

Abstract
Phosphatidylinositol 4-phosphate 5-kinase type I γ (PIPKIγ90) ubiquitination and subsequent degradation regulate focal adhesion assembly, cell migration, and invasion. However, it is unknown how upstream signals control PIPKIγ90 ubiquitination or degradation. Here we show that p70S6K1 (S6K1), a downstream target of mechanistic target of rapamycin (mTOR), phosphorylates PIPKIγ90 at Thr-553 and Ser-555 and that S6K1-mediated PIPKIγ90 phosphorylation is essential for cell migration and invasion. Moreover, PIPKIγ90 phosphorylation is required for the development of focal adhesions and invadopodia, key machineries for cell migration and invasion. Surprisingly, substitution of Thr-553 and Ser-555 with Ala promoted PIPKIγ90 ubiquitination but enhanced the stability of PIPKIγ90, and depletion of S6K1 also enhanced the stability of PIPKIγ90, indicating that PIPKIγ90 ubiquitination alone is insufficient for its degradation. These data suggest that S6K1-mediated PIPKIγ90 phosphorylation regulates cell migration and invasion by controlling PIPKIγ90 degradation.
Keywords: cell invasion, cell migration, phosphatidylinositol kinase, protein degradation, S6 kinase, PIP5K1C
Introduction
Cell migration and invasion are prerequisites for cancer metastasis (1, 2). Thus, the elucidation of the molecular mechanisms of cell migration and invasion is a compelling goal in cancer cell biology.
Phosphatidylinositol 4 phosphate 5-kinase type I γ (PIPKIγ90)2binds talin and localizes to focal adhesions (FAs) (3, 4). It catalyzes ATP-dependent phosphorylation of phosphatidylinositol 4-phosphate (PIP) to generate phosphatidylinositol 4,5-bisphosphate (PIP2), which binds and activates talin, vinculin, and focal adhesion kinase to mediate FA assembly (5, 6). PIP2 also binds many cytoskeletal proteins, such as neural Wiskott-Aldrich Syndrome protein, gelsolin, and profilin, to regulate actin polymerization (7–10). In addition, PIP2 is a precursor of several lipid second messengers, such as phosphatidylinositol 3,4,5-triphosphate (PIP3), inositol 1,4,5-triphosphate, and diacylglycerol. We have shown that depletion of PIPKIγ90 completely abolishes PIP3 production in HCT119 human colon cancer cells (11), indicating a critical role of PIPKIγ90 in lipid signaling. PIPKIγ90 is necessary for epithelial cell adherens junction assembly and progression through the E-cadherin-β-catenin signal pathway (12). PIPKIγ90 depletion inhibits cell proliferation, MMP9 secretion, and cell motility (13, 14).
PIPKIγ90 is essential for cell migration, invasion, and metastasis. It is required for focal adhesion assembly and disassembly, key steps in cell migration (11). Depletion of PIPKIγ90 inhibits growth factor-stimulated cell migration in MDA-MB-231 breast cancer cells and HeLa cervical cancer cells (14, 15). PIPKIγ90 knockdown also blocks the invasion of breast cancer and colon cancer cells (11, 16). Furthermore, PIPKIγ90-depleted 4T1 breast cancer cells show significant reduction in tumor progression and metastasis (13). PIPKIγ90 also regulates neutrophil migration by controlling cell polarity as well as rear retraction (17–19). PIPKIγ90 is a substrate for Src, which phosphorylates PIPKIγ90 at Tyr-644, enhancing its binding to talin and reducing talin-β integrin interaction (20). Talin, in turn, activates integrins and initiates FA assembly to regulate cell migration and invasion. In addition, phosphorylation of PIPKIγ90 at Tyr-639 by epidermal growth factor (EGF) receptor influences tumor cell migration and metastasis (13).
It has been demonstrated that the ubiquitin proteasome pathway regulates FA assembly and disassembly and, consequently, cell migration and invasion through ubiquitinating FA proteins (16, 21–26), and our research indicates that PIPKIγ90 is a key molecule that mediates the role of the ubiquitin proteasome pathway in this regard. Our published data indicate that PIPKIγ90 functions to regulate focal adhesion assembly and disassembly (11). We also demonstrated that PIPKIγ90 ubiquitination at Lys-97 by HECTD1, an E3 ubiquitin ligase that regulates cell migration, results in PIPKIγ90 degradation, thus controlling dynamic PIP2 production to mediate FA assembly/disassembly, cell migration, invasion, and metastasis (16). However, it is not clear how upstream signaling pathways control PIPKIγ90 ubiquitination or degradation during cell migration and invasion.
Ribosomal protein S6 kinase β 1 (also called p70S6K1 or S6K1), a serine-threonine kinase, is one of the mTOR pathway effectors. It is well known that S6K1 regulates cell growth, survival, and metabolism (27–31). Recent evidence indicates that it also regulates cancer cell invasion and metastasis (32, 33), but the molecular mechanisms behind these processes are less defined. In this study, we demonstrate that S6K1 phosphorylates PIPKIγ90 at Thr-553 and Ser-555 and that S6K1-mediated phosphorylation controls PIPKIγ90 degradation to regulate the development of FAs and invadopodia and, consequently, cell migration and invasion.
Results
The residues Thr-553 and Ser-555 of human PIPKIγ90 are consensus sites for Akt and S6K1 (Fig. 1A). To learn whether Akt1 and S6K1 phosphorylate PIPKIγ90, FLAG-PIPKIγ90 was co-transfected with an empty vector, constitutively active Akt1, and S6K1 (Myr-Akt1 and S6K1-F5A-E389-R3A) (34, 35). FLAG-PIPKIγ90 was immunoprecipitated, and PIPKIγ90 phosphorylation was detected with an anti-RXRXXpS/T motif antibody. Both Myr-Akt1 and S6K1-F5A-E389-R3A promoted PIPKIγ90 phosphorylation (Fig. 1B). To examine whether S6K1 phosphorylates PIPKIγ90 at Thr-553 and Ser-555 in vitro, HA-S6K1-F5A-E389-R3A was immunoprecipitated from CHO-K1 cells and incubated with purified recombinant GST-PIPKIγ90501–668, -PIPKIγ90501–668T553A, -PIPKIγ90501–668S555A, and -PIPKIγ90501–668T553A,S555A in a kinase reaction buffer containing ATP. The phosphorylation of these recombinant proteins was detected as described in Fig. 1B. Mutation at Thr-553 or Ser-555 caused a decrease in PIPKIγ90 phosphorylation, whereas mutation at both Thr-553 and Ser-555 abolished its phosphorylation (Fig. 1C). To determine whether S6K1 phosphorylates PIPKIγ90 at the same sites in cells, HA-S6K1-F5A-E389-R3A was co-transfected with FLAG-PIPKIγ90, PIPKIγ90T553A, PIPKIγ90S555A, and PIPKIγ90T553A,S555A into CHO-K1 cells. The phosphorylation of FLAG-PIPKIγ90 and the mutants was determined as described in Fig. 1B. Substitution of Thr-553 with Ala caused a significant reduction in PIPKIγ90 phosphorylation, and substitution of Ser-555 with Ala dramatically inhibited the phosphorylation, whereas substitution of both Thr-553 and Ser-555 completely abolished PIPKIγ90 phosphorylation (Fig. 1D). These data suggest that S6K1 phosphorylates PIPKIγ90 at residues Thr-553 and Ser-555.
FIGURE 1.

S6K1 phosphorylates PIPKIγ90 at Thr-553 and Ser-555. A, alignment of the Akt/S6K1 consensus sequences from different species. B, transfection of constitutively active Akt1 or S6K1 promoted PIPKIγ90 phosphorylation. FLAG-PIPKIγ90 was co-transfected with an empty vector, Myr-Akt1, and S6K1-F5A-E389-R3A into CHO-K1 cells. FLAG-PIPKIγ90 was immunoprecipitated, and PIPKIγ90 phosphorylation was detected with an anti-RXRXXpS/T motif antibody. a.u., arbitrary unit; *, p < 0.05. C, S6K1 phosphorylated PIPKIγ at Thr-553 and Ser-555 in vitro. Recombinant GST-PIPKIγ90501–668, -PIPKIγ90501–668T553A, -PIPKIγ90501–668S555A, and -PIPKIγ90501–668T553A,S555A were phosphorylated with constitutively active S6K1 that was immunoprecipitated from CHO-K1 cells. D, S6K1 phosphorylated PIPKIγ at Thr-553 and Ser-555 in CHO-K1 cells. HA-S6K1-F5A-E389-R3A was co-transfected with FLAG-PIPKIγ90, -PIPKIγ90T553A, -PIPKIγ90S555A, and -PIPKIγ90T553A,S555A into CHO-K1 cells. Data are presented as mean ± S.E. of three independent experiments. **, p < 0.01; ***, p < 0.001 versus WT. E, EGF and HGF stimulated PIPKIγ phosphorylation. MDA-MB-231 cells stably expressing FLAG-PIPKIγ90 were serum-starved and stimulated with EGF (20 ng/ml), HGF (50 ng/ml), SCF (20 ng/ml), and PDGF (20 ng/ml) for 20 min. FLAG-PIPKIγ90 was immunoprecipitated (IP), and phosphorylation was detected with an anti-RXRXXpS/T motif antibody. Data are presented as mean ± S.E. of three independent experiments. *, p < 0.05; **, p < 0.01 versus control (Ctrl). F, HGF-stimulated PIPKIγ phosphorylation was inhibited by Akt and S6K1 inhibitors. MDA-MB-231 cells stably expressing FLAG-PIPKIγ90 were serum-starved, treated with Akt inhibitor VIII and the S6K1 inhibitors DG2 (10 μm) or PF4708671 (10 μm), and then stimulated with HGF for 20 min. Data are presented as mean ± S.E. of four independent experiments. *, p < 0.05 versus HGF. G, HGF-stimulated PIPKIγ phosphorylation was suppressed by depletion of S6K1. S6K1 in MDA-MB-231 cells stably expressing FLAG-PIPKIγ90 was depleted using lentiviruses that express S6K1 shRNAs. Cell were serum-starved and then stimulated with HGF (20 ng/ml) for 20 min. Data are presented as mean ± S.E. of three independent experiments. *, p < 0.05.
To find out whether EGF or HGF stimulates PIPKIγ90 phosphorylation at residues Thr-553 and Ser-555, MDA-MB-231 cells stably expressing FLAG-PIPKIγ90 were serum-starved and stimulated with EGF, HGF, SCF, and PDGF. FLAG-PIPKIγ90 was immunoprecipitated with anti-FLAG-agarose beads, and PIPKIγ90 phosphorylation was detected with an anti-RXRXXpS/T motif antibody. EGF and HGF stimulated PIPKIγ90 phosphorylation, whereas SCF and PDGF did not (Fig. 1E). Similar results were observed in MDA-MB-468 cells (supplemental Fig. S1A). HGF and EGF stimulated Akt and S6K1 activation in a time-dependent manner, whereas SCF and PDGF had no obvious effects (Fig. 1E and supplemental Fig. S1, B and C). Because both S6K1 and Akt were activated by HGF or EGF in MDA-MB-231 cells, we tested whether S6K1 or Akt mediate PIPKIγ90 phosphorylation. MDA-MB-231 cells that stably express FLAG-PIPKIγ90 were treated with Akt inhibitor VIII or the S6K1 inhibitors DG2 and PF4708671 and then challenged with HGF. Akt inhibitor VIII inhibited HGF-stimulated Akt, S6K1, and PIPKIγ90 phosphorylation. The S6K1 inhibitors DG2 and PF4708671 did not influence Akt and S6K1 activation but inhibited S6K1 activity (as indicated by the reduction in ribosomal protein S6 phosphorylation) and PIPKIγ90 phosphorylation (Fig. 1F). To further examine whether S6K1 phosphorylates PIPKIγ90 in cells, MDA-MB-231 cells that stably express FLAG-PIPKIγ90 were infected with lentiviruses that express S6K1 shRNAs or empty vector. The resulted cells were stimulated with vehicle or HGF. S6K1 knockdown significantly inhibited HGF-induced PIPKIγ90 phosphorylation (Fig. 1G). These results indicate that PIPKIγ90 is a substrate for S6K1.
We showed previously that depletion of PIPKIγ90 using shRNA inhibited the migration of MDA-MB-231 cells and that re-expression of PIPKIγ90 restored the migration of PIPKIγ90-depleted cells (16). Based on these data, we decided to test the effect of phosphorylation site mutants PIPKIγ90T553A,S555A and PIPKIγ90T553E,S555E on cell migration. MDA-MB-231 cells that express PIPKIγ90 shRNA were infected with retroviruses expressing codon-modified ZZ-PIPKIγ90, -PIPKIγ90T553A,S555A, and -PIPKIγ90T553E,S555E (Fig. 2A), and cell migration was determined by time-lapse cell migration assays as described previously (16). As shown in Fig. 2B, cells that express PIPKIγ90T553A,S555A had a reduction in cell migration whereas those expressing PIPKIγ90 or PIPKIγ90T553E,S555E did not. Further analysis indicated that PIPKIγ90T553A,S555A inhibited cell migration by disrupting the directionality (Fig. 2C). This result implies that PIPKIγ90 phosphorylation regulates cell migration basically by modulating the directionality of the migrating cells.
FIGURE 2.

PIPKIγT553A,S555A inhibited cell migration. A, expression of ZZ-PIPKIγ, -PIPKIγT553A,S555A, or -PIPKIγT553E,S555E in PIPKIγ-depleted MDA-MB-231 cells. B, migration tracks of MDA-MB-231 cells that express a control (Ctrl) shRNA and PIPKIγ-depleted MDA-MB-231 cells that stably express ZZ-PIPKIγ, -PIPKIγT553A,S555A, or -PIPKIγT553E,S555E. C, statistic results of velocity, net distance, total path, and directionality of cells that express a control shRNA and PIPKIγ-depleted cells that stably express ZZ-PIPKIγ, -PIPKIγT553A,S555A, or -PIPKIγT553E,S555E. The data are expressed as mean ± S.E. of more than 40 cells from three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001. D, PIPKIγ-depleted MDA-MB-231 cells that stably express FLAG-PIPKIγ or -PIPKIγ90T553A,S555A were plated on fibronectin, fixed, and co-stained with anti-PIPKIγ and anti-paxillin antibodies. The images of PIPKIγ and paxillin were acquired using a TIRF microscope. E, the area distribution of paxillin at FAs in cells that stably express FLAG-PIPKIγ90WT or -PIPKIγT553A,S555A. Data are mean ± S.E. of three independent experiments. In each experiment, FAs of 20 cells from each group were analyzed and plotted. *, p < 0.05; ***, p < 0.001 versus WT.
Because PIPKIγ90 is a master regulator of FAs (11, 16), key machineries for cell migration, we examined whether the phosphorylation site mutant PIPKIγ90T553A,S555A influences FA formation. To this end, PIPKIγ90-depleted MDA-MB-231 cells that stably express FLAG-PIPKIγ90WT and -PIPKIγ90T553A,S555Awere plated on fibronectin, fixed, and co-stained with PIPKIγ90 and paxillin antibodies using PIPKI90-depleted cells as a control. FAs were viewed with a TIRF microscope. PIPKIγ90WT was co-localized with paxillin at FAs, whereas PIPKIγ90T553A,S555A was deficient in localizing to FAs (Fig. 2D). Cells expressing PIPKIγ90T553A, S555A had a significant reduction in FA formation in comparison with the WT (Fig. 2, D and E), suggesting that PIPKIγ90 phosphorylation may regulate cell migration through modulating FA assembly.
To assess the potential role of PIPKIγ90 phosphorylation in cancer cell invasion, the Matrigel-invasive capabilities of PIPKIγ90-depleted MDA-MB-231 cells that express ZZ-PIPKIγ90, ZZ-PIPKIγ90T553A,S555A, or ZZ-PIPKIγ90T553E,S555Ewere measured. Re-expression of PIPKIγ90WT in PIPKIγ90-depleted cells restored cell invasion to an extent comparable with the invasion of cells expressing empty pLKO.1 vector, and that of PIPKIγ90T553E, S555E partially rescued cell invasion. In contrast, re-expression of PIPKIγ90T553A, S555A only slightly enhanced cell invasion (Fig. 3, A and B). Similar results were observed when PIPKIγ90 and the mutants were expressed in parental MDA-MB-231 cells (supplemental Fig. S2), suggesting a dominant negative function of PIPKIγ90T553A,S555A. To explore the role of S6K1 in cell invasion, we examined the effect of the S6K1 inhibitor DG2 on the invasion of MDA-MB-231 cells. We found that S6K1 inhibition impaired invasion of the cells (Fig. 3C). In particular, 10 μm S6K1 inhibitor DG2 significantly decreased the invasive potential of the cells by ∼90% (in the absence of HGF) and 80% (in the presence of HGF). To further examine the requirement for S6K1 in cell invasion, this kinase was depleted in MDA-MB-231 cells using S6K1 shRNA (Fig. 3D). Cells transfected with S6K1 shRNA could not invade efficiently compared with cells expressing shRNA control (Fig. 3E). S6K1-depleted cells, even in the presence of HGF, could not invade normally compared with cells expressing shRNA control. Akt1, another protein kinase that potentially phosphorylates PIPKIγ90, was also depleted in MDA-MB-231 cells by using two different shRNAs. Depletion of Akt1 caused a slight reduction in the phosphorylation of S6K1 and S6 ribosomal protein (Fig. 3F). Depletion of Akt1 in MDA-MB-231 cells did not exhibit a significant reduction in invasive ability. As shown in Fig. 3G, depletion of Akt1 slightly reduced HGF-induced invasion of MDA-MB-231 cells. However, in the absence of HGF, cells expressing Akt1 shRNAs had higher number of invaded cells compared with cells with shRNA control, implying that Akt1 is not mandatory for the invasion of MDA-MB-231 cells. To further examine the role of S6K1-mediated PIPKIγ90 phosphorylation in cell invasion, the effects of the S6K1 inhibitor DG2 on the invasion of PIPKIγ-depleted cells that express ZZ-PIPKIγ90, -PIPKIγ90T553A,S555A, or -PIPKIγ90T553E,S555E were examined. DG2 significantly inhibited the invasion of cells expressing PIPKIγ90 but had only marginal effects on the invasion of cells expressing PIPKIγ90T553A,S555A or -PIPKIγ90T553E,S555E (Fig. 3H). These results indicate that S6K1-mediated PIPKIγ90 phosphorylation regulates cell invasion.
FIGURE 3.

S6K1-mediated PIPKIγ90 phosphorylation is essential for the invasion. A, PIPKIγ90 and PIPKIγ90T553E,S555E restored the invasive capacity of PIPKIγ-depleted cells but PIPKIγT553A,S555A did not. PIPKIγ-depleted MDA-MB-231 cells were infected with retroviruses that express codon-modified ZZ-PIPKIγ90, -PIPKIγ90T553A,S555A, or PIPKIγ90T553E,S555E and then selected with neomycin. Cells that express shRNA control (Ctrl) were used as controls. w/o, without. B, quantification of the experiment in A. White column, without HGF; gray column, 20 ng/ml HGF. Data are presented as mean ± S.E., n = 3. *, p < 0.05; **, p < 0.01 versus shRNA A1. C, inhibition of the invasion of MDA-MB-231 cells in the absence (white columns) and presence (gray columns) of HGF by the S6K1 inhibitor DG2. Data are mean ± S.E. of three independent experiments. **, p < 0.01. D, S6K1 and Akt activation in MDA-MB-231 cells expressing a control shRNA or S6K1 shRNAs. E, depletion of S6K1 by shRNA inhibited the invasion of MDA-MB-231 cells. Data are presented as mean ± S.E. of three independent experiments. **, p < 0.01; ***, p < 0.001. F, S6K1 and ribosomal protein S6 phosphorylation in MDA-MB-231 cells expressing a control shRNA or Akt1 shRNAs. G, Akt1 knockdown did not significantly affect the invasion of MDA-MB-231 cells. White columns, without HGF; pink columns, with HGF. The data are expressed as mean ± S.E. of three independent experiments. H, effects of the S6K1 inhibitor DG2 on the invasion of PIPKI-depleted MDA-MB-231 cells that express ZZ-PIPKIγ90, -PIPKIγ90T553A,S555A, or -PIPKIγ90T553E,S555E. Cell invasion was performed in the presence of DG2 (black columns, 10 μm) or vehicle (white columns) with HGF (20 ng/ml) in the lower chambers. Data are mean ± S.E. of three independent experiments. *, p < 0.05; ***, p < 0.001.
Because of the crucial role of matrix metalloproteinase-mediated matrix degradation in cell invasion (36–38), we set out to determine whether the S6K1-PIPKIγ90 pathway regulates matrix degradation. To examine whether the phosphorylation-deficient mutants of PIPKIγ90 influence matrix degradation, we examined the gelatin degradation activity of PIPKIγ90-depleted MDA-MB-231 cells that were rescued with PIPKIγ90WT, PIPKIγ90T553A,S555A, and PIPKIγ90T553E,S555E. Glass-bottom dishes were coated with Alexa 488-conjugated gelatin. The coated dishes were then dried, fixed with glutaraldehyde, and reduced with sodium borohydride. The cells were plated on dishes and treated with HGF. The cells were fixed and stained with cortactin, an invadopodium marker. Matrix degradation was examined by TIRF microscopy. Cells expressing PIPKIγ90WT had similar matrix degradation activity compared with cells expressing shRNA control. However, cells with PIPKIγ90T553A,S555A had significantly lower matrix degradation activity, whereas cells expressing PIPKIγ90T553E,S555E showed a slight reduction in degraded areas (Fig. 4, A and B). To further corroborate these findings, we tested the effect of S6K1 inhibition on matrix degradation. Similar to invasion, S6K1 inhibition affected this function and considerably decreased the gelatin degradation (Fig. 4C). These data suggest that S6K1-mediated PIPKI90 phosphorylation regulates matrix degradation.
FIGURE 4.

S6K1-mediated PIPKIγ phosphorylation is crucial for matrix degradation. A, effect of PIPKIγT553A,S555A and PIPKIγT553E,S555E on gelatin degradation in PIPKIγ-depleted cells. PIPKIγ-depleted MDA-MB-231 cells that express FLAG-PIPKIγ90, -PIPKIγ90T553A,S555A, or -PIPKIγ90T553E,S555E were resuspended in DMEM containing 1% FBS and HGF, plated on Alexa 488 gelatin-coated glass-bottom dishes, and cultured for 10 h. Scale bar = 20 μm. B, quantification of the experiment in A. Data are presented as mean ± S.E. of three independent experiments. *, p < 0.05; **, p < 0.01 versus shRNA control (Ctrl). AU, arbitrary unit. C, inhibition of invadopodium formation in MDA-MB-231 cells by the S6K1 inhibitor DG2. Data are presented as mean ± S.E. of three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus control.
To examine the possible association of the S6K1 pathway with cancer metastasis, human breast cancer tissue array slides, including primary tumors and the matched metastatic tumors of lymph node tissues (US Biomax), were stained for phospho-S6 ribosomal protein (Ser(P)-235/236), a substrate of S6K1. Among the tissues from 50 subjects analyzed, phospho-S6 staining was positive in 20 cases of metastatic tumors (40%) and in six cases of the matched primary tumors (12%) (Fig. 5, A and B). Also, phospho-S6 staining in 15 cases of metastatic tumors (30%) was significantly higher than the staining in the matched primary tumors; one case was lower (2%), and 34 cases were unchanged (68%). These data suggest that activation of the S6K1 pathway positively correlates with human breast cancer metastasis (p < 0.001).
FIGURE 5.
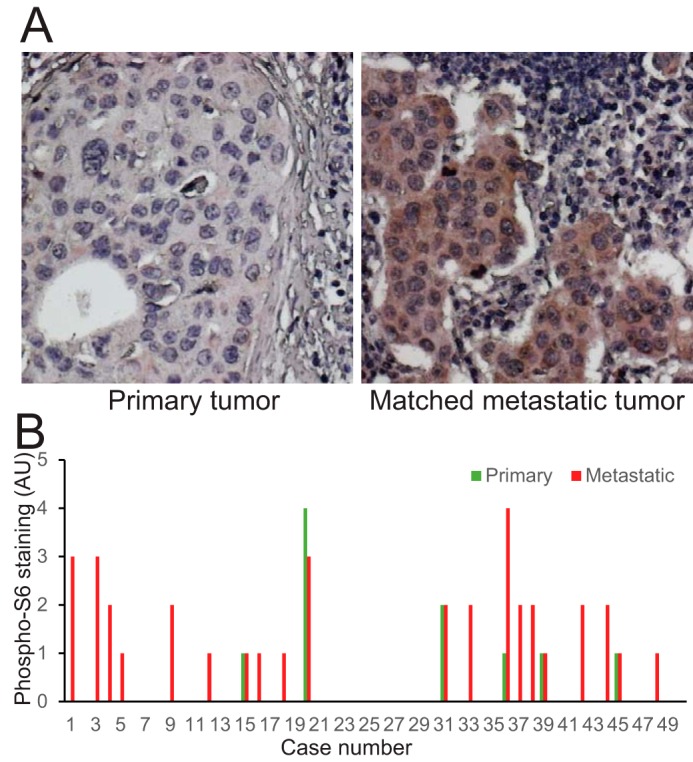
S6K1 activation correlates with breast cancer metastasis in human clinical specimens. A, human breast cancer primary tumors and the matched metastatic tumors of lymph node tissue were stained with anti-phospho-S6 ribosome protein antibody. B, the intensities of phospho-S6 staining were scored from 0–4, with 4 as the strongest. AU, arbitrary unit.
To measure the kinase activity of PIPKIγ90, ZZ-PIPKIγ90 was transfected into CHO-K1 cells and immunoprecipitated with IgG-conjugated-agarose beads or protein A-agarose using ZZ-PIPKIγ90K188,200R, a kinase-deficient mutant, as a negative control. The activities of PIPKIγ90 and mutants were measured by PIP2 production using PIP and [γ-32P]ATP as substrates. PI(4,5)P2 was separated by thin layer chromatography, imaged by autoradiography, and quantified by liquid scintillation counting. The kinase activity was detected in IgG-agarose beads that were incubated with ZZ-PIPKIγ90-transfected lysates but not in protein A-agarose beads incubated with the same lysate; very low activity was observed in IgG-agarose beads that were incubated with ZZ-PIPKIγ90K188,200R (supplemental Fig. S3A). To know whether mutation at Thr-553 and Ser-555 affects the activity of PIPKIγ90, ZZ-PIPKIγ90WT, -PIPKIγ90T553A,S555A, and -PIPKIγ90T553E,S555E were transfected into CHO-K1 cells and immunoprecipitated with IgG-agarose beads. The activities of PIPKIγ90 and mutants were measured using the same method. Substitution of Thr-553 and Ser-555 with alanine and glutamate did not affect PIPKIγ90 activity in vitro (supplemental Fig. S3B).
To determine whether PIPKIγ90 phosphorylation regulates its degradation, CHO-K1 cells were transfected with FLAG-PIPKIγ90WT, FLAG-PIPKIγ90T553A,S555A, and FLAG-PIPKIγ90T553E,S555E and treated with DMSO and carfilzomib, a specific proteasome inhibitor. As shown in Fig. 6A, PIPKIγ90T553A,S555A was not efficiently degraded and was more resistant to degradation than PIPKIγ90WT and PIPKIγ90T553E, S555E. To further confirm the stability of the T553A,S555A mutant, we determined the time course of PIPKIγ90 degradation. Avi-tagged PIPKIγ90WT and mutants were transfected into CHO-K1 cells with stable expression of BirA, and then labeled with biotin. Then, biotin was washed away and cells were split into dishes with media containing avidin. PIPKIγ90 and mutants were detected using Dylight 680 Streptavidin by harvesting the cells at different time points. PIPKIγ90T553A,S555A was more resistant to degradation in comparison to WT and PIPKIγ90T553E,S555E mutant (Fig. 6B) and had a significantly longer half-life than the WT and PIPKIγ90T553E,S555E (Fig. 6C).
FIGURE 6.

S6K1-mediated phosphorylation regulates PIPKIγ degradation. A, the steady-state levels of PIPKIγWT, PIPKIγT553A,S555A, and PIPKIγT553E,S555E in CHO-K1 cells that were transiently transfected with FLAG-PIPKIγWT, -PIPKIγT553A,S555A and -PIPKIγT553E,S555E, respectively, and treated with DMSO or carfilzomib (1 μm). B, substitution of Thr-553 and Ser-555 with Ala, but not Glu, inhibited degradation of PIPKIγ. CHO-K1 cells expressing BirA were transfected with Avi-PIPKIγWT, -PIPKIγT553A,S555A, and -PIPKIγT553E,S555E and labeled with biotin. The levels of PIPKIγ were detected by Western blotting using Dylight 680-streptavidin. C, time course of degradation of PIPKIγWT, PIPKIγT553A,S555A, and PIPKIγT553E,S555E in CHO-K1 cells. Data represent mean ± S.E. of three experiments. **, p < 0.01; ***, p < 0.001. D, CHO-K1 cells were transiently transfected with Dendra2-PIPKIγ90WT, -PIPKIγ90T553A,S555A, and -PIPKIγ90T553E,S555E and plated on fibronectin. The cells were irradiated for 2 min by a 408-nm laser to convert the Dendra2 fusion protein into red Dendra2 fusion protein. The intensities of the red fluorescence were recorded using time-lapse imaging. Scale bar = 20 μm. E, quantification of the degradation of Dendra2-PIPKIγ90WT, -PIPKIγ90T553A,S555A, and -PIPKIγ90T553E,S555E. Data are presented as mean ± S.E. of four independent experiments. F, the S6K1 inhibitor DG2 or PF4708671 stabilizes PIPKIγ90WT. CHO-K1 cells were transfected with Dendra2-PIPKIγ90WT. 24 h post-transfection, cells were treated with DG2 (10 μm) or PF4708671 (10 μm) for 30 min and then irradiated for 2 min using a 408-nm laser. Data are presented as mean ± S.E. of three experiments. G, the S6K1 inhibitor DG2 (10 μm) had little effect on the degradation of Dendra2-PIPKIγ90T553A,S555E. Data are presented as mean ± S.E. of three experiments.
To further demonstrate the role of S6K1-mediated PIPKIγ90 phosphorylation in PIPKIγ90 degradation, CHO-K1 cells were transfected with Dendra2-PIPKIγ90, -PIPKIγ90T553A,S555A, and -PIPKIγ90T553E,S555E and plated on fibronectin-coated glass-bottom dishes. The cells were irradiated by a 408-nm laser to convert the Dendra2 fusion protein into its red fluorescence form. The red fluorescence protein degradation was recorded by time-lapse imaging at 10-min intervals. Dendra2-PIPKIγT553A,S555A was more stable/resistant to degradation, with a half-life of >4 h, in comparison with the WT and T553E,S555E mutant of PIPKIγ90, which both showed a relatively higher rate of degradation, with half-lives of 2.5 and 3 h, respectively (Fig. 6, D and E). To examine the role of S6K1 in regulating PIPKIγ90 degradation, CHO-K1 cells that expressed Dendra2-PIPKIγ90 were treated with the S6K1 inhibitors DG2 (10 μm) or PF4708671 (10 μm), and the degradation of Dendra2-PIPKIγ90 was analyzed. As shown in Fig. 6F, S6K1 inhibition caused a significant increase in the stability of Dendra2-PIPKIγWT compared with the control. However, DG2 had no effect on the degradation of Dendra-PIPKIγ90T553E,S555E (Fig. 6G). These results further support the concept that S6K1-mediated phosphorylation of PIPKIγ90 facilitates its degradation.
This prompted us to examine the ubiquitination of PIPKIγ90 and these mutants. To this end, Avi-ubiquitin was co-transfected with ZZ-PIPKIγ90, -PIPKIγ90T553A,S555A, or -PIPKIγ90T553E,S555E into CHO-K1 cells expressing BirA, labeled with biotin, and immunoprecipitated with IgG-agarose. Ubiquitination was detected with Dylight 680 streptavidin. Substitution of Thr-553 and Ser-555 with Ala caused an increase in PIPKIγ90 ubiquitination, whereas substitution with Glu had no significant change compared with the WT protein (Fig. 7A), indicating that PIPKIγ90 ubiquitination is not sufficient for its degradation.
FIGURE 7.

PIPKIγ90 degradation is required for cancer cell-mediated matrix degradation. A, ubiquitination of PIPKIγWT, -PIPKIγT553A,S555A, and -PIPKIγT553E,S555E. Avi-ubiquitin (Avi-Ub) was co-transfected with ZZ-PIPKIγWT, -PIPKIγT553A,S555A, and -PIPKIγT553E,S555E into CHO-K1 cells expressing BirA. The cells were labeled with biotin, and the ZZ-tagged proteins were immunoprecipitated with IgG-agarose. The ubiquitination was detected using Dylight 680-streptavidin. Data are representative of two independent experiments. B, the steady-state levels of PIPKIγ in MDA-MB-231 cells that express empty pLKO.1 vector or S6K1 shRNAs, treated with DMSO or carfilzomib (Carf, 5 μm). Data are presented as mean ± S.E. of three independent experiments. *, p < 0.05. Ctrl, control. C, the steady-state levels of PIPKIγ in MDA-MB-231 cells that express empty pLKO.1 vector or Akt1 shRNA, treated with DMSO or bortezomib/carfilzomib (B+C, 1 μm each). Data are presented as mean ± S.E. of three independent experiments. D, the expression levels of PIPKIγ in MDA-MB-231 cells expressing a control shRNA or PIPKIγ shRNA A1 and the PIPKIγ-depleted cells that stably express ZZ-PIPKIγ and -PIPKIγK97R. E, PIPKIγWT restored gelatin degradation in PIPKIγ-depleted MDA-MB-231 cells but PIPKIγK97R, a ubiquitination-deficient mutant, did not. Scale bar = 20 μm. F, quantification of the experiment in E. Data are mean ± S.E. of three independent experiments. *, p < 0.05. AU, arbitrary unit.
To compare the roles of S6K1 and Akt1 in PIPKIγ90 degradation, we examined the steady-state levels of PIPKIγ90 in S6K1-depleted MDA-MB-231 cells. The level of PIPKIγ90 in S6K1-depleted cells was significantly higher than that in cells expressing a control shRNA (Fig. 7B). Treatment with carfilzomib resulted in a significant increase in PIPKIγ90 level in cells expressing control shRNA but not in S6K1-depleted cells. However, depletion of Akt1 by expressing its shRNA had no significant effect on the steady-state levels of PIPKIγ90 (Fig. 7C). These results suggest that S6K1-mediated phosphorylation facilitates PIPKIγ90 degradation.
Our previous published results indicate that PIPKIγ90 ubiquitination at lysine 97 and subsequent degradation are necessary for breast cancer cell invasion (16). To examine the role of PIPKIγ90 degradation in matrix degradation, we compared the matrix degradation activities of PIPKIγ90-depleted MDA-MB-231 cells that express codon-modified ZZ-PIPKIγ90 or ZZ-PIPKIγ90K97R using normal and PIPKIγ90-depleted MDA-MB-231 cells as controls (Fig. 7D). PIPKIγ90K97R is an ubiquitination- and degradation-resistant mutant. Depletion of PIPKIγ90 inhibited matrix degradation, and re-expression of PIPKIγ90 restored matrix degradation in PIPKIγ90-depleted cells whereas that of PIPKIγ90K97R did not (Fig. 7, E and F), further supporting the hypothesis that dynamic PIPKIγ90 degradation is essential for extracellular matrix degradation.
Discussion
The ubiquitin proteasome pathway regulates FA assembly and disassembly and, consequently, cell migration and invasion by ubiquitinating FA proteins (16, 21–26), and we recently demonstrated that PIPKIγ90 ubiquitination and subsequent degradation control FA dynamics to regulate cell migration and invasion (16). In this study, we demonstrated that S6K1-mediated PIPKIγ90 phosphorylation regulates PIPKIγ90 degradation to control the development of FAs and invadopodia and, consequently, cell migration and invasion.
We demonstrated that PIPKIγ90 is a substrate for S6K1. We showed that S6K1 phosphorylated PIPKIγ90 when they were co-transfected into CHO-K1 cells (Fig. 1B) and that substitution of the Thr-553 and Ser-555 sites with alanine abolished PIPKIγ90 phosphorylation by S6K1 in vitro and in cells (Fig. 1, C and D). We also revealed that PIPKIγ90 phosphorylation was stimulated by HGF and EGF and that HGF-stimulated phosphorylation was inhibited by the S6K1 inhibitors DG2 and PF4708671, Akt inhibitor VIII, as well as S6K1 knockdown (Fig. 1, E–G). The S6K1 inhibitors DG2 and PF4708671 caused 68% and 45% reduction in PIPKIγ90 phosphorylation in HGF-stimulated MDA-MB-231 cells, respectively. Akt inhibitor VIII suppressed 85% of PIPKIγ90 phosphorylation. The related higher efficiency of Akt1 inhibitor is probably due to its inhibition of both Akt and S6K1 activation. Thus, we estimated that S6K1 mediated approximately 50–70% of Thr-553 and Ser-555 phosphorylation in HGF-stimulated MDA-MB-231 cells. Endogenous PIPKIγ90 phosphorylation has not been examined because of reagent limitation. Nevertheless, these results indicate that PIPKIγ90 is a substrate for S6K1 in the system we used.
When we started writing this manuscript, Le et al. (39) reported that Akt1 phosphorylated PIPKIγ90 at Ser-555. Indeed, PIPKIγ90 was phosphorylated when it was co-transfected with Akt1 (Fig. 1B), and HGF-stimulated PIPKIγ90 phosphorylation was inhibited by Akt inhibitor VIII (Fig. 1F), suggesting that Akt1 is also a potential protein kinase that phosphorylates PIPKIγ90. However, depletion of Akt1 did not significantly inhibit the invasion of MDA-MB-231 cells (Fig. 3G). This result is consistent with previous reports showing that Akt activation potentially blocks carcinoma motility, including migration and invasion in breast cancer cells (40–43). Therefore, although both S6K1 and Akt1 phosphorylate PIPKIγ90, S6K1 is functionally more relevant than Akt1 in regulating PIPKIγ90 phosphorylation and cell invasion in breast cancer cells.
It is generally believed that protein polyubiquitination is sufficient for protein degradation (44, 45), but our findings indicate that PIPKIγ90 ubiquitination alone is insufficient for its degradation. The phosphorylation-deficient mutant PIPKIγ90T553A,S555A cannot be degraded efficiently compared with the WT and T553E,S555E mutant (Fig. 6, B–E). Moreover, the S6K inhibitors DG2 and PF4708671 inhibited the degradation of PIPKIγ90 but not that of PIPKIγ90T553E,S555E. However, substitution of Thr-553 and Ser-555 with alanine did not suppress but, instead, enhanced PIPKIγ90 ubiquitination (Fig. 7A). Our data show that PIPKIγ90 binds to 14-3-3 proteins, a family of adaptor proteins that regulate protein degradation (46–48), in a phosphorylation-dependent manner.3 However, although a role for this interaction with 14-3-3 proteins may be involved, it remains unknown how S6K1-mediated phosphorylation regulates PIPKIγ90 degradation.
The suppressive role of the phosphorylation-deficient mutant PIPKIγ90T553A,S555A in cell migration provides a new evidence for the role of PIPKIγ90 degradation in cell migration. Previous studies have demonstrated the essential role of PIPKIγ90 in the regulation of cell migration (14–16). Our recent study indicates that PIPKIγ90 ubiquitination by HECTD1 and subsequent degradation control FA dynamics and cell migration. Here we show that the phosphorylation-deficient mutant PIPKIγ90T553A,S555A was resistant to degradation and inhibited migration behavior by suppressing directionality and net distance from origin in comparison with PIPKIγ90WT and PIPKIγ90T553E, S555E (Fig. 2). Because of the central role of FAs in cell migration, the FA defect in cells expressing PIPKIγ90T553A,S555A may contribute to its inhibition of cell migration (Fig. 2, D and E). The effect of PIPKIγ90T553A,S555A on FA formation is probably caused by its enhanced stability, which interferes with talin binding to β integrins and integrin activation. This is consistent with our previous finding that PIPKIγ90K97R, a degradation-resistant mutant, had a diminished FA assembly rates (16).
As a downstream target of mTOR, the role of S6K1 in regulating cell growth, survival, and metabolism has been well documented, whereas its role in cancer cell invasion and the downstream targets that mediate this process remain to be defined. Previous studies have established a crucial role of PIPKIγ90 in cancer cell invasion (11, 14, 16). In this study, we demonstrated that S6K1-mediated PIPKIγ90 phosphorylation at Thr-553 and Ser-555 is indispensable for breast cancer cell invasion. PIPKIγ90T553A,S555A-expressing cells had a remarkably decreased capability to invade through Matrigel. On the other hand, cells expressing the WT and PIPKIγ90T553E,S555E mutant had similar invasive abilities (Fig. 3, A and B). This discrepancy may, in part, be due to the negative charge of the carboxyl group on the glutamate side chain, which could mimic the negative charge on a phosphorylated threonine/serine of PIPKIγ90. However, alanine with a neutral methyl side chain could not restore normal function of PIPKIγ90 in cell invasion. Inhibition of S6K1 by the S6K1 inhibitor DG2 or depletion of S6K1 using shRNAs considerably diminished the invasion of MDA-MB-231 cells (Fig. 3, C and E). Furthermore, inhibition of mTOR using rapamycin also inhibited cell invasion (49). However, depletion of Akt1 had a minimal effect on this function (Fig. 3G). Based on these findings and previous reports of the negative role of Akt1 in cell migration and invasion, we conclude that, although both S6K and Akt1 can phosphorylate PIPKIγ90, only S6K has a major positive role in regulating breast cancer cell invasion.
Matrix metalloproteinases-mediated matrix degradation is critical for cell invasion (36–38). However, the molecular mechanisms that regulate this process are not entirely understood. Our data show that PIPKIγ90T553A,S555A, a degradation-resistant mutant, had a significantly limited cellular ability to mediate gelatin degradation. In contrast, cells expressing the WT or PIPKIγ90T553E,S555E mutant had similar abilities to digest gelatin (Fig. 4, A and B). Moreover, PIPKIγ90K97R, which is an ubiquitination site mutant and is resistant to proteasome degradation, was unable to restore the matrix degradation in PIPKIγ90-depleted cells (Fig. 7, E and F). Furthermore, depletion of S6K1 by shRNA enhanced the stability of PIPKIγ90 (Fig. 7B) but significantly reduced the cellular capability to degrade the gelatin matrix (Fig. 4C). These data suggest that the S6K1-PIPKIγ90 pathway controls PIPKIγ90 degradation to regulate matrix degradation and cell invasion, probably through modulating the secretion of matrix metalloproteinases (13).
Spatial and temporary production of PIP2 is crucial for cell migration and invasion. This highly regulated PIP2 production is controlled by PIPKIγ90 ubiquitination and subsequent degradation. However, PIPKIγ90 ubiquitination alone is insufficient for its degradation; instead, the new data presented here show that S6K1-mediated PIPKIγ90 phosphorylation is also necessary for the degradation of ubiquitinated PIPKIγ90. S6K1 phosphorylates PIPKIγ90 at Thr-553 and Ser-555 to mediate the dynamic degradation of PIPKIγ90, thus controlling FA dynamics and matrix degradation and, consequently, cell migration and invasion. Our findings uncover a new paradigm for control of protein degradation, implying that a similar mechanism may also occur in other systems and processes.
Experimental Procedures
Reagents
IgG-agarose was described previously (50). The S6K1 inhibitor DG2 and anti-paxillin antibody (clone 5H11) were from Millipore. The S6K1 inhibitor PF4708671 was from ApexBio (Houston, TX). Akt inhibitor VIII was from Cayman Chemical Co. The anti-RXRXXpS/T motif antibody (23C8D2), anti-p70 S6 kinase antibody (49D7), anti-phospho-p70 S6 kinase (Thr(P)-389) antibody (9205), anti-S6 ribosomal protein antibody (5G10), anti-phospho-S6 ribosomal protein (Ser(P)-235/236) antibody (D57.2.2E), anti-Rsk2 antibody, and anti-phospho-Rsk2 and (Ser(P)-227) antibody were purchased from Cell Signaling Technology. The anti-PIPKIγ90 polyclonal antibody (MAO-R1), anti-Akt1 antibody (Tyr-89), and anti-phospho-Akt (Ser(P)-473) antibody (EP2109Y) were from Abcam. Anti-FLAG M2-agarose beads, anti-tubulin antibody, and pLKO1 lentivirus shRNAs that target PIPKIγ90, S6K1, and Akt1, respectively, were from Sigma. The PIPKIγ90 shRNA clone was TRCN0000037668 (A1). The S6K1 shRNA clones were TRCN0000003158 and TRCN0000003159. The Akt1 shRNA clones were TRCN0000010174 and TRCN0000039793. pBabe-Puro-Myr-FLAG-AKT1 was a gift from William Hahn (Addgene plasmid 15294). pRK7-HA-S6K1-F5A-E389 was a gift from John Blenis (Addgene plasmid 8988). DyLight 549 conjugated goat anti-mouse IgG (heavy+light chain) was from Thermo Scientific. Alexa 488-labeled gelatin and Alexa 647-phalloidin were from Life Technologies. Fibronectin was from Akron Biotech. HGF, EGF, PDGF, and SCF were from Prospec, Inc. Growth factor-reduced Matrigel was from BD Biosciences. Pfu Ultra was from Agilent Technologies. The Safectine RU50 transfection kit was purchased from Syd Labs (Malden, MA). DNA primers were synthesized by Integrated DNA Technologies.
Plasmid Construction
pZZ-PIPKIγ90 and the codon-modified plasmids pZZ-PIPKIγ90 and pBabe-ZZ PIPKIγ90 were described previously (16, 50). The codon-modified plasmids pZZ-PIPKIγ90T553A,S555A and -PIPKIγ90T553E,S555E were generated by Pfu Ultra-based PCR using the codon-modified pZZ-PIPKIγ90 as a template and 5′-cgg tac agg cgg cgc gca cag gcg gct gga cag gat ggc agg-3′/5′-cct gcc atc ctg tcc agc cgc ctg tgc gcg ccg cct gta ccg-3′ and 5′-cgg tac agg cgg cgc gaa cag gag tct gga cag gat ggc agg-3′/5′-cct gcc atc ctg tcc aga ctc ctg ttc gcg ccg cct gta ccg-3′ as primers, respectively. The codon-modified pBabe-ZZ-PIPKIγ90T553A,S555A and pBabe-ZZ-PIPKIγ90T553E,S555E were made by sequentially digesting the codon-modified pZZ-PIPKIγ90T553A,S555A and -PIPKIγ90T553E,S555E with Age1, blunting with Klenow, and digesting with Sal1. The smaller fragments were subcloned into the pBabe-neo vector that had been treated with BamH1, Klenow, and Sal1. pFLAG-PIPKIγ90 was generated by PCR amplifying PIPKIγ90 using pEGFP-PIPKIγ90 as a template and 5′-aat tat aga tct atg gag ctg gag gta ccg gac gag-3′/5′-ata tat gaa ttc tta tgt gtc gct ctc gcc gtc gga-3′ as primers. The PCR products were digested with BglII and EcoR1 and inserted into the pFLAG-C1 vector cut with the same enzymes. pFLAG-PIPKIγ90T553A, -PIPKIγ90S555A, and PIPKIγ90T553A,S555A were generated by Pfu Ultra-based PCR using pFLAG-PIPKIγ90 as a template and 5′-cgg tac agg cgg cgc gca cag tcg tct gga cag gat ggc agg-3′/5′-cct gcc atc ctg tcc aga cga ctg tgc gcg ccg cct gta ccg-3′, 5′-cgg tac agg cgg cgc aca cag gcg tct gga cag gat ggc agg-3′/5′-cct gcc atc ctg tcc aga cgc ctg tgt gcg ccg cct gta ccg-3′, and 5′-cgg tac agg cgg cgc gca cag tcg tct gga cag gat ggc agg-3′/5′-cct gcc atc ctg tcc aga cga ctg tgc gcg ccg cct gta ccg-3′ as primers, respectively. pDendra2-PIPKIγ90WT, -PIPKIγ90T553A,S555A, and -PIPKIγ90T553E,S555E were generated by digesting the fragments from pFLAG-PIPKIγ90 and the Thr-553 and Ser-555 mutants using BglII and EcoRI and subcloning into pDendra2 vectors. pGEX-4T-3-PIPKIγ90501–668, -PIPKIγ90501–668T553A, - PIPKIγ90501–668S555A, and PIPKIγ90501–668T553A,S555A were constructed by PCR-amplifying the fragments encoding residues 501–668 using primers 5′-aat ttg gat ccg agg acg aag gcc ggc c-3′/5′-ata tat gaa ttc tta tgt gtc gct ctc gcc gtc gga-3′ and templates pFLAG-PIPKIγ90, -PIPKIγ90T553A, - PIPKIγ90S555A, and PIPKIγ90T553A,S555A, respectively. The PCR products were digested with BamH1 and EcoR1 and inserted into the pGEX-4T-3 vector digested with the same enzymes. All plasmids were sequenced by Eurofins MWG Operon (Huntsville, AL).
Cell Culture and Transfection
CHO-K1 cells, MDA-MB-231 and MDA-MB-468 human breast cancer cells, and 293T human embryonic kidney cells were from the American Type Culture Collection and were maintained in DMEM (Sigma) containing 10% FBS, penicillin (100 units/ml), and streptomycin (100 μg/ml). CHO-K1 and 293T cells were transfected with Safectine RU50 according to the protocol of the manufacturer.
Preparation of Viruses and Cell Infection
293T cells were transfected with the pBabe retroviral or pLKO1 lentiviral system using Safectine RU50 transfection reagent according to the protocol of the manufacturer. The virus particles were applied to overnight cultures of breast cancer cells for infection. Cells that stably express pLKO1 lentiviral shRNAs were obtained by selecting the infected cells with 1 μg/ml puromycin, and cells that were infected with pBabe retroviruses were stabilized by growing infected cells in the presence of 0.7 mg/ml neomycin for 10 days.
PIPKIγ90 Phosphorylation
FLAG-PIPKIγ90 (or mutants) was co-transfected with an empty vector or a plasmid expressing active kinase into CHO-K1 cells. The cells were lysed with radioimmune precipitation assay buffer (50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1% IPEGAL, 0.5% deoxycholate, and 5 mm EDTA) containing protease inhibitor mixture and phosphatase inhibitor mixture. FLAG-PIPKIγ90 was immunoprecipitated with anti-FLAG-agarose beads. The immune complexes were analyzed by SDS-polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane. PIPKIγ90 phosphorylation was detected with an anti-RXRXXpS/T motif antibody. To detect PIPKIγ90 phosphorylation in breast cancer cells, cells stably expressing FLAG-PIPKIγ90 were treated with Akt or S6K1 inhibitor and then stimulated with growth factors. FLAG-PIPKIγ90 was immunoprecipitated, and PIPKIγ90 phosphorylation was detected as described above.
PIPKIγ90 Degradation
CHO-K1 cells stably expressing BirA were transfected with Avi-PIPKIγ90, Avi-PIPKIγ90T553A,S555A, and Avi-PIPKIγ90T553E,S555E. The cells were incubated with 500 μm biotin for 2 h, washed with PBS, and cultured in normal culture medium containing 200 μg/ml Avidin. The cells were lysed at different time points, and the levels of biotin-labeled PIPKIγ90 (or mutants) were detected with Dylight 680-streptavidin.
Live Cell Imaging and Dendra2-PIPKIγ90 Degradation
CHO-K1 cells were transiently transfected with Dendra2-PIPKIγWT, -PIPKIγT553A,S555A, and -PIPKIγT553E,S555E and cultured in fibronectin-coated glass-bottom dishes. Time-lapse live cell imaging was conducted on a Nikon A1 R microscope. Before excitation, there should not be any red Dendra2-emission signal visible. Photoconversion was performed at ×100 magnification with near-UV irradiation (408 nm) for 120 s. Green-to-red photoconversion was monitored in real time using a 561-nm channel. Images were captured at 20-min intervals and analyzed using NIS-Elements software.
Ubiquitination Assays
Avi-ubiquitin was co-transfected with ZZ-PIPKIγ90, -PIPKIγ90T553A,S555A, and -PIPKIγ90T553E,S555Eand co-transfected with an ubiquitin ligase or an empty vector into CHO-K1 cells stably expressing EGFP-BirA (50). 24 h post-transfection, cells were incubated with 500 μm biotin, 1 μm bortezomib, and 1 μm carfilzomib for 6 h and then scraped in PBS. The cells were spun down, lysed with 150 μl of 1× SDS sample buffer (without 2-mercaptoethanol) containing protease inhibitor mixture and bortezomib/carfilzomib and boiled immediately. The lysates were cleared, diluted to 1 ml, and incubated with rabbit IgG-Sepharose beads at 4 °C for 2 h to precipitate ZZ-tagged PIPKIγ90 (or the mutants). The beads were washed and analyzed by SDS-PAGE and Western blotting as above. The ubiquitination of the ZZ domain fusion protein was detected with Dylight 680-Streptavidin, whereas the expression of the ZZ domain fusion protein was probed with Dylight 680-rabbit IgG.
In Vitro PIPKIγ90 Activity Assays
PIPKIγ90 activity was measured as described previously (11). Briefly, pZZ-PIPKIγ90, pZZ-PIPKIγ90K188,200R, pZZ-PIPKIγ90T553A,S555A, and pZZ-PIPKIγ90T553E,S555E were transiently expressed in CHO-K1 cells and immunoprecipitated with IgG-agarose beads (50). The beads were washed and incubated with 100 μl of a kinase buffer containing 100 μm PI(4)P for 30 min at 37 °C. PIP2 formed in these assays was extracted as described previously (51) and separated by silicon TLC. PIP2 was visualized by autoradiography and quantitated by a Beckman liquid scintillation counter.
Cell Migration Assays
Cells were treated with trypsin and resuspended in DMEM containing 1% FBS and 10 ng/ml EGF, plated at low densities on glass-bottom dishes (Cellvis) coated with 5 μg/ml fibronectin, and cultured for 3 h in a CO2 incubator. Cell motility was measured with a Nikon Biostation IMQ. Cell migration was tracked for 6 h. Images were recorded every 10 min. The movement of individual cells was analyzed with NIS-Elements AR (Nikon) as described previously (16).
Focal Adhesion Staining
MDA-MB-231 cells were infected with lentiviruses that express PIPKIγ shRNA (A1) to deplete endogenous PIPKIγ, infected with retroviruses that express pBabe-FLAG-PIPKIγ90WT or FLAG-PIPKIγ90T553A,S555A, and selected with neomycin (0.7 mg/ml). The cells were trypsinized and plated on glass-bottom dishes that had been precoated with fibronectin (5 μg/ml). The cells were cultured for 4 h. The cells were fixed with 4% paraformaldehyde for 15 min, permeabilized for 15 min with 0.5% Triton X-100, and then blocked with 5% BSA in PBS for 1 h. The cells were then incubated with a rabbit polyclonal anti-PIPKIγ antibody and a mouse monoclonal anti-paxillin antibody, washed with PBS, and then incubated with a Dylight480-labeled goat anti-rabbit and a Dylight550-labeled goat anti-mouse secondary antibody. After washing with PBS, the images of PIPKIγ and paxillin were acquired with a Nikon Eclipse Ti TIRF microscope equipped with a ×60, 1.45 numerical aperture objective, CoolSNAP HQ2 charge-coupled device camera (Roper Scientific). Focal adhesion area distribution was analyzed with Nis-Elements.
Invasion Assays
One hundred microliters of Matrigel (1:30 dilution in serum-free DMEM) was added to each Transwell polycarbonate filter (6-mm diameter, 8-μm pore size, Costar) and incubated with the filters at 37 °C for 6 h. Breast cancer cells were trypsinized and washed three times with DMEM containing 1% FBS. The cells were resuspended in DMEM containing 1% FBS at a density of 5 × 105 cells/ml. The cell suspensions (100 μl) were seeded into the upper chambers, and 600 μl of DMEM containing 50 ng/ml HGF were added to the lower chambers. The cells were allowed to invade for 12 h (or as indicated) in a CO2 incubator, fixed, stained, and quantitated as described previously (11).
Gelatin Degradation Assays
Gelatin degradation assays were performed as described previously (52). Briefly, glass-bottom dishes were coated with warm Alexa 488-conjugated gelatin (0.2 mg/ml) in PBS containing 2% sucrose. The coated dishes were dried, fixed with prechilled glutaraldehyde solution (0.5%), washed with PBS, and then reduced with 5 mg/ml of sodium borohydride in PBS. The dishes were washed extensively with PBS and then incubated with DMEM containing 10% FBS and antibiotics for 1 h. Cells were plated at low density to the dishes and cultured for 12 h, fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100 and stained with cortactin or Alexa 647 phalloidin. Images were acquired using a TIRF microscope and analyzed with NIS Elements software.
Gel Data Quantification
Gel data were quantified by analyzing inverted images using ImageJ as described previously (21). Data from different experiments were normalized to controls. If values from different experiments had a high variation, then datasets were further normalized by dividing the numbers in a dataset with a factor (e.g. 2) so that the biggest values from different experiments were similar.
Author Contributions
N. J., Q. Z., L. L., W. L., L. Q., and J. X. performed experiments and data analysis. T. G. contributed reagents and participated in discussions. N. J. wrote the paper. C. H. directed the research, performed experiments, and wrote the paper.
Supplementary Material
Acknowledgments
We thank Dr. Andrew Morris for critical reading of the manuscript.
This work was supported by American Cancer Society Research Scholar Grant RSG-13-184-01-CSM (to C. H.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Figs. S1–S3.
N. Jafari, Q. Zheng, L. Li, W. Li, L. Qi, J. Xiao, T. Gao, and C. Huang, unpublished data.
- PIPKIγ90
- phosphatidylinositol 4-phosphate 5-kinase type I γ
- FA
- focal adhesion
- HGF
- hepatocyte growth factor
- PIP
- phosphatidylinositol 4-phosphate
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- PIP3
- phosphatidylinositol 3,4,5-triphosphate
- SCF
- stem cell factor
- mTOR
- mechanistic target of rapamycin
- S6K1
- p70S6K1
- TIRF
- total internal reflection fluorescence.
References
- 1. Locascio A., and Nieto M. A. (2001) Cell movements during vertebrate development: integrated tissue behaviour versus individual cell migration. Curr. Opin. Genet. Dev. 11, 464–469 [DOI] [PubMed] [Google Scholar]
- 2. Yamaguchi H., Wyckoff J., and Condeelis J. (2005) Cell migration in tumors. Curr. Opin. Cell Biol. 17, 559–564 [DOI] [PubMed] [Google Scholar]
- 3. Di Paolo G., Pellegrini L., Letinic K., Cestra G., Zoncu R., Voronov S., Chang S., Guo J., Wenk M. R., and De Camilli P. (2002) Recruitment and regulation of phosphatidylinositol phosphate kinase type 1 γ by the FERM domain of talin. Nature 420, 85–89 [DOI] [PubMed] [Google Scholar]
- 4. Ling K., Doughman R. L., Firestone A. J., Bunce M. W., and Anderson R. A. (2002) Type I γ phosphatidylinositol phosphate kinase targets and regulates focal adhesions. Nature 420, 89–93 [DOI] [PubMed] [Google Scholar]
- 5. Gilmore A. P., and Burridge K. (1996) Regulation of vinculin binding to talin and actin by phosphatidyl-inositol-4–5-bisphosphate. Nature 381, 531–535 [DOI] [PubMed] [Google Scholar]
- 6. Goñi G. M., Epifano C., Boskovic J., Camacho-Artacho M., Zhou J., Bronowska A., Martín M. T., Eck M. J., Kremer L., Gräter F., Gervasio F. L., Perez-Moreno M., and Lietha D. (2014) Phosphatidylinositol 4,5-bisphosphate triggers activation of focal adhesion kinase by inducing clustering and conformational changes. Proc. Natl. Acad. Sci. U.S.A. 111, E3177–E3186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Miki H., Miura K., and Takenawa T. (1996) N-WASP, a novel actin-depolymerizing protein, regulates the cortical cytoskeletal rearrangement in a PIP2-dependent manner downstream of tyrosine kinases. EMBO J. 15, 5326–5335 [PMC free article] [PubMed] [Google Scholar]
- 8. Rohatgi R., Ho H.-Y., and Kirschner M. W. (2000) Mechanism of N-Wasp activation by Cdc42 and phosphatidylinositol 4,5-bisphosphate. J. Cell Biol. 150, 1299–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Janmey P. A., and Stossel T. P. (1987) Modulation of gelsolin function by phosphatidylinositol 4,5-bisphosphate. Nature 325, 362–364 [DOI] [PubMed] [Google Scholar]
- 10. Lassing I., and Lindberg U. (1985) Specific interaction between phosphatidylinositol 4,5-bisphosphate and profilactin. Nature 314, 472–474 [DOI] [PubMed] [Google Scholar]
- 11. Wu Z., Li X., Sunkara M., Spearman H., Morris A. J., and Huang C. (2011) PIPKIγ Regulates focal adhesion dynamics and colon cancer cell invasion. PLoS ONE 6, e24775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ling K., Bairstow S. F., Carbonara C., Turbin D. A., Huntsman D. G., and Anderson R. A. (2007) Type Iγ phosphatidylinositol phosphate kinase modulates adherens junction and E-cadherin trafficking via a direct interaction with μ 1B adaptin. J. Cell Biol. 176, 343–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen C., Wang X., Xiong X., Liu Q., Huang Y., Xu Q., Hu J., Ge G., and Ling K. (2015) Targeting type Iγ phosphatidylinositol phosphate kinase inhibits breast cancer metastasis. Oncogene 34, 4635–4646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sun Y., Turbin D. A., Ling K., Thapa N., Leung S., Huntsman D. G., and Anderson R. A. (2010) Type I γ phosphatidylinositol phosphate kinase modulates invasion and proliferation and its expression correlates with poor prognosis in breast cancer. Breast Cancer Res. 12, R6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sun Y., Ling K., Wagoner M. P., and Anderson R. A. (2007) Type I γ phosphatidylinositol phosphate kinase is required for EGF-stimulated directional cell migration. J. Cell Biol. 178, 297–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li X., Zhou Q., Sunkara M., Kutys M. L., Wu Z., Rychahou P., Morris A. J., Zhu H., Evers B. M., and Huang C. (2013) Ubiquitylation of phosphatidylinositol 4-phosphate 5-kinase type I γ by HECTD1 regulates focal adhesion dynamics and cell migration. J. Cell Sci. 126, 2617–2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lokuta M. A., Senetar M. A., Bennin D. A., Nuzzi P. A., Chan K. T., Ott V. L., and Huttenlocher A. (2007) Type Iγ PIP kinase is a novel uropod component that regulates rear retraction during neutrophil chemotaxis. Mol. Biol. Cell 18, 5069–5080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xu W., Wang P., Petri B., Zhang Y., Tang W., Sun L., Kress H., Mann T., Shi Y., Kubes P., and Wu D. (2010) Integrin-induced PIP5K1C kinase polarization regulates neutrophil polarization, directionality, and in vivo infiltration. Immunity 33, 340–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tang W., Zhang Y., Xu W., Harden T. K., Sondek J., Sun L., Li L., and Wu D. (2011) A PLCβ/PI3Kγ-GSK3 signaling pathway regulates cofilin phosphatase slingshot2 and neutrophil polarization and chemotaxis. Dev. Cell 21, 1038–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ling K., Doughman R. L., Iyer V. V., Firestone A. J., Bairstow S. F., Mosher D. F., Schaller M. D., and Anderson R. A. (2003) Tyrosine phosphorylation of type Iγ phosphatidylinositol phosphate kinase by Src regulates an integrin-talin switch. J. Cell Biol. 163, 1339–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huang C., Rajfur Z., Yousefi N., Chen Z., Jacobson K., and Ginsberg M. H. (2009) Talin phosphorylation by Cdk5 regulates Smurf1-mediated talin head ubiquitylation and cell migration. Nat. Cell Biol. 11, 624–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huang C. (2010) Roles of E3 ubiquitin ligases in cell adhesion and migration. Cell Adh. Migr. 4, 10–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Deng S., and Huang C. (2014) E3 ubiquitin ligases in regulating stress fiber, lamellipodium, and focal adhesion dynamics. Cell Adh. Migr. 8, 49–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rafiq K., Guo J., Vlasenko L., Guo X., Kolpakov M. A., Sanjay A., Houser S. R., and Sabri A. (2012) c-Cbl ubiquitin ligase regulates focal adhesion protein turnover and myofibril degeneration induced by neutrophil protease cathepsin G. J. Biol. Chem. 287, 5327–5339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Iioka H., Iemura S., Natsume T., and Kinoshita N. (2007) Wnt signalling regulates paxillin ubiquitination essential for mesodermal cell motility. Nat. Cell Biol. 9, 813–821 [DOI] [PubMed] [Google Scholar]
- 26. Sekine Y., Tsuji S., Ikeda O., Sugiyma K., Oritani K., Shimoda K., Muromoto R., Ohbayashi N., Yoshimura A., and Matsuda T. (2007) Signal-transducing adaptor protein-2 regulates integrin-mediated T cell adhesion through protein degradation of focal adhesion kinase. J. Immunol. 179, 2397–2407 [DOI] [PubMed] [Google Scholar]
- 27. Fenton T. R., and Gout I. T. (2011) Functions and regulation of the 70 kDa ribosomal S6 kinases. Int. J. Biochem. Cell Biol. 43, 47–59 [DOI] [PubMed] [Google Scholar]
- 28. Han S., Khuri F. R., and Roman J. (2006) Fibronectin stimulates non–small cell lung carcinoma cell growth through activation of akt/mammalian target of rapamycin/S6 kinase and inactivation of LKB1/AMP-activated protein kinase signal pathways. Cancer Res. 66, 315–323 [DOI] [PubMed] [Google Scholar]
- 29. Shamji A. F., Nghiem P., and Schreiber S. L. (2003) Integration of growth factor and nutrient signaling: implications for cancer biology. Mol. Cell 12, 271–280 [DOI] [PubMed] [Google Scholar]
- 30. Shi Z.-M., Wang J., Yan Z., You Y.-P., Li C.-Y., Qian X., Yin Y., Zhao P., Wang Y.-Y., Wang X.-F., Li M.-N., Liu L.-Z., Liu N., and Jiang B.-H. (2012) MiR-128 inhibits tumor growth and angiogenesis by targeting p70S6K1. PLoS ONE 7, e32709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xu Q., Liu L.-Z., Qian X., Chen Q., Jiang Y., Li D., Lai L., and Jiang B.-H. (2012) MiR-145 directly targets p70S6K1 in cancer cells to inhibit tumor growth and angiogenesis. Nucleic Acids Res. 40, 761–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Khotskaya Y. B., Goverdhan A., Shen J., Ponz-Sarvise M., Chang S.-S., Hsu M.-C., Wei Y., Xia W., Yu D., and Hung M.-C. (2014) S6K1 promotes invasiveness of breast cancer cells in a model of metastasis of triple-negative breast cancer. Am. J. Transl. Res. 6, 361–376 [PMC free article] [PubMed] [Google Scholar]
- 33. Hsieh A. C., Liu Y., Edlind M. P., Ingolia N. T., Janes M. R., Sher A., Shi E. Y., Stumpf C. R., Christensen C., Bonham M. J., Wang S., Ren P., Martin M., Jessen K., Feldman M. E., et al. (2012) The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485, 55–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schalm S. S., and Blenis J. (2002) Identification of a conserved motif required for mTOR signaling. Curr. Biol. 12, 632–639 [DOI] [PubMed] [Google Scholar]
- 35. Boehm J. S., Zhao J. J., Yao J., Kim S. Y., Firestein R., Dunn I. F., Sjostrom S. K., Garraway L. A., Weremowicz S., Richardson A. L., Greulich H., Stewart C. J., Mulvey L. A., Shen R. R., Ambrogio L., et al. (2007) Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell 129, 1065–1079 [DOI] [PubMed] [Google Scholar]
- 36. Deryugina E. I., and Quigley J. P. (2006) Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 25, 9–34 [DOI] [PubMed] [Google Scholar]
- 37. Gialeli C., Theocharis A. D., and Karamanos N. K. (2011) Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 278, 16–27 [DOI] [PubMed] [Google Scholar]
- 38. Brown G. T., and Murray G. I. (2015) Current mechanistic insights into the roles of matrix metalloproteinases in tumour invasion and metastasis. J. Pathol. 237, 273–281 [DOI] [PubMed] [Google Scholar]
- 39. Le O. T., Cho O. Y., Tran M. H., Kim J. A., Chang S., Jou I., and Lee S. Y. (2015) Phosphorylation of phosphatidylinositol 4-phosphate 5-kinase γ by Akt regulates its interaction with talin and focal adhesion dynamics. Biochim. Biophys. Acta 1853, 2432–2443 [DOI] [PubMed] [Google Scholar]
- 40. Chin Y. R., and Toker A. (2010) The actin-bundling protein Palladin is an Akt1-specific substrate that regulates breast cancer cell migration. Mol. Cell 38, 333–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Irie H. Y., Pearline R. V., Grueneberg D., Hsia M., Ravichandran P., Kothari N., Natesan S., and Brugge J. S. (2005) Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial–mesenchymal transition. J. Cell Biol. 171, 1023–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu H., Radisky D. C., Nelson C. M., Zhang H., Fata J. E., Roth R. A., and Bissell M. J. (2006) Mechanism of Akt1 inhibition of breast cancer cell invasion reveals a protumorigenic role for TSC2. Proc. Natl. Acad. Sci. U.S.A. 103, 4134–4139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Toker A., and Yoeli-Lerner M. (2006) Akt signaling and cancer: surviving but not moving on. Cancer Res. 66, 3963–3966 [DOI] [PubMed] [Google Scholar]
- 44. Finley D., and Chau V. (1991) Ubiquitination. Annu. Rev. Cell Biol. 7, 25–69 [DOI] [PubMed] [Google Scholar]
- 45. Pickart C. M. (2000) Ubiquitin in chains. Trends Biochem. Sci. 25, 544–548 [DOI] [PubMed] [Google Scholar]
- 46. Muslin A. J., Tanner J. W., Allen P. M., and Shaw A. S. (1996) Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell 84, 889–897 [DOI] [PubMed] [Google Scholar]
- 47. Dar A., Wu D., Lee N., Shibata E., and Dutta A. (2014) 14-3-3 proteins play a role in the cell cycle by shielding Cdt2 from ubiquitin-mediated degradation. Mol. Cell Biol. 34, 4049–4061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Weiner H., and Kaiser W. M. (1999) 14-3-3 proteins control proteolysis of nitrate reductase in spinach leaves. FEBS Lett. 455, 75–78 [DOI] [PubMed] [Google Scholar]
- 49. Zheng Y., Rodrik V., Toschi A., Shi M., Hui L., Shen Y., and Foster D. A. (2006) Phospholipase D couples survival and migration signals in stress response of human cancer cells. J. Biol. Chem. 281, 15862–15868 [DOI] [PubMed] [Google Scholar]
- 50. Huang C., and Jacobson K. (2010) Detection of protein-protein interactions using nonimmune IgG and BirA-mediated biotinylation. BioTechniques 49, 881–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Honeyman T. W., Strohsnitter W., Scheid C. R., and Schimmel R. J. (1983) Phosphatidic acid and phosphatidylinositol labelling in adipose tissue: relationship to the metabolic effects of insulin and insulin-like agents. Biochem. J. 212, 489–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Qi L., Jafari N., Li X., Chen Z., Li L., Hytönen V. P., Goult B. T., Zhan C.-G., and Huang C. (2016) Talin2-mediated traction force drives matrix degradation and cell invasion. J. Cell Sci. 129, 3661–3674 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.