

Abstract
Progressive memory impairment such as that associated with depression, stroke, and Alzheimer's disease (AD) can interfere with daily life. In particular, AD, which is a progressive neurodegenerative disorder, prominently features a memory and learning impairment that is related to changes in acetylcholine and abnormal β-amyloid (Aβ) deposition in the brain. In the present study, we investigated the effects of dehydroevodiamine·HCl (DHED) on cognitive improvement and the related mechanism in memory-impaired rat models, namely, a scopolamine-induced amnesia model and a Aβ1-42-infused model. The cognitive effects of DHED were measured using a water maze test and a passive avoidance test in the memory-impaired rat models. The results demonstrate that DHED (10 mg/kg, p.o.) and Donepezil (1 mg/kg, p.o.) ameliorated the spatial memory impairment in the scopolamine-induced amnestic rats. Moreover, DHED significantly improved learning and memory in the Aβ1-42-infused rat model. Furthermore, the mechanism of these behavioral effects of DHED was investigated using a cell viability assay, reactive oxygen species (ROS) measurement, and intracellular calcium measurement in primary cortical neurons. DHED reduced neurotoxicity and the production of Aβ-induced ROS in primary cortical neurons. In addition, similar to the effect of MK801, DHED decreased intracellular calcium levels in primary cortical neurons. Our results suggest that DHED has strong protective effects against cognitive impairments through its antioxidant activity and inhibition of neurotoxicity and intracellular calcium. Thus, DHED may be an important therapeutic agent for memory-impaired symptoms.
Keywords: Alzheimer's disease, β-Amyloid, Cognitive function, Dehydroevodiamine, Scopolamine
INTRODUCTION
Memory loss that occurs gradually with aging is considered to be a normal process; however, progressive memory impairment such as that related to depression, stroke, and Alzheimer's disease (AD) can adversely interfere with daily life [1,2]. Specifically, AD, which is a progressive neurodegenerative disorder, is associated with pathogenic damage in the neocortex and hippocampus, and is therefore involves a global cognitive impairment that includes deficits in memory, orientation, judgment, and reasoning [3]. The pathology of AD also features abnormal β-amyloid (Aβ) deposits that form neuritic plaques and neurofibrillary tangles. The neuropathological changes in AD include cortical atrophy, neuronal and volume loss, disruption of neuronal pathways, and impairment of the cholinergic system [4,5]. The neurotoxic Aβ aggregates are the main causative factor of synaptic loss through oxidative stress and are closely related to the changes in memory and cognition. Furthermore, abnormal Aβ metabolism is related to an upregulation of calcium signaling and results in memory loss [6,7,8]. In addition to Aβ, acetylcholine (ACh) plays an essential role in proper cognitive function. An ACh deficit is associated with cognitive deficits in AD. Therefore, augmentation of cholinergic neurotransmission using acetylcholinesterase (AChE) inhibitors may be an important therapeutic approach to AD [9].
Animal models have been used to effectively investigate the pathophysiological mechanism of memory impairment [10]. For example, scopolamine-induced amnesic rats have been used to identify agents that have cognitive-enhancing activities [11]. Scopolamine acts as a competitive antagonist at muscarinic ACh receptors in the cholinergic system and disrupts cognitive functions such as learning and memory. In an Aβ-infused rat model, working memory and cognition are disrupted due to an increase in reactive oxygen species (ROS) and lipid peroxide in the cortex and hippocampus [12]. Aβ injection into the brain modulates synaptic plasticity and impairs memory formation and cognitive function [6]. Notably, some therapeutic candidates have been reported to alleviate memory deficits in an Aβ1-42-infused model [13,14,15].
Dehydroevodiamine·HCl (DHED), which is purified from Evodia rutaecarpa Bentham, can inhibit AChE [16,17]. Antiamnestic effects of DHED occur through an improvement of learning and memory and an inhibition of neuronal dysfunction in a scopolamine-induced amnesic rat model [18,19]. Furthermore, DHED has hypotensive and neuroprotective effects and modulates nitric oxide production [20,21]. Despite these well-studied effects of DHED on cognition, the underlying mechanism of these effects of DHED still remains unknown.
In the present study, we investigated the cognitive improvement and underlying mechanism that is related to DHED activity in 2 memory-impaired rat models, namely, a scopolamine-induced amnesia model and a Aβ1-42-infused model. The mechanism of DHED activity was elucidated using a cell viability assay, ROS measurement, and intracellular calcium measurement in primary cortical neurons.
METHODS
Chemicals and reagents
Dehydroevodiamine·HCl (DHED, Indolo[2',3':3,4] pyrido[2,1-b]quinazolinium-5,7,8,13-tetrahydro-14-methyl-5-oxo-chloride, CAS 67909-49-3) was synthesized and supplied by the Jeil Pharmaceutical Company (Seoul, Korea). DHED was suspended in 0.3% carboxymethylcellulose (CMC) solution for oral administration to rats, and dissolved in dimethylsulfoxide (DMSO) and diluted with phosphate-buffered saline (PBS) for application to the cell cultures and intraperitoneal injection into rats. The Aβ1-42 peptide was purchased from US Peptide (Rancho Cucamonga, CA) and incubated in 0.1 M PBS (pH=7.4) at 37℃ for 7 days for aging. All drugs were prepared immediately before use.
Animal preparation
Wistar rats (7-week-old) were housed in groups of 2 or 3 in clear Plexiglas cages containing sawdust. Animals were maintained on a 12-h light/dark cycle (light on at 8:00 a.m.), and given food and water ad libitum. Behavioral experiments were carried out between 10:00 a.m. and 6:00 p.m. in a soundproof laboratory. All experiments were performed in accordance with the Guidelines for Animal Experiments of the Ethics Committee of the Seoul National University.
Scopolamine-induced amnesia rat model
We used an established scopolamine-induced amnesia rat model [18,22]. This model was tested in a water maze for 6 days. The testing procedure is described below in the description of the water maze test. For training, each animal received 2 trials per day for 5 consecutive days and was allowed to swim for 90 sec. Rats were given scopolamine (1 mg/kg, i.p.) at 1 h before the first test. A single dose of DHED (10 mg/kg, p.o.) or Donepezil (1 mg/kg, p.o.) was given to rats 30 min after scopolamine treatment. The second and third test was conducted 4 h and 24 h after scopolamine treatment, respectively. DHED or Donepezil was suspended in 0.3% CMC solution using an electric homogenizer to inject rats orally.
Aβ1-42 infused rat model
The surgical techniques that we used have been described previously [23,24,25,26]. A solvent of 35% acetonitrile plus 0.1% trifluoroacetic acid was used as the vehicle for the Aβ1-42 peptide (Aβ1-42, US peptide, Rancho Cucamonga, CA). For intracerebroventricular (ICV) infusion, rats were stereotaxically implanted with an osmotic pump (Alzet model 1007D, Alzet Co., CA) for continuous infusion of Aβ1-42. This procedure strongly improved the reproducibility and reliability in yielding an animal model of AD with impaired memory. The osmotic pump contained 90 µL of Aβ1-42 solution. The infusion rate via an infusion cannula (stainless steel, 0.35 mm in diameter) was 0.5 µL/h, and the total amount infused was approximately 4.2 nM of Aβ1-42. The rats, except for those in the control group, were lightly anesthetized with sodium pentobarbital (50 mg/kg BW, i.p.). The skull was exposed and a hole (right, relative to bregma; 0.8 posterior, ??.4 lateral, and +2.6 mm below the cortical surface) was drilled according to the atlas of Paxinos and Watson (1986) using a stereotaxic frame. The guide cannula was secured with dental cement and stainless steel skull screws. DHED was suspended in 0.3% CMC solution using an electric homogenizer to inject rats orally. During infusion, rats were injected with DHED (10 mg/kg, p.o.) for 21 days.
Water maze test
The water maze test was carried out to evaluate the effects of DHED on spatial memory. The testing procedure is described in Yamada et al. [27]. The experimental apparatus consisted of a circular water tank (140 cm in diameter and 45 cm high). An invisible platform (15 cm in diameter and 35 cm high) was placed 1.5 cm below the surface of the water. The water temperature was 21~23℃. The pool maze was located in a large test room with many external clues (e.g. pictures, lamps, etc.). These cues were visible from the pool maze for animals to use for spatial orientation. The position of the cues remained unchanged throughout the task. Data collection was automated using a video image motion analyzer (Ethovision, Noldus Information Technology h.v., Netherlands). For descriptive data collection, the pool was subdivided into 4 equal quadrants that were formed using imaging lines. For the reference memory test, each animal received 4 trials per day for 5 consecutive days. At the start of a trial the animal was placed randomly at 1 of 4 fixed starting points facing the wall (designated North, South, East, and West) and allowed to swim for 60 sec or until it escaped the task by finding the platform. The platform was located in the middle of one quadrant and equidistant from the center and edge of the pool throughout the test period. In each training session, the latency to escape onto the hidden platform was recorded. If the animal found the platform then it was allowed to remain there for 15 sec before returning to its home cage. If the animal was unable to find the platform within 60 sec then the training was terminated and a maximum score of 60 sec was assigned. After 48 h of the final trial, the animals received a spatial probe trial, where the platform was removed from the tank and the animals were allowed to swim for 60 sec. We measured the swim time and path length spent in the target quadrant where the platform was located during the training.
Passive avoidance test
A step-through type passive avoidance test apparatus (Model PACS-30, Columbus Instruments Int., USA) that is described in Shen et al. [28] was used to evaluate the effects of DHED on learning and memory. The shuttle box was divided into 2 chambers of equal size (23.5×15.5×15.5 cm) and separated using a guillotine door (6.5×4.5 cm). The light chamber was equipped with an illuminator, and animals can enter the dark chamber through the guillotine door. Animals were initially placed in the light chamber with the door open. Animals displayed an explorative behavior and then entered the dark compartment. Upon entering the dark compartment, the door was closed automatically. Training was repeated until the animals entered the dark compartment within 20 sec (training trial). After the training trial, the rats were placed in the illuminated chamber. When the rat entered the dark chamber an electrical foot shock (0.5 mA) was delivered for 3 sec through the grid floor and the door was closed automatically (acquisition trial). The animals were placed in the light chamber again at 24 h after the acquisition trial, and the latency time to enter the dark chamber was measured for 300 sec (retention trial). If an animal did not enter the dark chamber within the cut-off time (300 sec) then it was assigned a latency value of 300 sec.
Primary cell culture
For primary neuronal culture, the cerebral cortex was dissected from embryonic day 18 Sprague-Dawley (SD) rat embryos and dissociated using gentle trituration. Cells were plated in 35-mm dishes that were coated with polyethylene (0.2 mg/ml in sodium borate buffer, pH 8.3) at a density of 2×105 cells per dish. After overnight incubation in Dulbecco's Modified Eagle Medium (DMEM, GibcoBRL, NY, USA) that was supplemented with 10% fetal bovine serum (FBS, GibcoBRL, NY, USA), the media was replaced with serum-free defined medium for neurons [DMEM supplemented with 2 mM of glutamine, 1 mM of pyruvate, penicillin-streptomycin-amphotericin B mixture (GibcoBRL, NY, USA), 5 mM of HEPES, 0.5% glucose, 10 µg/ml of insulin, 30 nM of sodium selenite, 20 nM of progesterone, 100 µM of putrescine, and 20 ng/ml of transferrin]. The cultured cells were incubated at 37℃ in 5% CO2, and the media was replaced every other day. Experiments were performed after 14~15 days. The growth medium was replaced with 2% FBS medium and tested with the peptides or drugs for the indicated times.
Cell viability assay
WST-1 was used to evaluate cell viability as described previously [26]. WST-1-metabolizing activity was determined according to the manufacturer's instructions (Roche, IN, USA). Primary cortical neurons were plated in a 96-well plate at a density of 1×105 cells/well. DHED was introduced into the media of primary cortical neurons 4 h before treatment with 25 µM of Aβ1-42 or 1 µM of staurosporine. This colorimetric assay measures the metabolic activity of viable cells. Briefly, after incubating cells that were treated with various reagents, 10 µl of WST-1 was added to the culture media. The culture was incubated at 37℃ in a humidified atmosphere of 95% air and 5% CO2 for 4 h. The absorbance of the reaction product was measured with an ELISA reader (Bio-Rad, Germany) at a wavelength of 450 nm.
Measurement of ROS generation
ROS in cortical neurons were assayed using dye 2',7'-dichlorofluoroscein diacetate (DCFH-DA Molecular Probes, OR, USA). Cells were washed with HEPES-buffered saline (HBS) and incubated in the dark for 1 h in HBS containing 200 µM of DCFH-DA. Upon incubation, cells take up DCFH-DA, where intracellular esterase cleaves the molecule into DCFH. DCFH isoxidized into DCF in the presence of reactive oxygen species. The total fluorescence was measured using a spectrofluorometer (Molecular Devices Co., CA, USA) at an emission wavelength of 488 nm and an excitation wavelength of 524 nm [29].
Calcium imaging
Fluo-4 AM was used to measure intracellular calcium levels as described previously [30,31]. Stock solutions of 5-mM Fluo- 4 AM (Molecular Probes, USA) in DMSO and 20% pluoronic F-127 (Molecular Probes, USA) were prepared and stored in 3 µl aliquots at ??0℃. Primary cortical neurons were incubated with Fluo-4 AM (final concentration of 5 µM) for 10 min in the incubator at 37℃. Cells were superfused with Hanks' Balanced Salt solution (HBSS, pH=7.1, Invitrogen Co., USA) and gassed with oxygen at 37℃. For imaging of the Fluo-4 fluorescence, excitation light was provided using an argon laser at 488 nm and the emission was filtered with a 515 nm long pass filter. Images were acquired using the photomultiplier of a Zeiss LSM 510 confocal microscope. For continuous monitoring of Fluo- 4 fluorescence, time series of images were obtained at regular intervals (10 sec). Fluorescence was measured and normalized to the average baseline fluorescence that was collected over 10 sec. After the injection of vehicle, 10 µM of MK-801, or 10 µM of DHED in media, the change in fluorescence for each experiment was measured for 5 min. After 5 min of the measurement, 1.2 mM of CaCl2 and 50 mM of KCl were inserted in the media, and the change in fluorescence for each experiment was measured for another 5 min. For analysis, the fluorescence intensity was averaged off-line after image acquisition using the Zeiss LSM imaging software.
Statistical analysis
Data were expressed as the mean±SE or as a percent of the control value±S.E. One-way ANOVA and two-way ANOVA tests were applied to study the relationship between the different variables. A p<0.05 was considered to be significant.
RESULTS
DHED ameliorates spatial memory deficits in scopolamine-injected rats
Scopolamine-injected rats performed significantly worse on the water maze test (Fig. 1). Scopolamine (1 mg/kg, i.p.) was given to rats after 5 days of training trials. DHED (10 mg/kg, p.o.) or Donepezil (1 mg/kg, p.o.) was administrated 30 min after the injection of scopolamine. In this study, the concentrations of DHED were determined by previous studies [18,32]. In the first test (F3,14=4.89, p=0.0157), the latency for scopolamine-injected rats (60.79±10.21 sec) was longer than that of the control group (18.75±8.42 sec). However, this longer latency after scopolamine injection was decreased after DHED (20.43±8.89 sec) or Donepezil (16.07±9.22 sec) treatment. Similarly, the latency for scopolamine-injected rats (40.75±18.42 sec) was longer than that of the control group (11.21±4.22 sec) in the second test (F3,14=3.95, p=0.0311). However the longer latency after scopolamine injection was decreased after DHED (5.63±1.45 sec) or Donepezil (4.75±0.52 sec) treatment. The difference between the 4 groups in the third test was not significant.
Fig. 1. Effect of DHED or Donepezil on spatial memory impairment in scopolamine-injected rats.
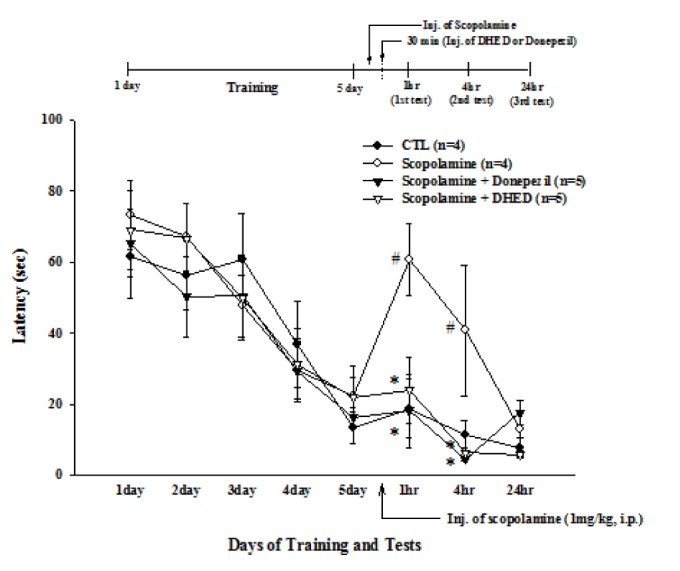
Training trials were carried out twice a day for 5 consecutive days. Scopolamine (1 mg/kg, i.p.) was given to rats immediately after the last training trial. DHED (10 mg/kg, p.o.) or Donepezil (1 mg/kg, p.o.) was given to rats 30 min after scopolamine administration. The trial tests were carried out at 1 h (1st), 4 h (2nd), and 24 h (3rd) after scopolamine administration. Latency is the time until a rat arrived at the platform. Note that at 1 h and 4 h the scopolamine-induced long latency was significantly decreased after DHED or Donepezil treatment. Data represents mean±standard error (SE). #p<0.05 compared with control and *p<0.05 compared with the scopolamine-injected group, one-way ANOVA.
DHED ameliorates learning and memory impairment in Aβ1-42-infused rats
To investigate whether DHED administration improves learning and memory impairment after Aβ1-42 infusion, we used the water maze test (Fig. 2A, 2B) and the passive avoidance (Fig. 2C) test. Aβ1-42 (0.6 nmol/day, i.c.v.) was infused into the brain ventricle of each rat for 7 days using brain infusion kits and mini-osmotic pumps. Synchronously, DHED (10 mg/kg, p.o.) was administrated for 21 days. DHED improved Aβ1-42-induced spatial memory impairments, as shown in the water maze test (Fig. 2). The changes in escape latency during training trials for each group are shown in Fig. 2A. There was no significant difference between the training trials on days 1~3. On day 5 of the training test (F2,29=5.60, p=0.0047), the latency for Aβ1-42-infused rats (17.0±3.2 sec) was longer than that of the control group (7.2±1.1 sec). There was no significant difference between the training trials of Aβ1-42-infused rats and DHED-treated rats. However, the latency for DHED-treated rats (4.4±0.6 sec) was shorter than that of Aβ1-42-infused rats (14.0±2.7 sec) and recovered to that of the control group (4.5±0.4 sec) on day 5 of the training test (F2,29=14.39, p<0.0001). On day 7, a 60-sec spatial probe trial was carried out to examine whether the animal had learned the position of the platform that was in zone 1 (Fig. 2B). The platform was removed for the test. In the control group (F3,48=20.04, p<0.0001), the time spent in zone 1 (21.28±0.94 sec) was longer than that spent in other zones (zone 2, 14.78±0.53; zone 3, 12.98±0.90; zone 4, 11.00±0.78 sec). In addition, the pattern of the probe test in the Aβ1-42-infused rats was different from that in the control group. In the Aβ1-42-infused rats, the time spent in zone 1 (17.02±0.86 sec) was significantly different from the time spent in zone 3 (9.57±1.07 sec vs. zone 1, p=0.0402), but not the time spent in zone 2 (16.01±1.41 sec) and zone 4 (17.40±2.43 sec). However, the pattern of the probe test in the DHED-treated rats was similar to that in the control group. In DHED-treated rats (F3,36=10.30, p<0.0001), the spent time in zone 1 (21.41±1.20 sec) was longer than that spent in other zones (zone 2, 14.29±0.87; zone 3, 11.94±1.40; zone 4, 12.37±1.26 sec). In addition, the times spent in zone 1 were significantly different among the 3 groups (F2,29=3.69, p=0.0374). Furthermore, the passive avoidance test was carried out after the final treatment of DHED. As shown in Fig. 2C, the latency for Aβ1-42-infused rats (96.98±32.10 sec) was shorter than that of the control group (244.30±29.67 sec) in the passive avoidance test (F2,29=4.96, p=0.0141). However, the Aβ1-42-decrease in latency was increased after DHED (204.85±38.69 sec) treatment.
Fig. 2. Effect of DHED on memory impairment in Aβ1-42-infused rats.

(A) In Aβ1-42-infused rats, the training trial of the water maze test was performed after repeated administrations of DHED (10 mg/kg, p.o.) for 21 days. On trial day 5, latency was significantly increased in Aβ1-42-infused rats compared to the control group. However, latency was significantly decreased after DHED treatment compared to vehicle-treated Aβ1-42-infused rats. *p<0.05 compared to Aβ1-42-infused rats, one-way ANOVA. (B) The probe trial of the water maze test was carried out on day 2 after the final training trial. Note that the time spent in the platform quadrant (zone 1) was significantly decreased in the Aβ1-42-infused group compared to the control group. However, this time was significantly increased after DHED treatment compared to the Aβ1-42-infused only rats. Data represents mean±SE. #p<0.05 compared with Aβ1-42-infused rats and *p<0.05 compared with time in zone 1, one-way ANOVA. (C) In Aβ1-42-infused rats, which were generated via an infusion of the Aβ1-42 peptide into the lateral ventricle, a passive avoidance test was performed after repeated administrations of DHED (10 mg/kg, p.o.) for 21 days. Note that latency was decreased significantly in the Aβ1-42-infused group compared to the control group. However, latency was significantly increased after DHED treatment compared to the Aβ1-42-infused rats. Data represents mean±SE. *p<0.05 compared with the Aβ1-42-infused rats, one-way ANOVA.
DHED reduces Aβ1-42-induced neurotoxicity and the production of reactive oxygen species
As shown in Fig. 3A, cell viability was significantly reduced by approximately 60% after treatment with the Aβ1-42 peptide (25 µM). Moreover, pretreatment with DHED (0.5, 1, 2, and 4 µM) at 4 h before treatment with the Aβ1-42 peptide significantly enhanced cell viability in a dose-dependent manner (2 µM, 55.89±8.23; 4 µM, 57.84±4.58% vs. Aβ1-42-treated neurons, p<0.0001). In addition, pretreatment with 50 µM of vitamin E at 4 h before treatment with the Aβ1-42 peptide enhanced cell viability (70.06±5.93%). As shown in Fig. 3B, we investigated whether pretreatment with DHED could affect the production of reactive oxygen species (ROS) after exposing primary cultured neurons to Aβ1-42 peptide. ROS production was significantly increased by approximately 140% after exposure to the Aβ1-42 peptide compared with the control. However, pretreatment with DHED (1, 2, and 4 µM) decreased ROS production in a dose-dependent manner (1 µM, 123.21±4.84; 2 µM, 118.36±5.20; 4 µM, 115.23±4.41% vs. Aβ1-42-treated neurons, p=0.0002). In addition, pretreatment with 50 µM of vitamin E decreased ROS production (116.22±8.80%).
Fig. 3. Effect of DHED on Aβ1-42-induced neurotoxicity and reactive oxygen species (ROS).

(A) Treatment with Aβ1-42 peptide (25 µM) markedly induced neurotoxicity in primary neurons. However, the cell viability was significantly increased in a dose-dependent manner after pretreatment with DHED (0.5, 1, 2, and 4 µM) compared with the Aβ1-42-only-treated group (*p<0.05). In addition, cell viability was significantly increased after pretreatment with 50 µM of vitamin E compared with the Aβ1-42-only-treated group (*p<0.05). DHED or vitamin E was administered at 4 h before treatment with Aβ1-42. Data are expressed as a percent of the control value±SE. At least 2 experiments were carried out in triplicates. (B) ROS levels were significantly increased after treatment with the Aβ1-42 peptide. However, pretreatment with DHED (0.5, 1, 2, and 4 µM) significantly decreased ROS compared to the Aβ1-42-only-treated group (*p<0.05 and *p<0.01). In addition, pretreatment with 50 µM of vitamin E significantly decreased ROS compared to the Aβ1-42-only-treated group (*p<0.05). DHED or vitamin E was administered for 4 h before treatment with Aβ1-42. Data are expressed as a percent of the control value ± SE. At least 2 experiments were carried out in triplicates.
DHED reduces the KCI-dependent increase in intracellular calcium levels
As shown in Fig. 4, intracellular Ca2+ levels were significantly increased after treatment with 1.2-mM CaCl2 and 50-mM KCl (1.26±0.06 vs. baseline, p=0.0004). However, pretreatment with 10 µM of MK801 or 10 µM of DHED at 5 min before treatment with 1.2 mM of CaCl2 and 50 mM of KCl significantly decreased intracellular Ca2+ levels (MK801, 1.09±0.05; DHED, 1.04±0.07 vs. vehicle-treated neurons, p=0.0048).
Fig. 4. Effect of DHED on intracellular Ca2+ levels in neurons.

(A) Intracellular Ca2+ was assessed in neurons using confocal microscopy. Primary cortical neurons were treated with 1.2 mM of CaCl2 and 50 mM of KCl after 30 min of vehicle, 10 µM of MK801, or 10 µM of DHED treatment. Intracellular Ca2+ levels were measured after CaCl2 and KCl treatment. Note that intracellular Ca2+ levels were increased in vehicle-treated neurons. However, intracellular Ca2+ levels were decreased after pretreatment with MK801 or DHED compared to vehicle-treated neurons. DIC, differential interference contrast. (B) Quantification of intracellular calcium levels for analysis is shown. Data represents the mean±SE. *p<0.05 compared with vehicle-treated neurons, one-way ANOVA.
DISCUSSION
Our results show that DHED ameliorated cognitive functions in memory-impaired rat models, including a scopolamine-induced amnesia model and an amyloid-infused model. The cognitive effects of DHED in these models were measured using the water maze test and passive avoidance test. Additionally, the mechanism of the effects of DHED was determined using a cell viability assay, ROS measurement, and intracellular calcium measurement in primary cortical neurons.
AD is a devastating neurological disorder that involves a progressive loss of cognitive function, including learning and memory functions. In addition, AD is associated with cholinergic dysfunction, such as a reduction in ACh release and increased AChE activity, as well as Aβ deposition. Furthermore, AD is associated with oxidative stress and neuroinflammation [23,33]. Scopolamine-induced amnesia in rats is an effective model of AD because it impairs spatial learning and memory through inhibiting cholinergic signaling and increasing Aβ accumulation, oxidative stress, and synaptic dysfunction [34,35]. Here, we demonstrate that DHED significantly increased scopolamine-induced memory loss similar to the effect of donepezil. Donepezil suppresses scopolamine-induced cognitive impairments, such as simple conditioning, attention, working memory, and spatial mapping [36]. Donepezil is well known as a cholinesterase inhibitor that is approved for AD treatment. Donepezil is effective in improving cognitive function [37,38]. Furthermore, the Aβ1-42-infused rat model that we used here shows the pathological hallmarks of AD, including amyloid deposition, robust neuroinflammation, synaptic marker changes, neuronal death, and memory impairments [23,33]. This model also exhibits impairments in nicotine- and K+-stimulated acetylcholine and/or dopamine release from the frontal cortex/hippocampus and striatum, respectively [24]. The Aβ1-42-infused rat model exhibits poor memory performance in water maze tests. Aβ1-42 infusion also impairs retention of long-term memory in a passive avoidance test [27]. Moreover, Aβ1-42-infused rats are a useful animal model for evaluating developmental processes at the early and/or middle stages of Alzheimer-like dementia [39]. Our study found that a consecutive administration of DHED (10 mg/kg, p.o.) for 21 days significantly improved Aβ1-42-induced cognitive impairments in rats.
We show that mechanisms such as a decrease in neurotoxicity, ROS, and intracellular calcium levels may underlie the cognitive improvement observed after DHED treatment in memory-impaired rat models, including a scopolamine-induced amnesia model and an amyloid-infused model. DHED had an inhibitory effect on Aβ1-42-induced neurotoxicity. The Aβ peptide that is generated from β-and γ-secretase activity on the amyloid precursor protein (APP) contributes to the neurodegenerative process of AD [3,40]. Pretreatment with DHED (1, 2, and 4 µM) for 4 h prior to Aβ1-42 (25 µM) significantly reduced cell death and ROS production. Aβ causes extensive degeneration and neuronal death through oxidative stress. Oxidative markers co-localize within amyloid deposits in the brains of AD patients [41] and transgenic AD mice [42]. In addition, oxygen free radicals are produced upon exposure of cells to Aβ. A recent study reported that oxidative stress promoted intracellular accumulation of the Aβ peptide through enhancing the amyloidogenic pathway [43]. In vivo and in vitro evidence suggests the possibility that antioxidants can prevent neurons from Aβ neurotoxicity [42,44]. Moreover, calcium regulates neural processes such as synaptic plasticity and apoptosis. A change in intracellular calcium levels is involved in the pathogenesis of AD [45,46]. In particular, increased intracellular calcium is related to cognitive impairment through neuronal degeneration such as Aβ accumulation and synaptic loss [47]. Aβ decreases dendritic spine density, suppresses long-term potentiation (LTP), facilitates long-term depression (LTD), and impairs learning and memory [48]. Oligomeric Aβ increased non-selectively calcium permeability, including an increase in calcium influx from the extracellular space, calcium leakage from intracellular calcium stores, and an increase in N-methyl-D-aspartate (NMDA) receptor-dependent calcium influx. Furthermore, Aβ increases an NMDA receptor-dependent calcium influx. NMDA receptor antibodies such as MK-801, which is a noncompetitive antagonist of the NMDA glutamate receptor, inhibit Aβ binding. MK-801 significantly inhibits the Aβ-induced increase in intracellular calcium levels [49]. Here we show that DHED treatment significantly attenuated intracellular calcium levels in primary cortical neurons. Thus, DHED potentially inhibits AChE and antagonizes the NMDA receptor in AD. β-glucan prevents cognitive decline through inhibiting acetylcholinesterase (AChE) in scopolamine-injected rats [9]. In addition, Diallyl disulfide, a compound of garlic, improves scopolamine-induced cognitive impairment in rat models through inhibiting ROS generation and AChE activity [50]. DHED associates with AChE inhibition and long-lasting facilitation of synaptic transmission through activation of muscarinic and NMDA receptors [51]. DHED is protective against immobilization stress-induced memory deficit and behavioral impairment [32]. DHED inhibits calyculin A-induced tau in AD-like rat brain slices [19]. Moreover, DHED decreases tau hyperphosphorylation and spatial memory impairment [16].
In conclusion, we show that DHED has strong protective effects on cognitive impairment through its antioxidant activity and inhibition of neurotoxicity and intracellular calcium in memory-impaired rat models. DHED might be a useful target for drug therapy development for impaired memory symptoms such as those in AD.
ACKNOWLEDGEMENTs
This study received financial support from the research fund of Dankook University in 2014.
Footnotes
Author contributions: K.Y.S.; acquisition and analysis of data and drafting of the manuscript. K.Y.K.; analysis and interpretation of data and drafting of the manuscript. Y.H.S.; critical revision of the manuscript.
CONFLICTS OF INTEREST: The authors declare no conflicts of interest.
References
- 1.Roussel M, Martinaud O, Henon H, Vercelletto M, Bindschadler C, Joseph PA, Robert P, Labauge P, Godefroy O, Group GS. The behavioral and cognitive executive disorders of stroke: the GREFEX study. PLoS One. 2016;11:e0147602. doi: 10.1371/journal.pone.0147602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Post RM. Epigenetic basis of sensitization to stress, affective episodes, and stimulants: implications for illness progression and prevention. Bipolar Disord. 2016;18:315–324. doi: 10.1111/bdi.12401. [DOI] [PubMed] [Google Scholar]
- 3.Suh YH, Checler F. Amyloid precursor protein, presenilins, and alpha-synuclein: molecular pathogenesis and pharmacological applications in Alzheimer's disease. Pharmacol Rev. 2002;54:469–525. doi: 10.1124/pr.54.3.469. [DOI] [PubMed] [Google Scholar]
- 4.Francis PT, Palmer AM, Snape M, Wilcock GK. The cholinergic hypothesis of Alzheimer's disease: a review of progress. J Neurol Neurosurg Psychiatry. 1999;66:137–147. doi: 10.1136/jnnp.66.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holston EC. The electrophysiological phenomenon of alzheimer's disease: a psychopathology theory. Issues Ment Health Nurs. 2015;36:603–613. doi: 10.3109/01612840.2015.1015696. [DOI] [PubMed] [Google Scholar]
- 6.Guntupalli S, Widagdo J, Anggono V. Amyloid-β-induced dysregulation of AMPA receptor trafficking. Neural Plast. 2016;2016:3204519. doi: 10.1155/2016/3204519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang WJ, Zhang X, Chen WW. Role of oxidative stress in Alzheimer's disease. Biomed Rep. 2016;4:519–522. doi: 10.3892/br.2016.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Magi S, Castaldo P, Macri ML, Maiolino M, Matteucci A, Bastioli G, Gratteri S, Amoroso S, Lariccia V. Intracellular calcium dysregulation: implications for Alzheimer's disease. Biomed Res Int. 2016;2016:6701324. doi: 10.1155/2016/6701324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haider A, Inam W, Khan SA, Hifza, Mahmood W, Abbas G. β-glucan attenuated scopolamine induced cognitive impairment via hippocampal acetylcholinesterase inhibition in rats. Brain Res. 2016;1644:141–148. doi: 10.1016/j.brainres.2016.05.017. [DOI] [PubMed] [Google Scholar]
- 10.Pistollato F, Ohayon EL, Lam A, Langley GR, Novak TJ, Pamies D, Perry G, Trushina E, Williams RS, Roher AE, Hartung T, Harnad S, Barnard N, Morris MC, Lai MC, Merkley R, Chandrasekera PC. Alzheimer disease research in the 21st century: past and current failures, new perspectives and funding priorities. Oncotarget. 2016;7:38999–39016. doi: 10.18632/oncotarget.9175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soodi M, Naghdi N, Hajimehdipoor H, Choopani S, Sahraei E. Memory-improving activity of Melissa officinalis extract in naïve and scopolamine-treated rats. Res Pharm Sci. 2014;9:107–114. [PMC free article] [PubMed] [Google Scholar]
- 12.Hashimoto M, Tozawa R, Katakura M, Shahdat H, Haque AM, Tanabe Y, Gamoh S, Shido O. Protective effects of prescription n-3 fatty acids against impairment of spatial cognitive learning ability in amyloid β-infused rats. Food Funct. 2011;2:386–394. doi: 10.1039/c1fo00002k. [DOI] [PubMed] [Google Scholar]
- 13.Yun HM, Kim HS, Park KR, Shin JM, Kang AR, il Lee K, Song S, Kim YB, Han SB, Chung HM, Hong JT. Placenta-derived mesenchymal stem cells improve memory dysfunction in an Aβ1-42-infused mouse model of Alzheimer's disease. Cell Death Dis. 2013;4:e958. doi: 10.1038/cddis.2013.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi Y, Kim HS, Shin KY, Kim EM, Kim M, Kim HS, Park CH, Jeong YH, Yoo J, Lee JP, Chang KA, Kim S, Suh YH. Minocycline attenuates neuronal cell death and improves cognitive impairment in Alzheimer's disease models. Neuropsychopharmacology. 2007;32:2393–2404. doi: 10.1038/sj.npp.1301377. [DOI] [PubMed] [Google Scholar]
- 15.Joo Y, Kim HS, Woo RS, Park CH, Shin KY, Lee JP, Chang KA, Kim S, Suh YH. Mefenamic acid shows neuroprotective effects and improves cognitive impairment in in vitro and in vivo Alzheimer's disease models. Mol Pharmacol. 2006;69:76–84. doi: 10.1124/mol.105.015206. [DOI] [PubMed] [Google Scholar]
- 16.Decker M. Novel inhibitors of acetyl- and butyrylcholinesterase derived from the alkaloids dehydroevodiamine and rutaecarpine. Eur J Med Chem. 2005;40:305–313. doi: 10.1016/j.ejmech.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 17.Unsworth WP, Kitsiou C, Taylor RJ. An expedient protecting-group-free total synthesis of (±)-dievodiamine. Org Lett. 2013;15:3302–3305. doi: 10.1021/ol4013469. [DOI] [PubMed] [Google Scholar]
- 18.Park CH, Lee YJ, Lee SH, Choi SH, Kim HS, Jeong SJ, Kim SS, Suh YH. Dehydroevodiamine.HCl prevents impairment of learning and memory and neuronal loss in rat models of cognitive disturbance. J Neurochem. 2000;74:244–253. doi: 10.1046/j.1471-4159.2000.0740244.x. [DOI] [PubMed] [Google Scholar]
- 19.Fang J, Liu R, Tian Q, Hong XP, Wang SH, Cao FY, Pan XP, Wang JZ. Dehydroevodiamine attenuates calyculin A-induced tau hyperphosphorylation in rat brain slices. Acta Pharmacol Sin. 2007;28:1717–1723. doi: 10.1111/j.1745-7254.2007.00655.x. [DOI] [PubMed] [Google Scholar]
- 20.Peng JH, Zhang CE, Wei W, Hong XP, Pan XP, Wang JZ. Dehydroevodiamine attenuates tau hyperphosphorylation and spatial memory deficit induced by activation of glycogen synthase kinase-3 in rats. Neuropharmacology. 2007;52:1521–1527. doi: 10.1016/j.neuropharm.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 21.Yang MC, Wu SL, Kuo JS, Chen CF. The hypotensive and negative chronotropic effects of dehydroevodiamine. Eur J Pharmacol. 1990;182:537–542. doi: 10.1016/0014-2999(90)90052-8. [DOI] [PubMed] [Google Scholar]
- 22.van der Staay FJ, Bouger PC. Effects of the cholinesterase inhibitors donepezil and metrifonate on scopolamine-induced impairments in the spatial cone field orientation task in rats. Behav Brain Res. 2005;156:1–10. doi: 10.1016/j.bbr.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 23.Nitta A, Fukuta T, Hasegawa T, Nabeshima T. Continuous infusion of beta-amyloid protein into the rat cerebral ventricle induces learning impairment and neuronal and morphological degeneration. Jpn J Pharmacol. 1997;73:51–57. doi: 10.1254/jjp.73.51. [DOI] [PubMed] [Google Scholar]
- 24.Itoh A, Nitta A, Nadai M, Nishimura K, Hirose M, Hasegawa T, Nabeshima T. Dysfunction of cholinergic and dopaminergic neuronal systems in beta-amyloid protein--infused rats. J Neurochem. 1996;66:1113–1117. doi: 10.1046/j.1471-4159.1996.66031113.x. [DOI] [PubMed] [Google Scholar]
- 25.Oka J, Suzuki E, Kondo Y. Endogenous GLP-1 is involved in beta-amyloid protein-induced memory impairment and hippocampal neuronal death in rats. Brain Res. 2000;878:194–198. doi: 10.1016/s0006-8993(00)02741-4. [DOI] [PubMed] [Google Scholar]
- 26.Shin KY, Lee GH, Park CH, Kim HJ, Park SH, Kim S, Kim HS, Lee KS, Won BY, Lee HG, Choi JH, Suh YH. A novel compound, maltolyl p-coumarate, attenuates cognitive deficits and shows neuroprotective effects in vitro and in vivo dementia models. J Neurosci Res. 2007;85:2500–2511. doi: 10.1002/jnr.21397. [DOI] [PubMed] [Google Scholar]
- 27.Yamada K, Tanaka T, Han D, Senzaki K, Kameyama T, Nabeshima T. Protective effects of idebenone and alpha-tocopherol on beta-amyloid-(1-42)-induced learning and memory deficits in rats: implication of oxidative stress in beta-amyloid-induced neurotoxicity in vivo. Eur J Neurosci. 1999;11:83–90. doi: 10.1046/j.1460-9568.1999.00408.x. [DOI] [PubMed] [Google Scholar]
- 28.Shen Z, Wang G, Lin SZ. Two-way shuttlebox avoidance conditioning and brain NADH in rats. Physiol Behav. 1990;48:515–517. doi: 10.1016/0031-9384(90)90292-c. [DOI] [PubMed] [Google Scholar]
- 29.Wang H, Joseph JA. Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radic Biol Med. 1999;27:612–616. doi: 10.1016/s0891-5849(99)00107-0. [DOI] [PubMed] [Google Scholar]
- 30.Nahm WK, Philpot BD, Adams MM, Badiavas EV, Zhou LH, Butmarc J, Bear MF, Falanga V. Significance of N-methyl-D-aspartate (NMDA) receptor-mediated signaling in human keratinocytes. J Cell Physiol. 2004;200:309–317. doi: 10.1002/jcp.20010. [DOI] [PubMed] [Google Scholar]
- 31.Roychowdhury S, Noack J, Engelmann M, Wolf G, Horn TF. AMPA receptor-induced intracellular calcium response in the paraventricular nucleus is modulated by nitric oxide: calcium imaging in a hypothalamic organotypic cell culture model. Nitric Oxide. 2006;14:290–299. doi: 10.1016/j.niox.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 32.Kim HJ, Shin KY, Chang KA, Ahn S, Choi HS, Kim HS, Suh YH. Dehydroevodiamine·HCl improves stress-induced memory impairments and depression like behavior in rats. Korean J Physiol Pharmacol. 2014;18:55–59. doi: 10.4196/kjpp.2014.18.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Craft JM, Van Eldik LJ, Zasadzki M, Hu W, Watterson DM. Aminopyridazines attenuate hippocampus-dependent behavioral deficits induced by human beta-amyloid in a murine model of neuroinflammation. J Mol Neurosci. 2004;24:115–122. doi: 10.1385/JMN:24:1:115. [DOI] [PubMed] [Google Scholar]
- 34.Bartus RT, Dean RL, 3rd, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982;217:408–414. doi: 10.1126/science.7046051. [DOI] [PubMed] [Google Scholar]
- 35.Bartus RT. On neurodegenerative diseases, models, and treatment strategies: lessons learned and lessons forgotten a generation following the cholinergic hypothesis. Exp Neurol. 2000;163:495–529. doi: 10.1006/exnr.2000.7397. [DOI] [PubMed] [Google Scholar]
- 36.Lindner MD, Hogan JB, Hodges DB, Jr, Orie AF, Chen P, Corsa JA, Leet JE, Gillman KW, Rose GM, Jones KM, Gribkoff VK. Donepezil primarily attenuates scopolamine-induced deficits in psychomotor function, with moderate effects on simple conditioning and attention, and small effects on working memory and spatial mapping. Psychopharmacology (Berl) 2006;188:629–640. doi: 10.1007/s00213-006-0556-3. [DOI] [PubMed] [Google Scholar]
- 37.Unzeta M, Esteban G, Bolea I, Fogel WA, Ramsay RR, Youdim MB, Tipton KF, Marco-Contelles J. Multi-target directed Donepezil-like ligands for Alzheimer's disease. Front Neurosci. 2016;10:205. doi: 10.3389/fnins.2016.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kotani S, Yamauchi T, Teramoto T, Ogura H. Donepezil, an acetylcholinesterase inhibitor, enhances adult hippocampal neurogenesis. Chem Biol Interact. 2008;175:227–230. doi: 10.1016/j.cbi.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 39.Nakamura S, Murayama N, Noshita T, Annoura H, Ohno T. Progressive brain dysfunction following intracerebroventricular infusion of beta(1-42)-amyloid peptide. Brain Res. 2001;912:128–136. doi: 10.1016/s0006-8993(01)02704-4. [DOI] [PubMed] [Google Scholar]
- 40.Kuperstein I, Broersen K, Benilova I, Rozenski J, Jonckheere W, Debulpaep M, Vandersteen A, Segers-Nolten I, Van Der Werf K, Subramaniam V, Braeken D, Callewaert G, Bartic C, D'Hooge R, Martins IC, Rousseau F, Schymkowitz J, De Strooper B. Neurotoxicity of Alzheimer's disease Aβ peptides is induced by small changes in the Aβ42 to Aβ40 ratio. EMBO J. 2010;29:3408–3420. doi: 10.1038/emboj.2010.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Subbarao KV, Richardson JS, Ang LC. Autopsy samples of Alzheimer's cortex show increased peroxidation in vitro. J Neurochem. 1990;55:342–345. doi: 10.1111/j.1471-4159.1990.tb08858.x. [DOI] [PubMed] [Google Scholar]
- 42.Pappolla MA, Chyan YJ, Omar RA, Hsiao K, Perry G, Smith MA, Bozner P. Evidence of oxidative stress and in vivo neurotoxicity of beta-amyloid in a transgenic mouse model of Alzheimer's disease: a chronic oxidative paradigm for testing antioxidant therapies in vivo. Am J Pathol. 1998;152:871–877. [PMC free article] [PubMed] [Google Scholar]
- 43.Misonou H, Morishima-Kawashima M, Ihara Y. Oxidative stress induces intracellular accumulation of amyloid beta-protein (Abeta) in human neuroblastoma cells. Biochemistry. 2000;39:6951–6959. doi: 10.1021/bi000169p. [DOI] [PubMed] [Google Scholar]
- 44.Behl C, Davis J, Cole GM, Schubert D. Vitamin E protects nerve cells from amyloid beta protein toxicity. Biochem Biophys Res Commun. 1992;186:944–950. doi: 10.1016/0006-291x(92)90837-b. [DOI] [PubMed] [Google Scholar]
- 45.LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat Rev Neurosci. 2002;3:862–872. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- 46.Liang J, Kulasiri D, Samarasinghe S. Ca2+ dysregulation in the endoplasmic reticulum related to Alzheimer's disease: a review on experimental progress and computational modeling. Biosystems. 2015;134:1–15. doi: 10.1016/j.biosystems.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 47.Birnbaum JH, Bali J, Rajendran L, Nitsch RM, Tackenberg C. Calcium flux-independent NMDA receptor activity is required for Aβ oligomer-induced synaptic loss. Cell Death Dis. 2015;6:e1791. doi: 10.1038/cddis.2015.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hermes M, Eichhoff G, Garaschuk O. Intracellular calcium signalling in Alzheimer's disease. J Cell Mol Med. 2010;14:30–41. doi: 10.1111/j.1582-4934.2009.00976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferreira IL, Bajouco LM, Mota SI, Auberson YP, Oliveira CR, Rego AC. Amyloid beta peptide 1-42 disturbs intracellular calcium homeostasis through activation of GluN2B-containing N-methyl-d-aspartate receptors in cortical cultures. Cell Calcium. 2012;51:95–106. doi: 10.1016/j.ceca.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 50.Manral A, Meena P, Saini V, Siraj F, Shalini S, Tiwari M. DADS analogues ameliorated the cognitive impairments of Alzheimer-like rat model induced by scopolamine. Neurotox Res. 2016;30:407–426. doi: 10.1007/s12640-016-9625-5. [DOI] [PubMed] [Google Scholar]
- 51.Park EJ, Suh YH, Kim JY, Choi S, Lee CJ. Long-lasting facilitation by dehydroevodiamine. HClof synaptic responses evoked in the CA1 region of rat hippocampal slices. Neuroreport. 2003;14:399–403. doi: 10.1097/00001756-200303030-00020. [DOI] [PubMed] [Google Scholar]