and ADM) and a membranal gene SPP1 were significantly down-regulated in lamb GCs (Table 5). This lower expression of extracellular matrix genes may be responsible for follicle development states and affect oocyte development competence in one-month-old lamb ovary.
CONCLUSION
In this study, RNA sequencing technology was employed in a whole genome approach to detect and analyze differences in gene expression in supersitimulated one-month-old lamb and adult ewe GCs. We found more than 300 genes to be differentially expressed. IPA analysis showed that IPA canonical pathways were involved in axonal guidance signaling. EGF, TBX2, beta-estradiol and other upstream regulators were activated, and TP53, let-7, PTEN and other upstream regulators were inhibited in lamb follicles. Among these regulators, many targeted extracellular matrix genes, such as EFEMP1, PTX3, ADM, and SPP1, were significantly down-regulated. The present study may contribute to a better understanding of the mechanisms governing the role of GCs in the growing follicle in prepubertal sheep.
Supplementary tables and figures
ACKNOWLEDGMENTS
This work was supported by the National Science Foundation of China (Grant No. U1203381, U1403184).
Footnotes
CONFLICT OF INTEREST
We certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.
REFERENCES
- 1.Scaramuzzi RJ, Baird DT, Campbell BK, et al. Regulation of folliculogenesis and the determination of ovulation rate in ruminants. Reprod Fertil Dev. 2011;23:444–67. doi: 10.1071/RD09161. [DOI] [PubMed] [Google Scholar]
- 2.Kennedy JP, Worthington CA, Cole ER. The post-natal development of the ovary and uterus of the Merino lamb. J Reprod Fertil. 1974;36:275–82. doi: 10.1530/jrf.0.0360275. [DOI] [PubMed] [Google Scholar]
- 3.Tassell R, Chamley WA, Kennedy JP. Gonadotrophin levels and ovarian development in the neonatal ewe lamb. Aust J Biol Sci. 1978;31:267–73. doi: 10.1071/bi9780267. [DOI] [PubMed] [Google Scholar]
- 4.Sonjaya H, Driancourt MA. Ovarian follicles during infancy in Romanov and Ile-de-France ewe lambs. J Reprod Fertil. 1987;81:241–8. doi: 10.1530/jrf.0.0810241. [DOI] [PubMed] [Google Scholar]
- 5.Rawlings NC, Evans AC, Honaramooz A, Bartlewski PM. Antral follicle growth and endocrine changes in prepubertal cattle, sheep and goats. Anim Reprod Sci. 2003;78:259–70. doi: 10.1016/s0378-4320(03)00094-0. [DOI] [PubMed] [Google Scholar]
- 6.Kelly JM, Kleemann DO, Walker SK. Enhanced efficiency in the production of offspring from 4- to 8-week-old lambs. Theriogenology. 2005;63:1876–90. doi: 10.1016/j.theriogenology.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 7.Gou KM, Guan H, Bai JH, et al. Field evaluation of juvenile in vitro embryo transfer (JIVET) in sheep. Anim Reprod Sci. 2009;112:316–24. doi: 10.1016/j.anireprosci.2008.05.008. [DOI] [PubMed] [Google Scholar]
- 8.Paramio MT, Izquierdo D. Current status of in vitro embryo production in sheep and goats. Reprod Domest Anim. 2014;49(Suppl 4):37–48. doi: 10.1111/rda.12334. [DOI] [PubMed] [Google Scholar]
- 9.Amiridis GS, Cseh S. Assisted reproductive technologies in the reproductive management of small ruminants. Anim Reprod Sci. 2012;130:152–61. doi: 10.1016/j.anireprosci.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 10.Ptak G, Loi P, Dattena M, Tischner M, Cappai P. Offspring from one-month-old lambs: studies on the developmental capability of prepubertal oocytes. Biol Reprod. 1999;61:1568–74. doi: 10.1095/biolreprod61.6.1568. [DOI] [PubMed] [Google Scholar]
- 11.Leoni GG, Bebbere D, Succu S, et al. Relations between relative mRNA abundance and developmental competence of ovine oocytes. Mol Reprod Dev. 2007;74:249–57. doi: 10.1002/mrd.20442. [DOI] [PubMed] [Google Scholar]
- 12.Morton KM. Developmental capabilities of embryos produced in vitro from prepubertal lamb oocytes. Reprod Domest Anim. 2008;43(Suppl 2):137–43. doi: 10.1111/j.1439-0531.2008.01153.x. [DOI] [PubMed] [Google Scholar]
- 13.Leoni GG, Palmerini MG, Satta V, et al. Differences in the kinetic of the first meiotic division and in active mitochondrial distribution between prepubertal and adult oocytes mirror differences in their developmental competence in a sheep model. PLoS One. 2015;10:e0124911. doi: 10.1371/journal.pone.0124911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamel M, Dufort I, Robert C, et al. Identification of differentially expressed markers in human follicular cells associated with competent oocytes. Hum Reprod. 2008;23:1118–27. doi: 10.1093/humrep/den048. [DOI] [PubMed] [Google Scholar]
- 15.Dias FC, Khan MI, Sirard MA, Adams GP, Singh J. Differential gene expression of granulosa cells after ovarian superstimulation in beef cattle. Reproduction. 2013;146:181–91. doi: 10.1530/REP-13-0114. [DOI] [PubMed] [Google Scholar]
- 16.Primig M. The bioinformatics tool box for reproductive biology. Biochim Biophys Acta. 2012;1822:1880–95. doi: 10.1016/j.bbadis.2012.05.018. [DOI] [PubMed] [Google Scholar]
- 17.Kang X, Liu G, Liu Y, Xu Q, Zhang M, Fang M. Transcriptome profile at different physiological stages reveals potential mode for curly fleece in Chinese tan sheep. PLoS One. 2013;8:e71763. doi: 10.1371/journal.pone.0071763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mortazavi A, Williams BA, Mccue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–8. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 20.Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26:136–8. doi: 10.1093/bioinformatics/btp612. [DOI] [PubMed] [Google Scholar]
- 21.Bonnet A, Cabau C, Bouchez O, et al. An overview of gene expression dynamics during early ovarian folliculogenesis: specificity of follicular compartments and bi-directional dialog. BMC Genomics. 2013;14:904. doi: 10.1186/1471-2164-14-904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 23.Clement F, Monniaux D. Multiscale modelling of ovarian follicular selection. Prog Biophys Mol Biol. 2013;113:398–408. doi: 10.1016/j.pbiomolbio.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 24.Hatzirodos N, Irving-Rodgers HF, Hummitzsch K, et al. Transcriptome profiling of granulosa cells of bovine ovarian follicles during growth from small to large antral sizes. BMC Genomics. 2014;15:24. doi: 10.1186/1471-2164-15-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Conti M, Hsieh M, Park JY, Su YQ. Role of the epidermal growth factor network in ovarian follicles. Mol Endocrinol. 2006;20:715–23. doi: 10.1210/me.2005-0185. [DOI] [PubMed] [Google Scholar]
- 26.Yamashita Y, Shimada M. The release of EGF domain from EGF-like factors by a specific cleavage enzyme activates the EGFR-MAPK3/1 pathway in both granulosa cells and cumulus cells during the ovulation process. J Reprod Dev. 2012;58:510–4. doi: 10.1262/jrd.2012-056. [DOI] [PubMed] [Google Scholar]
- 27.Khan DR, Guillemette C, Sirard MA, Richard FJ. Characterization of FSH signalling networks in bovine cumulus cells: a perspective on oocyte competence acquisition. Mol Hum Reprod. 2015;21:688–701. doi: 10.1093/molehr/gav032. [DOI] [PubMed] [Google Scholar]
- 28.Fujihara M, Comizzoli P, Keefer CL, Wildt DE, Songsasen N. Epidermal growth factor (EGF) sustains in vitro primordial follicle viability by enhancing stromal cell proliferation via MAPK and PI3K pathways in the prepubertal, but not adult, cat ovary. Biol Reprod. 2014;90:86. doi: 10.1095/biolreprod.113.115089. [DOI] [PubMed] [Google Scholar]
- 29.Hatzirodos N, Hummitzsch K, Irving-Rodgers HF, et al. Transcriptome profiling of granulosa cells from bovine ovarian follicles during atresia. BMC Genomics. 2014;15:40. doi: 10.1186/1471-2164-15-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao R, Wu WJ, Zhou XL, et al. Expression and preliminary functional profiling of the let-7 family during porcine ovary follicle atresia. Mol Cells. 2015;38:304–11. doi: 10.14348/molcells.2015.2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berkholtz CB, Lai BE, Woodruff TK, Shea LD. Distribution of extracellular matrix proteins type I collagen, type IV collagen, fibronectin, and laminin in mouse folliculogenesis. Histochem Cell Biol. 2006;126:583–92. doi: 10.1007/s00418-006-0194-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Donadeu FX, Fahiminiya S, Esteves CL, et al. Transcriptome profiling of granulosa and theca cells during dominant follicle development in the horse. Biol Reprod. 2014;91:111. doi: 10.1095/biolreprod.114.118943. [DOI] [PubMed] [Google Scholar]
- 33.Hiradate Y, Ohtake J, Hoshino Y, Tanemura K, Sato E. Adrenomedullin: a possible regulator of germinal vesicle breakdown. Biochem Biophys Res Commun. 2011;415:691–5. doi: 10.1016/j.bbrc.2011.10.139. [DOI] [PubMed] [Google Scholar]
- 34.Skinner MK, Schmidt M, Savenkova MI, Sadler-Riggleman I, Nilsson EE. Regulation of granulosa and theca cell transcriptomes during ovarian antral follicle development. Mol Reprod Dev. 2008;75:1457–72. doi: 10.1002/mrd.20883. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.