

Abstract
WZB117 (2-fluoro-6-(m-hydroxybenzoyloxy) phenyl m-hydroxybenzoate) inhibits passive sugar transport in human erythrocytes and cancer cell lines and, by limiting glycolysis, inhibits tumor growth in mice. This study explores how WZB117 inhibits the erythrocyte sugar transporter glucose transport protein 1 (GLUT1) and examines the transporter isoform specificity of inhibition. WZB117 reversibly and competitively inhibits erythrocyte 3-O-methylglucose (3MG) uptake with Ki(app) = 6 μm but is a noncompetitive inhibitor of sugar exit. Cytochalasin B (CB) is a reversible, noncompetitive inhibitor of 3MG uptake with Ki(app) = 0.3 μm but is a competitive inhibitor of sugar exit indicating that WZB117 and CB bind at exofacial and endofacial sugar binding sites, respectively. WZB117 inhibition of GLUTs expressed in HEK293 cells follows the order of potency: insulin-regulated GLUT4 ≫ GLUT1 ≈ neuronal GLUT3. This may explain WZB117-induced murine lipodystrophy. Molecular docking suggests the following. 1) The WZB117 binding envelopes of exofacial GLUT1 and GLUT4 conformers differ significantly. 2) GLUT1 and GLUT4 exofacial conformers present multiple, adjacent glucose binding sites that overlap with WZB117 binding envelopes. 3) The GLUT1 exofacial conformer lacks a CB binding site. 4) The inward GLUT1 conformer presents overlapping endofacial WZB117, d-glucose, and CB binding envelopes. Interrogating the GLUT1 mechanism using WZB117 reveals that subsaturating WZB117 and CB stimulate erythrocyte 3MG uptake. Extracellular WZB117 does not affect CB binding to GLUT1, but intracellular WZB117 inhibits CB binding. These findings are incompatible with the alternating conformer carrier for glucose transport but are consistent with either a multisubunit, allosteric transporter, or a transporter in which each subunit presents multiple, interacting ligand binding sites.
Keywords: glucose transport, ligand-binding protein, membrane transport, molecular docking, protein structure, competitive inhibition, facilitated diffusion, ligand binding, membrane transport protein
Introduction
Most cancer cells use anaerobic metabolism (glycolysis) to generate the ATP required for cellular processes and proliferation (1). This contrasts with normal, differentiated cells, which mostly use mitochondrial oxidative phosphorylation to sustain cellular function (2). The transition from aerobic to anaerobic metabolism is termed “the Warburg effect” and is driven by growth conditions, by mutations of proto-oncogenes and tumor-suppressor genes (2, 3). Vander Heiden et al. (1) have proposed that the metabolism of all proliferating cells (including cancer cells) is adapted to enhance nutrient uptake and nutrient incorporation into the biomass needed to produce a new cell. This would explain why the developing central nervous system is so susceptible to glucose deprivation resulting from glucose transport-deficiency at the blood-brain barrier (4) and why cancer cells are so sensitive to limited glucose availability (5).
This has prompted several groups to propose that suppressing anaerobic metabolism may offer an effective anti-cancer strategy. Three approaches have been used to limit glycolysis in cancer cells; that is glucose deprivation in vitro (6, 7), the in vitro and in vivo use of glycolysis inhibitors (8), and cellular glucose transport inhibition in vitro and in vivo (8, 9). All three approaches cause cancer cell death.
Glucose transport in most mammalian cells is catalyzed by one or more members of the GLUT2 (SLC2A) family of passive glucose transporters (10). GLUT1 (the blood brain barrier/erythroid glucose transporter) and, in some instances, GLUT3 (the neuronal glucose transporter) or GLUT12 expression is significantly increased in proliferating cells (11).
Successful strategies for passive glucose transport inhibition have exploited a range of molecules that bind at or close to either the exofacial (e.g. maltose, ethylidene glucose) or the endofacial sugar binding site (e.g. cytochalasin B; Refs. 12–14). A number of unrelated inhibitory molecules react with the GLUT1 purine binding site (e.g. ATP, AMP, caffeine; Ref. 15) or at other, less well defined sites (e.g. androgens, quercetin; Refs. 16 and 17). These and related molecules have been suggested as potential scaffolds for designing inhibitors of glucose uptake in cancer cells or cancer stem cells (8, 18–20).
Using such an approach, Zhang et al. (21) identified a class of polyphenolic compounds with sugar transport inhibitory potency. Among these, WBZ117 (2-fluoro-6-(m-hydroxybenzoyloxy)phenyl m-hydroxybenzoate) showed the highest affinity for sugar transport inhibition and inhibited tumor growth in a nude mouse model (9).
Although a promising strategy for cancer cell elimination, glucose transport inhibition in vivo may present several attendant complications. These include impaired insulin secretion, elevated blood glucose, diuresis, elevated glycation, impaired glucose transport across the blood-brain barrier, and reduced metabolic capacity in healthy cells that depends upon glycolysis for normal function. We, therefore, undertook a systematic analysis of WZB117 inhibition of glucose transport.
We found that WZB117 interacts with GLUT1 at the exofacial sugar binding site and thus acts as a reversible, competitive inhibitor of net glucose uptake and exchange glucose transport but as a noncompetitive inhibitor of sugar efflux from cells. WZB117 inhibits the insulin-sensitive glucose transport (GLUT4) with greater potency than its inhibition of either GLUT1 or GLUT3.
Results
Sugar transport in human erythrocytes is catalyzed by GLUT1 (22). Transport theory states that ligands binding reversibly at the exofacial sugar binding site act as competitive inhibitors of sugar uptake and as noncompetitive inhibitors of exit. Conversely, ligands binding reversibly at the endofacial sugar binding site act as noncompetitive inhibitors of sugar uptake and as competitive inhibitors of exit (23–25).
To understand whether WZB117 inhibits GLUT1 by binding at the exofacial or endofacial sugar binding site, we examined its effects on three modes of erythrocyte sugar transport: zero-trans 3MG uptake (uptake into cells lacking sugar), zero-trans 3MG exit (efflux from cells into medium lacking sugar), and equilibrium exchange 3MG uptake (unidirectional uptake of 3MG in cells where [3MG]i = [3MG]o).
WZB117 inhibits zero-trans 3MG (0.1 mm) uptake by erythrocytes in a dose-dependent manner with Ki(app) = 6.2 ± 1.6 μm (Fig. 1). CB (a known GLUT1 endofacial site ligand) inhibits 0.1 mm 3MG zero-trans uptake with Ki(app) = 0.3 ± 0.1 μm (see Fig. 4).
FIGURE 1.

Sensitivity of zero-trans 3MG (0.1 mm) uptake by erythrocytes to inhibition by WZB117. Ordinate: 3MG uptake in mmol/liter of cell water/min. Abscissa: WZB117 in μm. Each data point represents the mean ± S.E. of at least three separate measurements made in duplicate. The curves drawn through the points were computed by nonlinear regression assuming uptake is inhibited completely by inhibitor in a dose-dependent manner (see Equation 1). The results are: Ki(app) = 6.2 ± 1.6 μm, R2 = 0.946, standard error of regression = 0.0021 mmol/liter of cell water/min.
FIGURE 4.

CB and WZB117 stimulate 3MG uptake at subsaturating inhibitor concentrations. Ordinate: relative 3MG uptake. Abscissa: Inhibitor concentration in μm. Results are shown for WZB117-treated cells (●), for CB-treated cells (▵), and for cells exposed to CB plus 5 μm WZB117 (▴). Each data point is the mean ± S.E. of three or more duplicate measurements. The curves drawn through the points were computed by nonlinear regression according to Equation 4 or Equation 1 and have the following constants: WZB117-treated cells (●) Δ1 = 0.152 ± 0.056, K1 = 0.0014 ± 0.0013 μm, K2 = 11.0 ± 2.4 μm, R2 = 0.952, standard error of regression = 0.09; CB-treated cells (▵) Δ1 = 0.653 ± 0.115, K1 = 0.012 ± 0.011 μm, K2 = 0.29 ± 0.10 μm, R2 = 0.974, standard error of regression = 0.09; cells exposed to CB plus 5 μm WZB117 (▴) vo = 0.69 ± 0.02, K1 = 0.46 ± 0.07, R2 = 0.986, standard error of regression = 0.02. The arrows represent IC50 for CB inhibition of transport in the absence (solid lines) or presence (dashed lines) of 5 μm WZB117 respectively. Differences between transport in the absence and presence of each concentration of inhibitor were analyzed by ordinary one-way ANOVA using Dunnett's multiple comparisons test with a single pooled variance and a family-wise significance and confidence level of 0.01. CB (10 and 50 nm) and WZB117 (100 nm) significantly increased 3MG uptake. Transport inhibition is significant at WZB117 >5 μm and at CB >250 nm.
We assessed the reversibility of inhibition in two ways. The first method involved exposing RBCs to buffer containing or lacking WZB117 (3 μm) or CB (0.5 μm) for 10 min, then removing the inhibitors using a centrifugation/wash cycle and incubating the cells in inhibitor-free buffer for a further 15 min. We then measured 0.1 mm 3MG uptake in these cells. WZB117 (3 μm) or CB (0.5 μm) inhibit sugar uptake by 50% (Fig. 2). Removing the inhibitor before sugar uptake determinations restored sugar uptake to levels of 75% or greater of uninhibited controls (Fig. 2).
FIGURE 2.

Reversibility of transport inhibition by WZB117 and by CB. Ordinate: 3MG uptake in mmol/liter of cell water/min. Abscissa: experimental treatment. Control cells saw no inhibitor during sugar uptake; WZB117 and CB cells were exposed to inhibitor (3 μm WZB117 and 0.5 μm CB respectively) for 15 min on ice before measurement of 3MG uptake in the presence of inhibitor. WZB117 -washout and CB-washout cells were exposed to inhibitor (3 μm WZB117 and 0.5 μm CB, respectively) for 15 min on ice followed by inhibitor removal and exposure to inhibitor-free medium for 15 min on ice and finally measurement of 3MG uptake in the absence of inhibitor. Each symbol (●) represents the mean of duplicate measurements. The horizontal line and error bars represent the mean ± S.E. of each condition. Unpaired t test analysis indicates: +, transport under all treatments is significantly lower (p < 0.005) than in control cells; ++, transport after washout treatment is significantly greater (p < 0.005) than in the corresponding non-washout treatment.
WZB117 Is a Competitive Inhibitor of 3MG Uptake by Human Erythrocytes
The second method of assessing reversibility of inhibition evaluates the type of sugar transport inhibition produced by WZB117. Competitive inhibitors act by binding reversibly either to the active site of an enzyme or to a site whose occupancy occludes occupancy of the active site and vice versa (26).
Preincubation with WZB117 (7 μm) before initiation of transport measurements increased Km(app) for zero-trans 3MG uptake from 1.0 ± 0.2 mm to 2.8 ± 0.4 mm, but Vmax for 3MG uptake (0.59 ± 0.03 mmol/liter of cell water/min) was unaffected (Fig. 3). This result indicated that WZB117 is a competitive inhibitor of human erythrocyte sugar uptake. If WZB117 bound irreversibly to the active site, preincubation with the inhibitor would most likely have reduced Vmax for transport in addition to increasing Km(app).
FIGURE 3.

Effects of WZB117 and CB on Michaelis-Menten kinetics of zero-trans 3MG uptake (A), zero-trans 3MG exit (B and C), and equilibrium exchange 3MG uptake (D). Results are shown for control cells (○), WZB117-treated cells (●), and for CB-treated cells (▵). Each data point is the mean ± S.E. of three duplicate measurements. A, ordinate: 3MG uptake in mmol/liter of cell water/min. Abscissa: [3MG]o in mm. Curves drawn through the points were computed by nonlinear regression assuming Michaelis-Menten uptake kinetics (Equation 2) and have the following results: control cells (○),Vmax = 0.59 ± 0.03 mmol/liter of cell water/min, Km(app) = 1.05 ± 0.16 mm, R2 = 0.992, standard error of regression = 0.018 mmol/liter of cell water/min; WZB117 treatment (●): Vmax = 0.68 ± 0.04 mmol/liter of cell water/min, Km(app) = 2.77 ± 0.35 mm, R2 = 0.997, standard error of regression = 0.018 mmol/liter of cell water/min; CB treatment (▵): Vmax = 0.29 ± 0.05 mmol/liter of cell water/min, Km(app) = 1.67 ± 0.63 mm, R2 = 0.966, standard error of regression = 0.010 mmol/liter of cell water/min. B and C, ordinates: [3MG]i in mmol/liter of cell water. Abscissa: time In minutes. Curves drawn through the points were computed by nonlinear regression and numerical integration assuming Michaelis-Menten exit kinetics and have the following results: Control cells (○): Vmax = 2 mmol/liter of cell water/min, Km(app) = 15 mm; WZB117 treatment (●): Vmax = 0.8 mmol/liter of cell water/min, Km(app) = 15 mm; CB treatment (▵): Vmax = 2 mmol/liter of cell water/min, Km(app) = 47 mm. D, Hanes-Woolf plot of equilibrium exchange transport. Ordinate: [3MG]/rate of equilibrium exchange 3MG uptake in minutes. Abscissa: intracellular and extracellular [3MG] in mm. Lines drawn through the points were computed by nonlinear regression assuming that each line is described by Equation 3. The results are: control cells (○): Vmax = 13.39 ± 0.79 mmol/liter of cell water/min, Km(app) = 40.25 ± 3.57 mm, R2 = 0.990, standard error of regression = 0.17 min; WZB117 treatment (●): Vmax = 13.36 ± 2.02 mmol/liter of cell water/min, Km(app) = 82.25 ± 15.30 mm, R2 = 0.936, standard error of regression = 0.43 min; CB treatment (▵): Vmax = 8.87 ± 0.33 mmol/liter of cell water/min, Km(app) = 49.85 ± 2.55 mm, R2 = 0.996, standard error of regression = 0.16 min.
Conversely, CB (0.7 μm) reduces Vmax for 3MG uptake (0.29 ± 0.05 mmol/liter of cell water/min) but has no effect on Km(app) (Fig. 3A). This result confirms that CB is a noncompetitive inhibitor of uptake and is expected for an inhibitor which binds reversibly to the endofacial active site (25).
WZB117 Is a Noncompetitive Inhibitor of Net Sugar Exit
Because WZB117 competes for 3MG binding at or close to the exofacial sugar entry site, we hypothesized that WZB117 would act as a noncompetitive inhibitor of sugar exit (25). To this end, we performed sugar exit experiments where we measured the time course of zero-trans sugar exit from pre-loaded red cells into sugar-free external medium. The time course of exit was analyzed by simulating sugar exit by numerical integration using Berkeley Madonna software assuming Michaelis-Menten exit kinetics then adjusting simulated Michaelis-Menten parameters using the Levenberg-Marquardt algorithm until the fits between simulated and experimental exit data were optimal. Our analysis shows that WZB117 (0.7 μm) reduces Vmax for 3MG exit from 2 mmol/liter of cell water/min to 0.8 mmol/liter of cell water/min, whereas Km(app) for exit (15 mm) was unaffected (Fig. 3B). Consistent with published data (13), CB (0.7 μm) was competitive for sugar exit, increasing Km(app) to 47 mm with no effect on Vmax (Fig. 3C).
WZB117 Is a Competitive Inhibitor of Equilibrium Exchange Sugar Transport
Equilibrium exchange 3MG transport in human red blood cells ([3MG]i = [3MG]o) was competitively inhibited by WZB117. WZB117 (7 μm) increased Km(app) for 3MG exchange transport from 40.25 ± 3.57 mm to 82.25 ± 15.30 mm but had no effect on Vmax (13.39 ± 0.79 mmol/liter of cell water/min; Fig. 3D). CB (0.7 μm), however, was a mixed-type inhibitor of equilibrium exchange 3MG transport reducing Vmax (8.87 ± 0.33 mmol/liter of cell water/min) and increasing Km(app) (49.85 ± 2.56 mm; Fig. 3D).
Subsaturating Levels of CB and WZB117 Stimulate Sugar Uptake in Human Erythrocytes
Previous studies have shown that subsaturating concentrations of CB or exofacial inhibitors of GLUT1 produce a modest stimulation of erythrocyte sugar uptake (27, 28). Subsaturating levels of CB (≤0.1 μm) and WZB117 (0.1 μm) stimulate zero-trans 3MG uptake in human erythrocytes by up to 25% above untreated cells (Fig. 4). This effect is significant (p = 0.0075) at 10 and 50 nm CB and at 100 nm WZB117 (p < 0.001). CB and WZB117 may stimulate transport by reducing Km(app) for sugar uptake (Table 1), but the data are not conclusive.
TABLE 1.
Effects of subsaturating inhibitors on 3MG uptake
The Michaelis-Menten kinetics of 3MG (0.1–5 mm) uptake by RBCs was measured in paired experiments in the presence or absence of WZB117 (0.7 μm) or CB (0.025 μm) and analyzed by nonlinear regression using Equation 2. Each data point (uptake measured at each 3MG concentration) averaged three or more measurements made in duplicate. R2 for all fits was ≥0.994. Results are shown as the mean ± S.E. and as 95% confidence Intervals for Vmax and Km(app) under all conditions.
| Experimental condition |
Vmax |
Km(app) |
||
|---|---|---|---|---|
| Mean ± S.E. | 95% Confidence interval | Mean ± S.E. | 95% Confidence interval | |
| mmo/liter cell water/min | mm | |||
| Control | 0.63 ± 0.01 | 0.59–0.68 | 2.26 ± 0.12 | 1.92–2.68 |
| WZB117 0.7 μm | 0.55 ± 0.04 | 0.46–0.69 | 1.74 ± 0.26 | 1.08–2.91 |
| CB 0.025 μm | 0.60 ± 0.02 | 0.55–0.67 | 1.73 ± 0.13 | 1.36–2.23 |
Effect of WZB117 on CB Binding
CB is membrane-permeant and rapidly crosses the cell membrane to interact with the GLUT1 endofacial sugar binding site (23, 25). The simple carrier hypothesis for facilitated diffusion predicts that CB and WZB117 binding to GLUT1 are mutually exclusive (23–25). Accordingly, CB should competitively inhibit WZB117 inhibition of and binding to GLUT1. The IC50 for CB inhibition of 3MG uptake (0.35 μm) is not materially affected when WZB117 (5 μm) is present (Fig. 4).
WZB117 is without effect on [3H]CB binding to human RBCs but served as a low affinity inhibitor of CB binding to red cell membranes depleted of peripheral membrane proteins (Fig. 5; Ki(app) = 156 ± 24 μm). RBC membranes depleted of peripheral proteins are unsealed, and both membrane surfaces are accessible to buffer (29). Collectively, these results indicate: 1) CB binding to the GLUT1 endofacial sugar binding site is insensitive to WZB117 binding at the exofacial sugar binding site; 2) CB and WZB117 compete for binding at the GLUT1 endofacial sugar binding site; 3) RBCs are impermeable to WZB117 because inhibition of binding by a membrane-permeant WZB117 would have been evident in measurements with intact RBCs. In addition, these findings are consistent with the actions of d-glucose on CB binding to RBCs; extracellular d-glucose is without effect on binding, whereas intracellular d-glucose competitively inhibits binding (30).
FIGURE 5.

WZB117 is without effect on [3H]CB binding to human RBCs (●) but serves as a low affinity inhibitor of CB binding to red cell membranes depleted of peripheral membrane proteins (○). The lines drawn through the points were computed by linear regression. Ki(app) for inhibition of CB binding is computed as the -x-intercept. The results are: RBCs (●) Ki(app) = 373 ± 310 μm, R2 = 0.238, p value testing the null hypothesis that the overall slope is zero = 0.2765; membranes (○) Ki(app) = 150.4 ± 24.4 μm; R2 = 0.943, p value testing the null hypothesis that the overall slope is zero = 0.0058.
Isoform Specificity of Transport Inhibition
The GLUT (SLC2A) family of passive glucose transporters can show distinct, isoform-specific affinities for inhibitors. For example, the insulin-sensitive glucose transporter of fat and muscle (GLUT4) is inhibited by HIV protease inhibitors, but GLUT1 is not (31), whereas GLUT1, GLUT3, and GLUT4 are inhibited by CB and the intestinal fructose transporters GLUT5 and GLUT7 are not (32). We, therefore, stably transfected HEK293 cells with GLUT1, GLUT3, or GLUT4 and measured the concentration dependence of WZB117 inhibition of 2-deoxy-d-glucose (2DG, 0.1 mm) uptake at 37 °C. WZB117 inhibits GLUT1- and GLUT3-mediated 2DG uptake with Ki(app) ≈ 10 μm (Fig. 6A) but is a more potent inhibitor of GLUT4-mediated uptake (Ki(app) = 0.2 μm; Fig. 6A).
FIGURE 6.

WZB117 inhibition of sugar transport is GLUT isoform-specific. A, Dixon plot of transport inhibition by WZB117 in untransfected HEK293 cells (○) and in cells transfected with and expressing hGLUT1 (●), hGLUT3 (▵), or hGLUT4 (▴). Ordinate: 1/2DG uptake in min·μg protein/mol. Abscissa: WZB117 in μm. Lines drawn through the points were computed by linear regression and Ki(app) for WZB117 inhibition of transport computed as -x- intercept. The results are: untransfected cells (○), Ki(app) = 2.45 ± 0.66 μm, R2 = 0.975, standard error of regression = 3.66 × 1010 min·μg protein/mol; hGLUT1-transfected cells (●), Ki(app) = 5.71 ± 0.95 μm, R2 = 0.974, standard error of regression = 7.59 × 109 min·μg protein/mol; hGLUT3-transfected cells (▵), Ki(app) = 13.32 ± 5.12 μm, R2 = 0.862, standard error of regression = 8.82 × 109 min·μg protein/mol; hGLUT4-transfected cells (▴), Ki(app) = 0.23 ± 0.47 μm, R2 = 0.977, standard error of regression = 1.09 × 1011 min·μg protein/mol. B, effect of heterologous GLUT1 or GLUT4 expression on parental GLUT3 mRNA levels as detected by qPCR. For each condition the symbols (●) show the results of six separate measurements, and the horizontal lines plus error bars show their mean ± S.E. The conditions are untransfected (Control), GLUT1-transfected, and GLUT4-transfected cells. Results are normalized to one of the six GLUT3 message levels measured in untransfected cells. Ordinary one-way ANOVA shows that GLUT3 mRNA expression is significantly reduced in GLUT1-transfected and GLUT4-transfected cells relative to control cells (+++, p = 0.0001; ++, p = 0.0025). C, effect of heterologous GLUT1 or GLUT4 expression on parental GLUT3 expression. Results are shown for untransfected (UTF), GLUT1-transfected (G1), and GLUT4-transfected (G4) cells. Total GLUT3 and NaKATPase expression were assayed by obtaining whole cell lysates followed by SDS-PAGE of protein load-normalized samples and immunoblotting using protein-directed antibodies. Molecular mass markers are shown. D, quantitation of the effect of heterologous GLUT1 or GLUT4 expression on parental GLUT3 protein levels. Results (normalized to parental GLUT3 levels in untransfected cells as in Fig. 6 C) are shown for untransfected (Control), GLUT1-transfected (G1) and GLUT4-transfected (G3) cells. For each condition the symbols (●) show the results of four separate measurements, and the horizontal lines plus error bars show their mean ± S.E. Ordinary one-way ANOVA (+) shows that GLUT3 expression is significantly reduced in G1 and G4 cells relative to control cells (p = 0.0014). E, heterologous expression of GLUT1 and GLUT4 in HEK293 cells. Results are shown for untransfected (UTF), GLUT1-transfected (G1), and GLUT4-transfected (G4) cells. The GLUT1, GLUT4, and NaKATPase contents of protein load-normalized, whole cell lysates were assayed by immunoblot analysis using transporter specific (peptide-directed) antibodies (α-GLUT1, α-GLUT4, or α-NaKATPase IgGs) or with GLUT1 and GLUT4 by using α-myc IgGs. Molecular mass markers are shown. Heterologous expression of GLUT1 increased HEK293 cell GLUT1 expression over untransfected cells by 47.5- and 30.8-fold using-fold α-GLUT1 and α-myc IgGs, respectively. Heterologous expression of GLUT4 increased HEK293 cell GLUT4 expression over untransfected cells by 58.7- and 3.6-fold using α-GLUT4 and α-myc IgGs, respectively.
Heterologous expression of human GLUT1 or GLUT4 (Fig. 6E) significantly reduced endogenous GLUT3 mRNA (Fig. 6B) and protein (Fig. 6, C and D) levels in HEK293 cells as judged by qPCR and Western blotting analyses. This explains why high affinity, WZB117 inhibition of transport in GLUT4-transfected cells reduced transport more effectively than observed in untransfected cells (Fig. 6A). We hypothesize that endogenous GLUT3 expression is reduced in GLUT4- and GLUT1-transfected cells as a compensatory response to increased cellular sugar uptake.
Molecular Docking Studies
We undertook molecular docking studies of d-glucose, WZB117, and CB binding to a homology model of the exofacial (e2) open conformation of GLUT1 (hm-GLUT1-e2). We used the ligand-depleted chain A of the maltose-bound human GLUT3 (PDB code 4ZWC) structure (33) as the template to generate the homology model of the GLUT1-e2 (open) state. GLUT1 and GLUT3 share 65% sequence identity and 88% sequence similarity (10).
β-d-Glucose docks at three high affinity sites in hm-GLUT1-e2 (Fig. 7a; Fig. 8, a–f). We term these sites peripheral, intermediate, and core based upon their location within the exofacial, interstitium-exposed cavity of the GLUT1-e2 conformer. Eighteen β-d-Glc binding configurations were obtained at the core site. The affinity of binding (ΔG) ranged from −6 to −4 kcal/mol. Of these 18 possibilities, only 4 match the known stereospecificity of sugar inhibition of GLUT1-mediated l-sorbose entry in human erythrocytes (34). We illustrate one such complex in Fig. 8, e and f. β-d-Glc binding at this site is coordinated by hydrogen bonds to Gln161, Gln282, Asn317, and Glu380. Two additional β-d-Glc binding sites were detected in the exofacial cavity. β-d-Glc binding at the peripheral site (Fig. 8, a and b) and at the intermediate site (Fig. 8, c and d) presents fewer configurations, and in both instances C1 and its hydroxyl are solvent-exposed, whereas C6 and its hydroxyl are not. β-d-Glc binding at the intermediate site is coordinated by hydrogen bonds to Thr30, Asn34, Val69, Ser73, and Asn415. This orientation of sugar is incompatible with the stereochemistry of l-sorbose uptake inhibition described by Barnett et al. (34).
FIGURE 7.
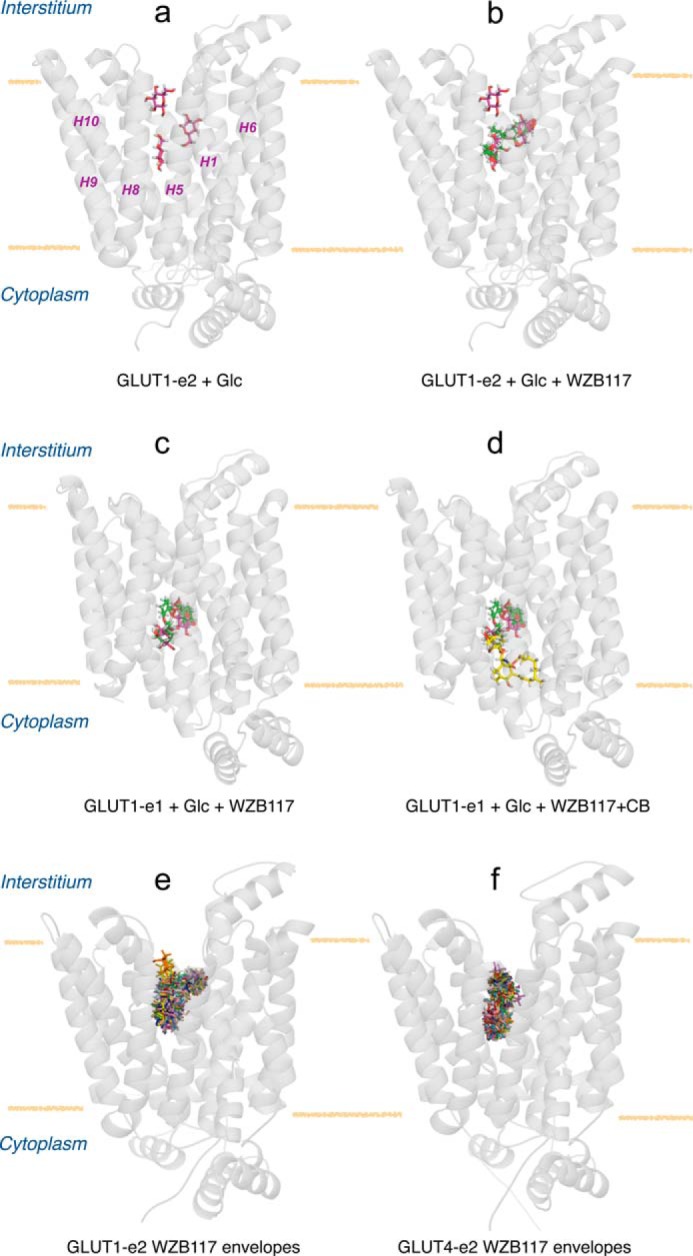
Molecular docking studies of ligand binding to hm-GLUT1 and hm-GLUT4. a, hm-GLUT1-e2 complexed with peripheral, intermediate, and core β-d-glucose molecules (red). GLUT1 is shown in transparent, schematic representation normal to the bilayer plane (horizontal yellow lines), membrane spanning α-helices (H1, H5, H6, H8, H9, and H10) are indicated, and locations of the interstitium and cytoplasm are highlighted. b, hm-GLUT1-e2 complexed with β-d-glucose molecules (red) and WZB117 (green). c, human GLUT1-e1 conformation complexed with intermediate and core β-d-glucose molecules (red) and WZB117 (green). d, human GLUT1-e1 conformation complexed with β-d-glucose molecules (red), WZB117 (green), and CB (yellow). e, hm-GLUT1-e2 conformation complexed with WZB117. The majority of computed binding sites (28 of 30) overlap with intermediate and core Glc binding sites. f, hm-GLUT4 e2 conformation complexed with WZB117. 30 potential binding sites are indicated; all overlap with peripheral and core Glc binding sites.
FIGURE 8.
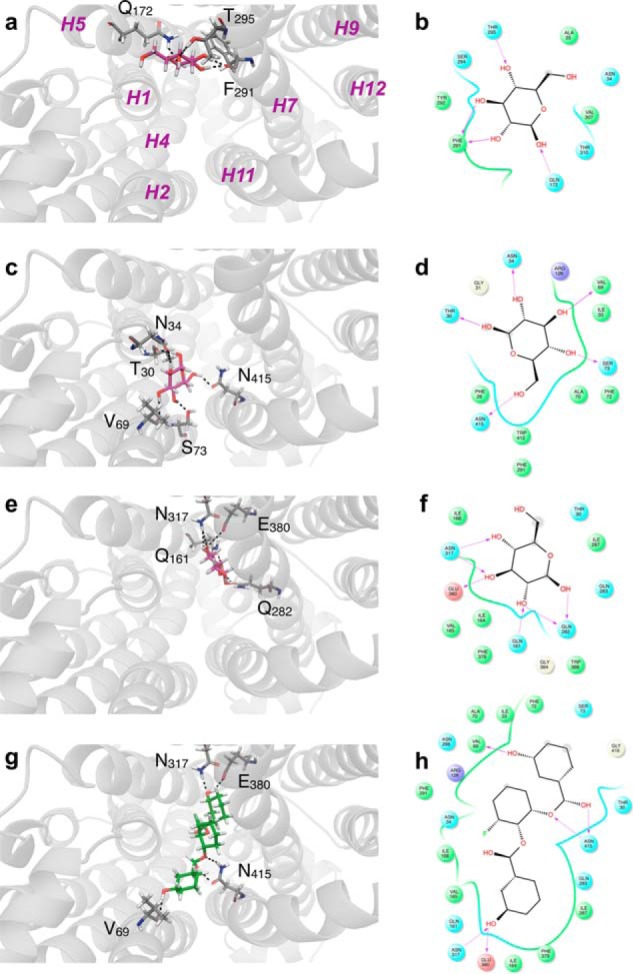
β-d-Glucose (a–f) and WZB117 (g and h) binding to hm-GLUT1-e2. The perspective is looking into the exofacial cavity of hm-GLUT1-e2 from the interstitium. The identities of several membrane spanning α helices (H1, H2, H4, H5, H7, H9, H11, and H12) are indicated in magenta in a. β-d-Glucose (in red) is shown docked to the peripheral (a and b), intermediate (c and d), and core (e and f) sites. Computed ΔG for ligand binding: peripheral, d-Glc binding site ΔG = −5.1 kcal/mol; intermediate d-Glc binding site ΔG = −5.1 kcal/mol; core binding site = −4.9 kcal/mol. WZB117 is shown in green (g), and its docking site overlaps with intermediate and core β-d-Glc binding sites. ΔG for WZB117 binding = −8.22 kcal/mol. Note that two additional configurations of WZB117-hm-GLUT1-e2 interactions were observed: one in which WZB117 interacts with and spans peripheral and core β-d-glucose binding sites (ΔG = −7.37 kcal/mol) and a second where WZB117interacts only with the peripheral and intermediate β-d-glucose binding sites (ΔG = −6.74 kcal/mol). Binding is shown as two representations: 1) three-dimensional in which GLUT1 is represented in transparent schematic format, Glc and WZB117 is in stick format, and H-bonds are represented as dashed lines (a, c, e, and g); 2) two-dimensional format in which Glc and WZB117 are shown as two-dimensional structures, coordinating GLUT1 residues are shown as circles, GLUT1 backbones are shown as ribbons, solvent-exposed regions of β-d-Glc and WZB117 are indicated by gray-shaded circles, and H-bonds and their directionality are represented as red arrows (b, d, f, and h).
WZB117 docking studies indicate that WZB117 can assume three positions in the exofacial cavity of hm-GLUT1-e2. When binding at its highest affinity site, WZB117 spanned the core and intermediate β-d-Glc binding sites (Fig. 7b and Fig. 8, g and h). Phenol ring 1 occupied the intermediate glucose binding site and was coordinated by hydrogen bonds to GLUT1 residues Val69 and Asn415. Phenol ring 2 occupied the core glucose binding site and was coordinated by hydrogen bonds to GLUT1 residues Asn317 and Glu380 (Fig. 8, g and h). The two lower affinity WZB117 binding sites comprise one in which WZB117 docks to and spans the peripheral and core d-Glc binding sites and a second where WZB117 interacted with the peripheral and intermediate d-Glc binding sites (not shown). CB did not dock at any high affinity sites in hm-GLUT1-e2 but did dock to the endofacial, GLUT1-e1 conformer (GLUT1-e1; Fig. 7d). The e1 GLUT1 conformation presented overlapping endofacial WZB117 and d-glucose sites that partially overlapped with the benzene ring of CB (Fig. 7, c and d).
Discussion
WZB117, a prototypic anticancer drug, inhibited human erythrocyte sugar transport by acting as a competitive inhibitor of sugar uptake and as a noncompetitive inhibitor of sugar exit. This suggests that WZB117 and extracellular glucose compete for binding to the same site on the erythrocyte sugar transporter. Molecular docking and ligand binding studies support this hypothesis by showing that extracellular d-glucose and WZB117 binding sites comprise overlapping amino side chains distinct from those involved in binding CB, an inhibitor that acts at the endofacial sugar binding site.
Sidedness of Inhibition
Several studies have demonstrated (regardless of the assumed carrier model) that reversible transport inhibitors binding at the exofacial sugar binding site of the passive glucose transporters serve as competitive inhibitors of net sugar uptake and equilibrium exchange transport and as noncompetitive inhibitors of net sugar exit (23–25). Conversely, reversible transport inhibitors binding at the endofacial sugar binding site are competitive inhibitors of net sugar exit and equilibrium exchange transport and are noncompetitive inhibitors of net sugar uptake (23–25). WZB117, therefore, fits the profile of a reversible inhibitor binding at the exofacial sugar binding site. CB behaves as a reversible inhibitor that binds at the endofacial sugar binding site, although the mixed-type inhibition of equilibrium exchange sugar transport and our molecular docking studies suggest that CB binding may involve more than simple interaction with the endofacial sugar binding site.
Determinants of Affinity and Isoform Specificity
Although several permutations of d-glucose and WZB117 binding sites were identified in GLUT1 docking studies, the highest affinity binding sites are consistent with the transport and ligand binding studies presented here and with previous studies of ligand binding to GLUT1 (12, 25, 30, 33, 35–37). Three exofacial d-glucose docking sites are present in the interstitium exposed exofacial cavity of hm-GLUT1-e2, peripheral, intermediate, and core. The stereochemistry of sugar binding at the peripheral and intermediate sites is incompatible with the findings of previous studies describing the stereochemistry of sugar transport inhibition by d-glucose analogs (34), whereas several configurations of sugar binding at the core site are compatible with the earlier studies. We, therefore, propose that the core d-glucose binding site may be the site at which hexose binding subsequently gives rise to sugar translocation.
WZB117 occupies three envelopes in GLUT1-e2. Envelope 1 comprises peripheral and core Glc binding sites, envelope 2 comprises intermediate and core Glc binding sites, and envelope 3 comprises peripheral and intermediate Glc sites. A majority (93%) of computed WZB117:GLUT1 complexes correspond to WZB117 binding within envelope 2. The remaining 7% are equally distributed between envelopes 1 and 3. WZB117 geometry is fully extended (linear) in envelope 1 but is “L” shaped in envelopes 2 and 3. Although each envelope would leave one available Glc binding site available for extracellular hexose, it is easy to see how Glc and WZB117 would compete for binding to GLUT1.
CB does not dock to hm-GLUT1-e2 but can be docked with high affinity to GLUT1-e1. d-Glucose and WZB117 also dock to GLUT1-e1 but bind at overlapping (and presumably mutually exclusive) sites deeper within the e1 cavity. These sites do overlap with the CB binding site, and the observed competitions between sugar, intracellular WZB117, and CB for binding to GLUT1 suggest that mutually exclusive binding could result from steric hindrance.
GLUT1 and GLUT3 are inhibited by WZB117 with Ki(app) of ∼5–10 μm. GLUT4, the insulin-sensitive glucose transporter of fat and muscle cells (38–40), is inhibited by WZB117 with a 30–100-fold greater potency.
The GLUT4-e2 conformer also presents peripheral, intermediate, and core Glc binding sites. However, the presence of Ile42 in GLUT4 transmembrane helix 1 (Thr30 in GLUT1 H1) leaves only linear envelope 1 (peripheral and core Glc sites) accessible to WZB117 (Fig. 7f). Molecular docking, therefore, suggests that WZB117 preferentially occupies envelope 2 in GLUT1-e2 and envelope 1 in GLUT4-e2. Km(app) for sugar transport and Ki(app) for transport inhibition are products of multiple steps in the catalytic cycle regardless of the presumed mechanism of transport (25, 41). It is not surprising, therefore, that WZB117 occupancy of different binding envelopes in two transporters characterized by very different transport kinetics (42) would result in significantly different Ki(app) for sugar transport inhibition.
Implications for Mechanism of Passive Sugar Transport
Four observations in the present study impact our understanding of glucose transport mechanism as follows. 1) As is the case for extracellular d-glucose (30), extracellular WZB117 does not affect CB binding to GLUT1. Hence, the transporter's exofacial sugar/WZB117 binding site and the endofacial CB binding site are not mutually exclusive. If CB binds only to the GLUT1-e1 conformer, which presents an endofacial sugar binding site, this behavior refutes the simple (alternating conformer) carrier model for sugar transport which predicts that inhibitor binding to the exofacial sugar binding site should competitively inhibit CB binding (43–45). 2) Extracellular WZB117 does not increase Ki(app) for CB noncompetitive inhibition of sugar uptake. Again, if CB binds only to the endofacial e1 conformer of GLUT1, this observation refutes the simple (alternating conformer) carrier model for sugar transport (43–45). 3) Like extracellular maltose and intracellular CB (27, 28), subsaturating extracellular WZB117 modestly stimulates sugar uptake. Transport stimulation by exofacial and endofacial transport inhibitors has been ascribed to cooperative interactions between co- existent exofacial and endofacial sugar binding sites present in the tetrameric GLUT1 transporter complex (27, 28). The transporter is proposed to comprise four subunits (GLUT1 proteins) in which each subunit behaves as an alternating conformer carrier but where intersubunit cooperativity results in two subunits presenting exofacial (e2) orientations and 2 subunits presenting endofacial (e1) conformations at any instant (46, 47). At subsaturating WZB117, there is a greater probability that only one of the two available GLUT1-e2 conformers is occupied by WZB117, and its occupancy is proposed to allosterically activate sugar uptake via the remaining, inhibitor-free e2 subunit (28). As WZB117 is further increased, the probability that inhibitor occupies both e2 subunits increases and leads to sugar uptake inhibition. An alternative hypothesis is that exofacial ligand-induced allostery is an intrasubunit phenomenon in which occupancy of the exofacial cavity by polyphenolic WZB117 increases GLUT1 affinity for, or transport of, the transported sugar through the same GLUT1 subunit (48). 4) Molecular docking studies reveal three exofacial d-glucose binding sites in the hm-GLUT1-e2 cavity, peripheral, intermediate, and core. This is consistent with previous observations that GLUT1 binds ≥2 mol of transported sugar per mol GLUT1 (49). Each envelope would leave one Glc binding site available for substrate. The results of this docking analysis, therefore, do not permit resolution of the mechanism of exofacial allostery (i.e. inter- or intrasubunit interactions).
Implications for Use as an Anticancer Agent
Although a promising strategy for cancer cell elimination, glucose transport inhibition using WZB117 could perturb organismal carbohydrate homeostasis in several important ways. Blood-brain and blood-tissue barrier GLUT1-dependent glucose transport will be depressed, perturbing peripheral and central neurometabolic function (50). Human, GLUT1-dependent, pancreatic beta cell insulin secretion in response to hyperglycemia will be diminished (51), and insulin-dependent, GLUT4-mediated skeletal muscle and adipose glucose transport will be prone to high affinity WZB117 inhibition thereby further compounding hyperglycemia. The net effects will include acute hyperglycemia and perturbations of neurometabolic function and insulin-dependent lipogenesis. These predictions are consistent with the reported hyperglycemia and lipodystrophy in mice after WZB117 treatment (9) and suggest that WZB117-treatment could induce metabolic syndrome/insulin resistance in individuals undergoing therapy.
The current study demonstrates that WZB117 inhibits facilitative sugar transport by competing with sugars for occupancy of the exofacial substrate binding site of the transporter. GLUT1, GLUT3, and GLUT4 are sensitive to transport inhibition, suggesting that sugar transport across the blood brain barrier in CNS astrocytes and neurons and in insulin-sensitive tissues is susceptible to inhibition (52). Most cancer cells rely on anaerobic glycolysis to generate the ATP and substrates required for cellular processes and proliferation (1). This contrasts with normal, differentiated cells, which mostly use mitochondrial oxidative phosphorylation to sustain cellular function (2). This could explain why sugar transport inhibition is a successful anti-cancer strategy in mice (9). However, human CNS development and function are especially dependent on glucose transport and metabolism (50), and the consequences of inhibition of glucose transport into and within the CNS could be catastrophic. We suggest, therefore, that a combination therapy of ketogenic diet (such as is used to manage GLUT1 deficiency; Refs. 4 and 53–55) plus sugar transport inhibition might be more successful.
Experimental Procedures
Reagents
([3H]Cytochalasin B and 3-O-[3H]methylglucose ([3H]3MG) were purchased from American Radiolabeled Chemicals (St. Louis, MO). Unlabeled 3MG, CB, and phloretin were purchased from Sigma. WZB117 was purchased from EMD Millipore (Billerica, MA).
Solutions
KCl medium was composed of 150 mm KCl, 5 mm HEPES, 4 mm EGTA, 5 mm MgCl2, pH 7.4. Solubilization buffer was composed of KCl medium with 0.5% Triton X-100. Phosphate-buffered saline was composed 140 mm NaCl, 10 mm Na2HPO4, 3.4 mm KCl, 1.84 mm KH2PO4, pH 7.3. Stop solution was composed of ice-cold KCl medium plus CB (CB; 10 μm) and phloretin (100 μm). Sample buffer contained 0.125 m Tris-HCl, pH 6.8, 4% SDS, 20% glycerol, and 50 mm DTT. Transfer buffer was composed of 12 mm Tris base, 96 mm glycine, 20% methanol.
Cells
De-identified whole human blood was purchased from Biological Specialty Corporation (Colmar, PA). HEK293 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin in a 37 °C humidified 5% CO2 incubator. Resealed, human red cell ghosts were made as described in Sage and Carruthers (56), and erythrocyte membranes were depleted of peripheral membrane proteins and resuspended in KCl medium at 4 mg protein/ml as described in Carruthers (57).
Heterologous Expression of GLUTs
GLUT1, GLUT3, and GLUT4 heterologous expression in HEK293 cells was as described previously (42, 58). Both GLUT1 and GLUT3 contain a myc-epitope in exofacial loop 1 (42). GLUT1, GLUT3, GLUT4, and NaKATPase expression in whole cell lysates was analyzed by Western blotting as previously described (42, 58).
Erythrocyte Sugar Transport Measurements
All human red cell sugar transport experiments were performed at 4 °C as previously described (15, 59, 60).
Zero-trans Uptake
Five volumes (100 μl) of 3MG uptake medium ± WZB117 or CB were added to 1 volume (20 μl) of sugar-depleted, 50% hematocrit (Ht) red cells, and sugar uptake allowed to proceed for 0.5–1 min at 4 °C. Uptake was stopped by adding 50 volumes (1 ml) of ice-cold stop solution containing 100 μm phloretin and 10 μm CB. Cells were washed in stop solution and lysed in 3% perchloric acid, and centrifugation-clarified lysates were assayed for radioactivity in duplicates using liquid scintillation counting.
Zero-trans Exit
Sugar-depleted packed RBCs were loaded with 10 mm 3MG by incubating with 20 mm 3MG (containing 10 μCi [3H]3MG/10 ml of cold 3MG for 1 h at 37 °C). One volume (0.5 ml) of sugar-loaded RBCs was added to 50 volumes of KCl medium ± CB or WBZ117 on a shaker with magnetic stirrer. Aliquots (0.5 ml) of the suspension were withdrawn at the indicated time intervals, washed in ice-cold stop solution, lysed, and assayed for remaining radioactivity.
Equilibrium Exchange Transport
Sugar-depleted RBCs were preloaded with 2.5–50 mm 3MG at 25 °C for 30 min. These cells were collected by centrifugation, incubated in 10 volumes of 3MG ± WZB117 or CB for 10 min at 4 °C, centrifuged again, and resuspended to 50% Ht in the appropriate 3MG and inhibitor medium. 3MG uptake medium (100 μl) ± WZB117 or CB was added to 20 μl of sugar-loaded cells (50% Ht), and the experiment was allowed to proceed for 0.5–1 min at 4 °C. Uptake was arrested by adding 50 volumes (1 ml) of ice-cold stop solution. Cells were centrifuged, and the pellet was washed in 1 ml of stop solution, collected by centrifugation, lysed in 3% perchloric acid, clarified, and assayed for radioactivity in duplicates using liquid scintillation counting.
HEK293 Cell Sugar Transport Measurements
All HEK293 cell sugar transport experiments were performed at 37 °C using 0.1 mm 2-deoxy-d-glucose as previously described (42, 58).
CB Binding
Equilibrium CB binding to red cells and to red cell membranes depleted of peripheral membrane proteins was performed as previously described (30). Briefly, 1 volume of sugar-depleted red cells (50% Ht) or red cell membranes (4 mg of membrane protein/ml) was suspended in one volume of ice-cold wash buffer 150 mm KCl, 5 mm HEPES, pH 7.4, containing 0.5 μm [3H]CB, 10 μm cytochalasin D, and varying concentrations of WZB117 (1–60 μm) for 15 min at 4 °C with constant end-over-end rotation. For total CB, 2 × 10 μl of the cell suspension were lysed in 100 μl of 3% perchloric acid and clarified by centrifugation, and the associated radioactivity was assayed by liquid scintillation counting. For free CB, the cell suspension was centrifuged at 10,000 × g for 30 s, and 10 μl of clarified supernatant were assayed in duplicate by scintillation counting. Bound CB was obtained as: total CB − free CB.
Quantitative RT-PCR
Expression levels of hGLUT3 mRNA were measured by qPCR using the QuaniTect SYBR Green PCR kit (Qiagen). Reactions were performed according to the manufacturer's protocol. Total RNA was isolated from HEK293 cells stably overexpressing hGLUT1 or hGLUT4 using the RNeasy kit and Qiashredder (Qiagen). cDNA was synthesized using the QuantiTect Reverse Transcriptase kit (Qiagen). qPCR reactions contained 100 ng of the indicated cDNA and 0.3 μm each human GLUT3-specific qPCR primers (G3, 5′-qF-AGCTCTCTGGGATCAATGCTGTGT and G3 3′qR-ATGGTGGCATAGATGGGCTCTTGA, where qF denotes qPCR forward and qR denotes qPCR reverse). Samples were run in triplicate on a Research PTC-200 Peltier Thermal Cycler with a Chromo4 real time PCR detector running Opticon Monitor 3 software (Bio-Rad). Results were analyzed by using the ΔΔCt method and normalized to an internal GAPDH marker.
Data Analysis
Linear and nonlinear regression analysis of data sets and statistical tests were accomplished using GraphPad Prism (Version 7.0a; La Jolla, CA).
Michaelis-Menten inhibition of sugar transport is assumed to be described by
| (Eq. 1) |
uptake, where vo is v measured in the absence of inhibitor I, [I] is the concentration of inhibitor and Ki(app) is that [I] producing 50% inhibition of uptake.
Michelis-Menten transport is assumed to be described by
| (Eq. 2) |
uptake, where Vmax is the maximum rate of 3MG transport, [3MG] is the concentration of 3MG, and Km(app) is that [3MG] where the rate of uptake is Vmax/2.
Hanes-Woolf analysis of transport assumes Michelis-Menten kinetics, and when data are plotted as [3MG]/v versus [3MG], the results fall on a straight line described by
| (Eq. 3) |
where Vmax is the maximum rate of 3MG transport, [3MG] is the concentration of 3MG, and Km(app) is [3MG], where the rate of uptake is Vmax/2.
Transport stimulation followed by inhibition by inhibitors was approximated first by normalizing all uptake to vo and then using the following model.
| (Eq. 4) |
Homology Modeling
The homology models of the e2 (open) state of GLUT1 and GLUT4 were generated using the maltose-bound human GLUT3 (PDB code 4ZWC) structure (33). GLUT1 and GLUT3 have 65% sequence identity and 88% sequence similarity (10). Ligands were removed, and chain A was used as the template for each modeled structure. Sequence alignments were generated using ClustalX. Homology models were built using Modeler-9.9 and analyzed using PROCHECK. The GLUT1-e1 structure (PDB code 4PYP; Ref. 61) was used directly.
Stochastic Docking
The GLUT1 crystal structure was obtained from the protein data bank using PDB code 4PYP. The structures for β-d-glucose and CB were obtained from ZINC. The WZB117 structure was generated using the three-dimensional structure generator Corina from Molecular Networks GmbH. Docking was performed using the Schrodinger software suite. The protein structure was preprocessed with the Protein Preparation Wizard, bond orders were assigned, hydrogens were added, and the H-bond network was optimized. The system was energy-minimized using the OPLS 2005 force field. Ligand structures were prepared with the LigPrep module, and the pKa of the ligands was calculated using the Epik module. Computational docking was performed by the GLIDE module in standard precision (SP) mode and default values for grid generation. Cavities for docking were calculated using the CastP server, and the grid was centered on the residues forming the cavity. No restraints were used during the docking.
Author Contributions
O. O. conducted most of the experiments, analyzed the results, and wrote most of the paper. K. L. undertook the molecular docking studies. A. S. assisted with the quantitative analysis of data. J. D. conducted the experiments on the isoform specificity of WZB117 inhibition of transport. A. C. conceived the idea for the project and co-wrote the paper with O. O.
This work was supported, in whole or in part, by National Institutes of Health Grants DK036081 and DK044888. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- GLUT
- glucose transport protein
- 2-DG
- 2-deoxy-d-glucose
- 3MG
- 3-O-methylglucose
- CB
- cytochalasin B
- WZB117
- (2-fluoro-6-(m-hydroxybenzoyloxy)phenyl m-hydroxybenzoate
- qPCR
- quantitative PCR
- e1
- endofacial
- e2
- exofacial
- Ht
- hematocrit
- ANOVA
- analysis of variance
- hm-GLUT1-e2
- homology model of the exofacial (e2) open conformation of GLUT1.
References
- 1. Vander Heiden M. G., Cantley L. C., and Thompson C. B. (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Palorini R., Cammarata F. P., Cammarata F., Balestrieri C., Monestiroli A., Vasso M., Gelfi C., Alberghina L., and Chiaradonna F. (2013) Glucose starvation induces cell death in K-ras-transformed cells by interfering with the hexosamine biosynthesis pathway and activating the unfolded protein response. Cell Death Dis. 4, e732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Warburg O. (1956) On the origin of cancer cells. Science 123, 309–314 [DOI] [PubMed] [Google Scholar]
- 4. De Giorgis V., and Veggiotti P. (2013) GLUT1 deficiency syndrome 2013: current state of the art. Seizure 22, 803–811 [DOI] [PubMed] [Google Scholar]
- 5. Pelicano H., Martin D. S., Xu R. H., and Huang P. (2006) Glycolysis inhibition for anticancer treatment. Oncogene 25, 4633–4646 [DOI] [PubMed] [Google Scholar]
- 6. Baracca A., Chiaradonna F., Sgarbi G., Solaini G., Alberghina L., and Lenaz G. (2010) Mitochondrial Complex I decrease is responsible for bioenergetic dysfunction in K-ras transformed cells. Biochim. Biophys. Acta 1797, 314–323 [DOI] [PubMed] [Google Scholar]
- 7. Chiaradonna F., Sacco E., Manzoni R., Giorgio M., Vanoni M., and Alberghina L. (2006) Ras-dependent carbon metabolism and transformation in mouse fibroblasts. Oncogene 25, 5391–5404 [DOI] [PubMed] [Google Scholar]
- 8. Xintaropoulou C., Ward C., Wise A., Marston H., Turnbull A., and Langdon S. (2015) A comparative analysis of inhibitors of the glycolysis pathway in breast and ovarian cancer cell line models. Oncotarget 6, 25677–25695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu Y., Cao Y., Zhang W., Bergmeier S., Qian Y., Akbar H., Colvin R., Ding J., Tong L., Wu S., Hines J., and Chen X. (2012) A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol Cancer Ther. 11, 1672–1682 [DOI] [PubMed] [Google Scholar]
- 10. Joost H. G., Bell G. I., Best J. D., Birnbaum M. J., Charron M. J., Chen Y. T., Doege H., James D. E., Lodish H. F., Moley K. H., Moley J. F., Mueckler M., Rogers S., Schürmann A., Seino S., and Thorens B. (2002) Nomenclature of the GLUT/SLC2A family of sugar/polyol transport facilitators. Am. J. Physiol. Endocrinol Metab 282, E974–E976 [DOI] [PubMed] [Google Scholar]
- 11. Ganapathy V., Thangaraju M., and Prasad P. D. (2009) k. Nutrient transporters in cancer: relevance to Warburg hypothesis and beyond. Pharmacol. Ther. 121, 29–40 [DOI] [PubMed] [Google Scholar]
- 12. Barnett J. E., Holman G. D., Chalkley R. A., and Munday K. A. (1975) Evidence for two asymmetric conformational states in the human erythrocyte sugar-transport system. Biochem. J. 145, 417–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Basketter D. A., and Widdas W. F. (1978) Asymmetry of the hexose transfer system in human erythrocytes: comparison of the effects of cytochalasin B, phloretin and maltose as competitive inhibitors. J. Physiol. 278, 389–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Devés R., and Krupka R. M. (1978) Cytochalasin B and the kinetics of inhibition of biological transport: a case of asymmetric binding to the glucose carrier. Biochim. Biophys. Acta 510, 339–348 [DOI] [PubMed] [Google Scholar]
- 15. Sage J. M., Cura A. J., Lloyd K. P., and Carruthers A. (2015) Caffeine inhibits glucose transport by binding at the GLUT1 nucleotide-binding site. Am. J. Physiol. Cell Physiol. 308, C827–C834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cunningham P., Afzal-Ahmed I., and Naftalin R. (2006) Docking studies show that d-glucose and quercetin slide through the transporter GLUT1. J. Biol. Chem. 281, 5797–5803 [DOI] [PubMed] [Google Scholar]
- 17. Naftalin R. J., Afzal I., Cunningham P., Halai M., Ross C., Salleh N., and Milligan S. R. (2003) Interactions of androgens, green tea catechins and the antiandrogen flutamide with the external glucose-binding site of the human erythrocyte glucose transporter GLUT1. Br. J. Pharmacol. 140, 487–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cairns R. A., Harris I. S., and Mak T. W. (2011) Regulation of cancer cell metabolism. Nat Rev Cancer 11, 85–95 [DOI] [PubMed] [Google Scholar]
- 19. Gambhir S. (2002) Molecular imaging of cancer with positron emission tomography. Nat. Rev. Cancer 2, 683–693 [DOI] [PubMed] [Google Scholar]
- 20. Shibuya K., Okada M., Suzuki S., Seino M., Seino S., Takeda H., and Kitanaka C. (2015) Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget 6, 651–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang W., Liu Y., Chen X., and Bergmeier S. (2010) Novel inhibitors of basal glucose transport as potential anticancer agents. Bioorg. Med. Chem. Lett. 20, 2191–2194 [DOI] [PubMed] [Google Scholar]
- 22. Mueckler M., Caruso C., Baldwin S. A., Panico M., Blench I., Morris H. R., Allard W. J., Lienhard G. E., and Lodish H. F. (1985) Sequence and structure of a human glucose transporter. Science 229, 941–945 [DOI] [PubMed] [Google Scholar]
- 23. Baker G. F., Basketter D. A., and Widdas W. F. (1978) Asymmetry of the hexose transfer system in human erythrocytes: experiments with non-transportable inhibitors. J. Physiol. 278, 377–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krupka R. M., and Devés R. (1981) An experimental test for cyclic versus linear transport models: the mechanisms of glucose and choline transport in erythrocytes. J. Biol. Chem. 256, 5410–5416 [PubMed] [Google Scholar]
- 25. Carruthers A., and Helgerson A. (1991) Inhibitions of sugar transport produced by ligands binding at opposite sides of the membrane: evidence for simultaneous occupation of the carrier by maltose and cytochalasin B. Biochemistry 30, 3907–3915 [DOI] [PubMed] [Google Scholar]
- 26. Segel I. H. (1975) Enzyme Kinetics, pp. 355–385, Wiley, New York [Google Scholar]
- 27. Cloherty E. K., Levine K. B., and Carruthers A. (2001) The red blood cell glucose transporter presents multiple, nucleotide-sensitive sugar exit sites. Biochemistry 40, 15549–15561 [DOI] [PubMed] [Google Scholar]
- 28. Hamill S., Cloherty E. K., and Carruthers A. (1999) The human erythrocyte sugar transporter presents two sugar import sites. Biochemistry 38, 16974–16983 [DOI] [PubMed] [Google Scholar]
- 29. Gorga F. R., and Lienhard G. E. (1982) Changes in the intrinsic fluorescence of the human erythrocyte monosaccharide transporter upon ligand binding. Biochemistry 21, 1905–1908 [DOI] [PubMed] [Google Scholar]
- 30. Helgerson A. L., and Carruthers A. (1987) Equilibrium ligand binding to the human erythrocyte sugar transporter: evidence for two sugar-binding sites per carrier. J. Biol. Chem. 262, 5464–5475 [PubMed] [Google Scholar]
- 31. Murata H., Hruz P. W., and Mueckler M. (2000) The mechanism of insulin resistance caused by HIV protease inhibitor therapy. J. Biol. Chem. 275, 20251–20254 [DOI] [PubMed] [Google Scholar]
- 32. Li Q., Manolescu A., Ritzel M., Yao S., Slugoski M., Young J. D., Chen X. Z., and Cheeseman C. I. (2004) Cloning and functional characterization of the human GLUT7 isoform SLC2A7 from the small intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 287, G236–G242 [DOI] [PubMed] [Google Scholar]
- 33. Deng D., Sun P., Yan C., Ke M., Jiang X., Xiong L., Ren W., Hirata K., Yamamoto M., Fan S., and Yan N. (2015) Molecular basis of ligand recognition and transport by glucose transporters. Nature 526, 391–396 [DOI] [PubMed] [Google Scholar]
- 34. Barnett J. E., Holman G. D., and Munday K. A. (1973) Structural requirements for binding to the sugar-transport system of the human erythrocyte. Biochem. J. 131, 211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Madej M. G., Sun L., Yan N., and Kaback H. R. (2014) Functional architecture of MFS D-glucose transporters. Proc. Natl. Acad. Sci. U.S.A. 111, E719–E727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cunningham P., and Naftalin R. (2013) Implications of aberrant temperature-sensitive glucose transport via the glucose transporter deficiency mutant (GLUT1DS) T295M for the alternate-access and fixed-site transport models. J. Membr. Biol. 246, 495–511 [DOI] [PubMed] [Google Scholar]
- 37. Manolescu A. R., Augustin R., Moley K., and Cheeseman C. (2007) A highly conserved hydrophobic motif in the exofacial vestibule of fructose transporting SLC2A proteins acts as a critical determinant of their substrate selectivity. Mol. Membr. Biol. 24, 455–463 [DOI] [PubMed] [Google Scholar]
- 38. James D. E., Brown R., Navarro J., and Pilch P. F. (1988) Insulin-regulatable tissues express a unique insulin-sensitive glucose transport protein. Nature 333, 183–185 [DOI] [PubMed] [Google Scholar]
- 39. Fukumoto H., Kayano T., Buse J. B., Edwards Y., Pilch P. F., Bell G. I., and Seino S. (1989) Cloning and characterization of the major insulin-responsive glucose transporter expressed in human skeletal muscle and other insulin-responsive tissues. J. Biol. Chem. 264, 7776–7779 [PubMed] [Google Scholar]
- 40. James D. E., Strube M., and Mueckler M. (1989) Molecular cloning and characterization of an insulin-regulatable glucose transporter. Nature 338, 83–87 [DOI] [PubMed] [Google Scholar]
- 41. Carruthers A. (1991) Mechanisms for the facilitated diffusion of substrates across cell membranes. Biochemistry 30, 3898–3906 [DOI] [PubMed] [Google Scholar]
- 42. Vollers S. S., and Carruthers A. (2012) Sequence Determinants of GLUT1-mediated accelerated-exchange transport - analysis by homology-scanning mutagenesis. J. Biol. Chem. 287, 42533–42544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Widdas W. F. (1952) Inability of diffusion to account for placental glucose transfer in the sheep and consideration of the kinetics of a possible carrier transfer. J. Physiol. 118, 23–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jardetzky O. (1966) Simple allosteric model for membrane pumps. Nature 211, 969–970 [DOI] [PubMed] [Google Scholar]
- 45. Lieb W. R., and Stein W. D. (1974) Testing and characterizing the simple carrier. Biochim. Biophys. Acta 373, 178–196 [DOI] [PubMed] [Google Scholar]
- 46. Graybill C., van Hoek A. N., Desai D., Carruthers A. M., and Carruthers A. (2006) Ultrastructure of human erythrocyte GLUT1. Biochemistry 45, 8096–8107 [DOI] [PubMed] [Google Scholar]
- 47. Zottola R. J., Cloherty E. K., Coderre P. E., Hansen A., Hebert D. N., and Carruthers A. (1995) Glucose transporter function is controlled by transporter oligomeric structure: a single, intramolecular disulfide promotes GLUT1 tetramerization. Biochemistry 34, 9734–9747 [DOI] [PubMed] [Google Scholar]
- 48. Cunningham P., and Naftalin R. (2014) Reptation-induced coalescence of tunnels and cavities in Escherichia coli XylE transporter conformers accounts for facilitated diffusion. J. Membr. Biol. 247, 1161–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Heard K. S., Fidyk N., and Carruthers A. (2000) ATP-dependent substrate occlusion by the human erythrocyte sugar transporter. Biochemistry 39, 3005–3014 [DOI] [PubMed] [Google Scholar]
- 50. Simpson I. A., Carruthers A., and Vannucci S. J. (2007) Supply and demand in cerebral energy metabolism: the role of nutrient transporters. J. Cereb. Blood Flow. Metab. 27, 1766–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Blodgett D. M., Nowosielska A., Afik S., Pechhold S., Cura A. J., Kennedy N. J., Kim S., Kucukural A., Davis R. J., Kent S. C., Greiner D. L., Garber M. G., Harlan D. M., and diIorio P. (2015) Novel observations from next-generation RNA sequencing of highly purified human adult and fetal islet cell subsets. Diabetes 64, 3172–3181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cura A. J., and Carruthers A. (2012) Role of monosaccharide transport proteins in carbohydrate assimilation, distribution, metabolism, and homeostasis. Compr. Physiol. 2, 863–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Brockmann K. (2009) The expanding phenotype of GLUT1-deficiency syndrome. Brain Dev. 31, 545–552 [DOI] [PubMed] [Google Scholar]
- 54. Klepper J. (2008) Glucose transporter deficiency syndrome (GLUT1DS) and the ketogenic diet. Epilepsia 49, 46–49 [DOI] [PubMed] [Google Scholar]
- 55. De Vivo D. C., Leary L., and Wang D. (2002) Glucose transporter 1 deficiency syndrome and other glycolytic defects. J. Child Neurol. 17, 3S15–3S23 [PubMed] [Google Scholar]
- 56. Sage J. M., and Carruthers A. (2014) Human erythrocytes transport dehydroascorbic acid and sugars using the same transporter complex. Am. J. Physiol. Cell Physiol. 306, C910–C917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Carruthers A. (1986) Anomalous asymmetric kinetics of human red cell hexose transfer: role of cytosolic adenosine 5′-triphosphate. Biochemistry 25, 3592–3602 [DOI] [PubMed] [Google Scholar]
- 58. De Zutter J. K., Levine K. B., Deng D., and Carruthers A. (2013) Sequence determinants of GLUT1 oligomerization: analysis by homology-scanning mutagenesis. J. Biol. Chem. 288, 20734–20744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Leitch J. M., and Carruthers A. (2009) α- and β-Monosaccharide transport in human erythrocytes. Am. J. Physiol. Cell Physiol. 296, C151–D161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cloherty E. K., Heard K. S., and Carruthers A. (1996) Human erythrocyte sugar transport is incompatible with available carrier models. Biochemistry 35, 10411–10421 [DOI] [PubMed] [Google Scholar]
- 61. Deng D., Xu C., Sun P., Wu J., Yan C., Hu M., and Yan N. (2014) Crystal structure of the human glucose transporter GLUT1. Nature 510, 121–125 [DOI] [PubMed] [Google Scholar]