

ABSTRACT
Hepatitis C virus (HCV) infects 2 to 3% of the world population and is a leading cause of liver diseases such as fibrosis, cirrhosis, and hepatocellular carcinoma. Many aspects of HCV study, ranging from molecular virology and antiviral drug development to drug resistance profiling, were supported by straightforward assays of HCV replication and infection. Among these assays, the HCV-dependent fluorescence relocalization (HDFR) system allowed live-cell visualization of infection without modifying the viral genome, but this strategy required careful recognition of the fluorescence relocalization pattern for its high fluorescence background in the cytoplasm. In this study, to achieve background-free visualization of HCV infection, a viral infection-activated split-intein-mediated reporter system (VISI) was devised. Uninfected Huh7.5.1-VISI cells show no background signal, while HCV infection specifically illuminates the nuclei of infected Huh7.5.1-VISI cells with either green fluorescent protein (GFP) or mCherry. Combining VISI-GFP and VISI-mCherry systems, we revisited HCV cell-to-cell transmission with clear-cut distinction of donor and recipient cells in a live-cell manner. Independently of virion assembly, exosomes have been reported to transfer HCV subgenomic RNA to initiate replication in uninfected cells, which suggested an assembly-free pathway. However, our data demonstrated that HCV structural genes and the p7 gene were essential for not only cell-free infectivity but also cell-to-cell transmission. Additionally, depletion of apolipoprotein E (ApoE) from donor cells but not from recipient cells significantly reduced HCV cell-to-cell transmission efficiency. In summary, we developed a background-free cell-based reporter system for convenient live-cell visualization of HCV infection, and our data indicate that complete HCV virion assembly machinery is essential for both cell-free and cell-to-cell transmission.
IMPORTANCE Hepatitis C virus (HCV) infects hepatocytes via two pathways: cell-free infection and cell-to-cell transmission. Structural modules of the HCV genome are required for production of infectious cell-free virions; however, the role of specific genes within the structural module in cell-to-cell transmission is not clearly defined. Our data demonstrate that deletion of core, E1E2, and p7 genes individually results in no HCV cell-to-cell transmission and that ApoE knockdown from donor cells causes less-efficient cell-to-cell transmission. Thus, this work indicates that the complete HCV assembly machinery is required for HCV cell-to-cell transmission. At last, this work presents an optimized viral infection-activated split-intein-mediated reporter system for easy live-cell monitoring of HCV infection.
KEYWORDS: HCV, cell-to-cell transmission, structural proteins
INTRODUCTION
Hepatitis C virus (HCV) infection is a leading cause of chronic liver disease. HCV infection affects 2 to 3% of the world population, and up to 80% of infected individuals develop a chronic infection which can lead to liver cirrhosis and hepatocellular carcinoma (1). Traditional interferon (IFN)-plus-ribavirin treatment is effective in only half of patients, while direct-acting antivirals (DAAs) have drastically improved the treatment efficacy (2, 3). However, due to the limited access to DAAs of patients worldwide, the emergence of DAA-resistant mutants, and side effects, a prophylactic vaccine is still needed to achieve complete eradication of HCV (4).
HCV is an enveloped positive-stranded RNA virus that belongs to the Hepacivirus genus of the Flaviviridae family (5). The HCV open reading frame (ORF) encodes a polyprotein of approximately 3,000 amino acids (aa), which is processed by host and viral proteases into 10 mature viral proteins: core; the envelope glycoproteins E1 and E2 (E1E2); the viroporin p7; and the nonstructural (NS) proteins NS2, NS3, NS4A, NS4B, NS5A, and NS5B. Core, E1E2, and p7 are essential for infectious cell-free virion production. Apart from the viral players in cell-free virion assembly, host apolipoproteins were found to be important for both infectious virion assembly and early steps of virus entry (6–12).
HCV uses two different transmission routes to infect hepatocytes: cell-free transmission and cell-to-cell transmission. HCV cell-free transmission starts from engagement of cell-free virions with several entry receptors (13, 14), including scavenger receptor class B type I (SRBI) (15), the tetraspanin CD81 (16), the tight junction proteins claudin-1 (CLDN1) (17) and occludin (OCLN) (18), and the receptor tyrosine kinases epidermal growth factor receptor (EGFR) (19), Niemann-Pick C1-like 1 cholesterol absorption receptor (NPC1L1) (20), syndecan 1 (SDC1) (21), and other lipoprotein receptors (22). After internalization, membrane fusion between viral and endosomal membranes induced by low pH leads to release of capsid in the cytosol. In contrast, HCV cell-to-cell transmission is resistant to anti-E2 neutralizing antibody, which assists dissemination and maintenance of DAA-resistant viral variants (23, 24). Additionally, HCV cell-to-cell transmission might be differently controlled by intracellular pH (25). Exosomes are reported to transfer genomic RNA to uninfected cells to evade antibody neutralization (26). Moreover, independently from virion production, exosomes were able to initiate replication in naive Huh7.5.1 cells (27). These observations suggested a virion-free infection pathway through cell-free transmission. However, the concept of virion-free infectivity is under debate because cell-free infection by subgenomic RNA-containing exosomes was not successful (28). Infectious HCV virion assembly requires host apolipoproteins (29, 30); however, dependence on host lipoproteins for HCV cell-to-cell transmission is controversial. One report stated that the lack of apolipoprotein E (ApoE) expression in a nonhepatic cell line blocked HCV cell-to-cell transmission (31). In contrast, knockdown of ApoE, ApoB, and microsomal triglyceride transfer protein (MTP) did not block efficient cell-to-cell transmission (32). Whether HCV assembly parts play an important role in cell-to-cell transmission has not been clearly determined due to the lack of a straightforward cell system which enables live-cell distinctions between donor and recipient cells during HCV cell-to-cell transmission.
In the last decade, the molecular biological study of HCV was advanced rapidly mainly owing to the establishment of the HCV cell culture system (HCVcc) (33–36). Detection of HCV infection often requires additional treatment of infected cells, such as fixation plus immunostaining or cell lysis plus quantitative reverse transcription-PCR (qRT-PCR) (37, 38). Live-cell detection of HCV infection was achieved by two means: (i) modification of the HCV genome by a fluorescence gene (38), which is accompanied by compromised virus fitness and genome stability, and (ii) an HCV-dependent fluorescence relocalization (HDFR) cell-based reporter system (39), which requires careful recognition of distinct fluorescence relocalization because of the high fluorescence signal in the cytoplasm. A more convenient and precise detection strategy will be beneficial in monitoring HCV cell-to-cell transmission in a live-cell manner.
In this study, taking advantage of a naturally occurring trans-splicing intein from Synechococcus elongatus (40), we devised a new viral infection-activated split-intein-mediated reporter system (VISI). Huh7.5.1-VISI cells show limited to no cytoplasm signal, while HCV infection specifically illuminates the nuclei of infected Huh7.5.1-VISI cells with either green fluorescent protein (GFP) or mCherry. Using VISI-GFP and VISI-mCherry systems, we revisited HCV cell-to-cell transmission regarding the role of structural genes and ApoE. We demonstrated that HCV structural genes and the p7 gene are essential for not only cell-free virion production but also cell-to-cell transmission. Additionally, depletion of ApoE from donor cells but not recipient cells significantly reduced HCV cell-to-cell transmission efficiency.
RESULTS
Development of a VISI reporter system for monitoring HCV infection.
To avoid the intensive mitochondrial fluorescent signal of the HDFR system and achieve more straightforward monitoring of HCV live-cell infection, we established a viral infection-activated split-intein-mediated cell reporter system (VISI). In the VISI system, we aim to minimize the mitochondrial fluorescent signal by splitting GFP into two halves (split site, aa 157/aa 158) (Fig. 1A). The N-terminal half of VISI encodes 3 repeats of simian virus 40 (SV40) nuclear localization sequences (NLS), N-terminal GFP, and N-intein (3XNLS-GFPn-INTEINn), while the C-terminal half of VISI expresses C-intein, C-terminal GFP, one NLS, and C-terminal IPS1 (INTEINc-GFPc-NLS-IPSc). Expression of HCV NS3-4A in Huh7.5.1-VISI-GFP cells could theoretically result in cleavage of IPSc, release of INTEINc-GFPc-NLS from mitochondria, and subsequent translocation into the nucleus. Inside the nucleus, protein splicing mediated by split intein occurs between 3XNLS-GFPn-INTEINn and INTEINc-GFPc-NLS, leading to formation of active GFP. Huh7.5.1 cells stably expressing both halves, named Huh7.5.1-VISI-GFP, show little to no green fluorescent signal in either the cytoplasm or the nucleus. Expression of HCV NS3-4A in Huh7.5.1-VISI-GFP cells through lentivirus transduction specifically activated a strong green fluorescent signal in nuclei (Fig. 1C and D). Western analysis demonstrated formation of intact GFP in nuclei of Huh7.5.1-VISI-GFP cells expressing NS3-4A (Fig. 1B and C). This observation suggested that NS3-4A expression activated translocation of the C terminus of VISI to the nucleus of Huh7.5.1-VISI-GFP cells where split-intein-mediated protein splicing resulted in maturation of intact GFP fused with 4 repeats of NLS and a residue segment from IPS1.
FIG 1.

Construction strategy of VISI system. (A) Schematic diagram of VISI system: fusion proteins of N-terminal and C-terminal halves of VISI are located in nucleus and associated with mitochondria, respectively; upon lentiviral transduction or HCV infection, NS3-4A cleavage activates transportation of the C-terminal piece from mitochondria to the nucleus where intact reporter protein (GFP*) is generated through split-intein splicing. GFP* represents reconstituted GFP which contains 4XNLS and IPS residues. The Huh7.5.1 cell line which contains the GFP-reported VISI system was named Huh7.5.1-VISI-GFP. (B and C) At 72 h postransduction, total cell lysates and cytosolic and nuclear fractions from Huh7.5.1-VISI-GFP cells were prepared for Western analysis. Detection of tubulin and lamin A/C served as fractionation control. Proteins are specified on the right of each panel. Positions of molecular mass standards are indicated on the left. (D) HCV NS3-4A expression in Huh7.5.1-VISI-GFP cell line activates GFP signal. Huh7.5.1-VISI-GFP cells were transduced with lentivirus mediating expression of HCV NS3-4A; an immunofluorescence assay of Huh7.5.1-VISI-GFP cells by using a specific NS3 antibody was conducted at 72 h posttransduction; GFP (autofluorescence in green) and NS3 (indirect immunofluorescence in red) were visualized. Nuclei were stained with DAPI (blue).
Next, the permissiveness of Huh7.5.1-VISI-GFP cells to authentic HCVcc infection was examined. Huh7.5.1-VISI-GFP cells were either electroporated with JcR2a-ΔE1E2 in vitro transcripts (Fig. 2A) or infected with Jc1 virus at a multiplicity of infection (MOI) of 5 (Fig. 2B to F). At different time points postelectroporation or postinfection, cells and/or culture supernatants were collected to test HCV genome replication, viral protein production, and infectious HCV production. Results demonstrated that Huh7.5.1-VISI-GFP cells supported efficient HCV genome replication (Fig. 2A) and infectious virion production (Fig. 2B) compared to parental Huh7.5.1 cells. Forty-eight hours after Lenti-NS3-4A transduction or HCVcc infection, Huh7.5.1-VISI-GFP cells were subjected to fluorescence-activated cell sorting (FACS) analysis (Fig. 2C). The GFP mean intensity of lentivirus-transduced or HCVcc-infected cells was observed to be higher than that in the untreated cells. At 72 h after HCVcc infection, nuclei of infected Huh7.5.1-VISI-GFP cells were observed with GFP signal (Fig. 2D). Moreover, Western analysis and live-cell monitoring of GFP signal demonstrated that kinetics of HCV core protein and NS3 expression are positively correlated with that of the intact GFP fusion protein (Fig. 2E and F), suggesting that the presence of the GFP signal directly indicates the viral protein expression level. Collectively, these results clearly demonstrated that harboring the VISI system in Huh7.5.1 cells caused no harm to the HCV life cycle and suggested that Huh7.5.1-VISI-GFP cells could be used for live-cell monitoring of HCV infection.
FIG 2.

Comparison of Huh7.5.1 and Huh7.5.1-VISI-GFP cell line in HCV genome replication, virus production, and viral protein expression. (A and B) The Huh7.5.1-VISI-GFP cell line supports efficient HCV genome replication and virus production. (A) Huh7.5.1 and Huh7.5.1-VISI-GFP cells were electroporated with JcR2a-ΔE1E2 RNA; luciferase activity was determined at the different time points postelectroporation. (B) Huh7.5.1 and Huh7.5.1-VISI-GFP cells were infected with HCVcc at an MOI of 5; the infectivity of HCV was measured by the TCID50 test at the different time points postinfection. Data represent the means from three independent assays; error bars represent standard deviations from the means. (C) Huh7.5.1-VISI-GFP cells were either transduced with Lenti-NS3-4A or infected with HCVcc; FACS analysis based on GFP intensity was performed 48 h posttreatment. (D) Activation of GFP signal in Huh7.5.1-VISI-GFP cell line by HCV infection. Immunofluorescence analysis of Huh7.5.1-VISI-GFP cell line at 72 h after Jc1 infection (MOI of 1). GFP autofluorescence (green) was visualized specifically in NS3-positive cells (red); nuclei were stained with DAPI (blue). (E) HCV viral protein expression in Huh7.5.1 and Huh7.5.1-VISI-GFP cells. At different time points after HCV infection, cells were collected and lysed in SDS-PAGE sample buffer; expression levels of HCV protein (core and NS3), GFP, and tubulin were detected by using Western analysis. Proteins are specified on the right of each panel. Positions of molecular mass standards are indicated on the left. Numbers indicate relative band intensity. (F) Huh7.5.1-VISI-GFP cells were infected with Jc1 RNA; fluorescence and bright-field images were taken at different time points after infection and merged.
Evaluation of anti-HCV drugs in Huh7.5.1-VISI-GFP cells.
Huh7.5.1-VISI-GFP cells indicate HCV infection by autofluorescence. The VISI system could offer a more convenient method for HCV drug screening and evaluation. To test the specificity of the HCV infection-activated fluorescent signal and check whether Huh7.5.1-VISI-GFP cells are suitable to evaluate anti-HCV drug activity, HCV-infected Huh7.5.1-VISI-GFP cells were treated with interferon (IFN) and simeprevir, respectively (Fig. 3A and B). At 24, 48, and 72 h after anti-HCV treatment, Huh7.5.1-VISI-GFP cells were subjected to immunofluorescence assays (Fig. 3A). IFN and simeprevir inhibition of HCV replication, as indicated by cytoplasmic NS3 staining, is well correlated with the autofluorescence located in the nuclei (Fig. 3B). In HCV-infected Huh7.5.1-GFP cells, inhibition of HCV replication by simeprevir did not reduce GFP signal (Fig. 3C). Additionally, simeprevir dose responses measured by GFP signal and NS3 signal showed no significant difference (Fig. 3D). This observation clearly indicated that activation of GFP signal in nuclei was HCV infection specific and that Huh7.5.1-VISI-GFP cells represented a convenient test system to evaluate anti-HCV drug.
FIG 3.

GFP signal activated by HCV infection could be eliminated by anti-HCV drugs. (A) Specific anti-HCV drugs (IFN-α/simeprevir) can terminate the GFP signal activated by established HCV infection. HCV infection was established by passaging of Huh7.5.1-VISI-GFP infected cells; these infected cells were treated with either IFN-α (1,000 IU/ml) or simeprevir (HCV NS3 inhibitor, 2 μM) for 24 h, 48 h, and 72 h. The immunofluorescence assay was conducted as described above. (B) HCV infection was established by passaging Huh7.5.1-GFP infected cells; these infected cells were treated with simeprevir for 72 h. The immunofluorescence assay was conducted as described above. (C) Statistical analysis of relative percentage of GFP-positive cells at different time points after anti-HCV treatment shows that the HCV-infected Huh7.5.1-VISI-GFP cells could reflect a real-time antiviral effect of anti-HCV drugs. (D) Statistical analysis of relative percentages of GFP-positive cells and NS3-positive cells at 72 h after simeprevir treatment with different dosages. Percentages of HCV-infected cells counted by GFP autofluorescence or NS3 immunofluorescence are shown. Data represent the means from three independent experiments; error bars represent standard deviations from the means.
Application of Huh7.5.1-VISI cells for live-cell visualization of HCV cell-to-cell transmission.
It has been well established that HCV infects hepatocytes through two different routes: (i) cell-free particle-mediated transmission and (ii) neutralizing antibody-resistant cell-to-cell transmission. The impact of HCV structural proteins or p7 deletions on HCV cell-to-cell transmission, however, has not been clearly resolved. Difficulty in differentiation between infected cell clusters from authentic cell-to-cell transmission and those from cell division is a major reason. To this end, we sought to use Huh7.5.1-VISI cells to revisit HCV cell-to-cell transmission. To achieve a clear-cut distinction between infection donor and recipient cells in cell-to-cell transmission assays, a Huh7.5.1-VISI-mCherry cell line was further generated using a previously described strategy. Similarly to Huh7.5.1-VISI-GFP cells, HCV infection lights the nuclei of Huh7.5.1-VISI-mCherry cells with a red fluorescent signal (Fig. 4B), and Huh7.5.1-VISI-mCherry cells are permissive to HCV replication and infectious virus production (Fig. 4A). Next, we combined Huh7.5.1-VISI-GFP cells and Huh7.5.1-VISI-mCherry cells to monitor HCV cell-to-cell transmission. Huh7.5.1-VISI-GFP cells were electroporated with Jc1 in vitro transcripts and then individualized and mixed with Huh7.5.1-VISI-mCherry cells at a ratio of 1:30. Cells were cultured in the presence of AR3A antibody to neutralize cell-free infectivity (Fig. 5A). Antibody-containing medium was replaced every 24 h. After 72 h of incubation, green fluorescent cells denoting donor cells and red fluorescent cells indicating recipient cells were examined (Fig. 5B and C). Additionally, culture supernatant was subjected to a 50% tissue culture infective dose (TCID50) test to rule out the presence of residual cell-free infectivity (Fig. 5D). Results demonstrated that AR3A antibody neutralization led to no detectable cell-free infectivity. Without neutralizing antibody or with control antibody, donor cell-free foci were observed, whereas with neutralizing antibody AR3A, only cell-to-cell transmission-based foci (specified by red recipient cells surrounding green donor cells) were observed (Fig. 5C). A representative cell-to-cell transmission-based focus is composed of 4 donor cells and 27 recipient cells (Fig. 5B). Moreover, antibody neutralization of cell-free infectivity did not significantly change the focus size. Overall, these results showed that the combination of Huh7.5.1-VISI-GFP cells and Huh7.5.1-VISI-mCherry cells offered a clear-cut method to monitor HCV cell-to-cell transmission.
FIG 4.

Establishment of Huh7.5.1-VISI-mCherry cell line to monitor HCV infection. (A) The Huh7.5.1-VISI-mCherry cell line supports efficient HCV genome replication and virus production. (Left) Huh7.5.1 and Huh7.5.1-VISI-mCherry cells were electroporated with JcR2a-ΔE1E2 RNA; luciferase activity was determined at the different time points postelectroporation. RLU, relative light units. (Right) Huh7.5.1 and Huh7.5.1-VISI-mCherry cells were infected with HCV; infectivity of HCV was measured by the TCID50 test at different time points postinfection. Data represent the means from three independent assays; error bars represent standard deviations from the means. (B) Activation of mCherry signal in Huh7.5.1-VISI-mCherry cell line by HCV infection. Immunofluorescence analysis of Huh7.5.1-VISI-mCherry cell line at 72 h after Jc1 infection (MOI of 1). mCherry autofluorescence (red) was visualized specifically in NS3-positive cells (green); nuclei were stained with DAPI (blue).
FIG 5.

Live-cell monitoring of HCV cell-to-cell transmission. (A) Schematic diagram of HCV cell-to-cell transmission system. Huh7.5.1-VISI-GFP cells electroporated with the HCV genome were used as donor cells; Huh7.5.1-VISI-mCherry cells were used as recipient cells. Donor cells were mixed at a 1:30 ratio with recipient cells. To neutralize cell-free transmission, HCV-neutralizing antibody AR3A or control antibody (HCV anti-E2 monoclonal antibody) was added to cocultures. After incubation for 96 h, images were taken under a fluorescence microscope. (B to D) Huh7.5.1-VISI-mCherry cells were cocultured with Huh7.5.1-VISI-GFP cells electroporated with Jc1 RNA in the presence of neutralizing (AR3A) or control (control) antibody or without antibody (Mock). (B) Numbers of HCV-positive donor cells (Huh7.5.1-VISI-GFP cells) or HCV-positive recipient cells (Huh7.5.1-VISI-mCherry cells) per focus after coculture for 96 h were counted. In the scatter plot, green triangles represent number of HCV-positive donor cells per focus; red dots represent number of HCV-positive recipient cells per focus; horizontal lines represent the median for 40 randomly selected foci. Statistical significance was determined by Student's t test. (C) HCV cell-to-cell transmission phenomena were observed by fluorescence microscopy after coculture for 72 h. The green fluorescence represents HCV-positive donor cells, and the red fluorescence represents HCV-positive recipient cells. The foci indicated by yellow arrows indicate virus spreading by cell-to-cell transmission with centered donor cells; the foci indicated by white arrows indicate virus spreading by cell-free transmission without donor cells. (D) Infectivity of supernatants from cocultures was determined by TCID50 titration on Huh7.5.1 cells. Means and standard deviations from three independent assays are shown (ND, not detected).
HCV structural genes and p7 gene are essential for HCV cell-to-cell transmission.
HCV cell-to-cell transmission is resistant to antibody neutralization and is suggested to be mediated by an exosome carrying replication-competent genome RNA. The structure module of the HCV genome (core gene, E1E2 gene, and p7 gene) is essential for cell-free infectious virion production. However, the role of these genes in HCV cell-to-cell transmission was controversial. Therefore, we examined the role of these genes in HCV cell-to-cell transmission using the VISI system. We generated a panel of Jc1 deletion mutants: core deletion, E1E2 deletion, and p7 deletion (Fig. 6A). Subgenomic JFH1 and Jc1 in vitro transcripts without core, E1E2, or p7 were used to electroporate Huh7.5.1-VISI-GFP donor cells. At 48 h postelectroporation, NS5 expression suggested that deletion of core, E1E2, or p7 did not affect HCV replication (Fig. 6B). In culture supernatant, HCV genome RNA was detected for all deletion mutants (Fig. 6E), suggesting that exosome-mediated RNA secretion does occur. However, only wild-type Jc1 released efficient core protein and infectious cell-free virions (Fig. 6C and D). After electroporation, individualized donor cells were also mixed with Huh7.5.1-VISI-mCherry recipient cells and cocultured as described above. At 72 h postcoculture in the presence of neutralizing antibody, cell-to-cell transmission was observed by fluorescence microscopy (Fig. 6G). The numbers of donor cells and recipient cells of each HCV-positive focus were counted (Fig. 6F). Results showed that comparable numbers of donor cells were observed for all. However, positive cell-to-cell transmission was observed only for Jc1 but not for any of the other deletion mutants. We additionally modified coculture conditions by mixing donor and recipient cells at a ratio of 1:1 and seeded a total of 1E7 cells, which still produced no single red recipient cell from these deletion mutants. These data clearly demonstrated that structural genes and the p7 gene were required for successful HCV cell-to-cell transmission.
FIG 6.

Essential role of HCV structural proteins and p7 in HCV cell-to-cell transmission. (A) Schematic representation of wild type (WT) and deletion constructs. Huh7.5.1-VISI-GFP cells were electroporated with wild-type or deletion mutant HCV RNA. (B) Western analysis of Huh7.5.1-VISI-GFP cell lysates harvested at 0 h or 48 h after electroporation. Expression levels of HCV proteins (E2, core, and NS5A) and tubulin were detected by using Western analysis. Proteins are specified on the right of each panel. Positions of molecular mass standards are indicated on the left. (C) Supernatants were harvested at 24 h, 48 h, and 72 h postelectroporation; extracellular infectivity was determined by TCID50 titration on Huh7.5.1 cells. Means and standard deviations from three independent assays are shown (ND, not detected). (D) The relative core protein level of supernatants harvested at 48 h was determined by enzyme-linked immunosorbent assay. Results are presented relative to supernatants from cells electroporated with the wild-type HCV genome and expressed as means and standard deviations from three independent assays. The dashed line represents the detection limit. (E) The HCV RNA secretion level of supernatants harvested at 48 h and 72 h postelectroporation was determined by RT-PCR. Means and standard deviations from three independent assays are shown. The dashed line represents the detection limit. (F and G)Huh7.5.1-VISI-mCherry cells were cocultured with Huh7.5.1-VISI-GFP cells electroporated with wild-type or deficient-type HCV RNA in the presence of neutralizing antibody. (F) Numbers of HCV-positive donor cells (Huh7.5.1-VISI-GFP cells) or HCV-positive recipient cells (Huh7.5.1-VISI-mCherry cells) per focus after coculture for 96 h were counted. In the scatter plot, green triangles represent number of HCV-positive donor cells per focus; red dots represent number of HCV-positive recipient cells per focus; horizontal lines represent the median for 40 randomly selected foci. (G) HCV cell-to-cell transmission phenomena were observed by fluorescence microscopy after coculture for 72 h. The green fluorescence represents HCV-positive donor cells, and the red fluorescence represents HCV-positive recipient cells.
To rule out the possibility that VISI modification of Huh7.5.1 cells changes the cell permissiveness to HCV cell-to-cell transmission, we further used parental Huh7.5.1 cells as both donor and recipient cells (Fig. 7). Subgenomic JFH1 and Jc1 in vitro transcripts without core, E1E2, or p7 were used to electroporate Huh7.5.1 cells. Individual electroporated cells were cocultured with naive Huh7.5.1 cells at a ratio of 1:30. After coculture for 72 h, cells were fixed and subjected to immunohistochemistry assays by using NS5A-specific antibody. The results showed that structural gene deletion, p7 deletion mutants, and subgenomic JFH1 produced foci with a mean cell number of only 4, which is equivalent to the number of divided donor cells found by using Huh7.5.1-VISI-GFP and Huh7.5.1-VISI-mCherry cells. Collectively, these results strongly indicated that structural proteins and p7 were essential not only for cell-free infectious virion production but also for cell-to-cell transmission.
FIG 7.
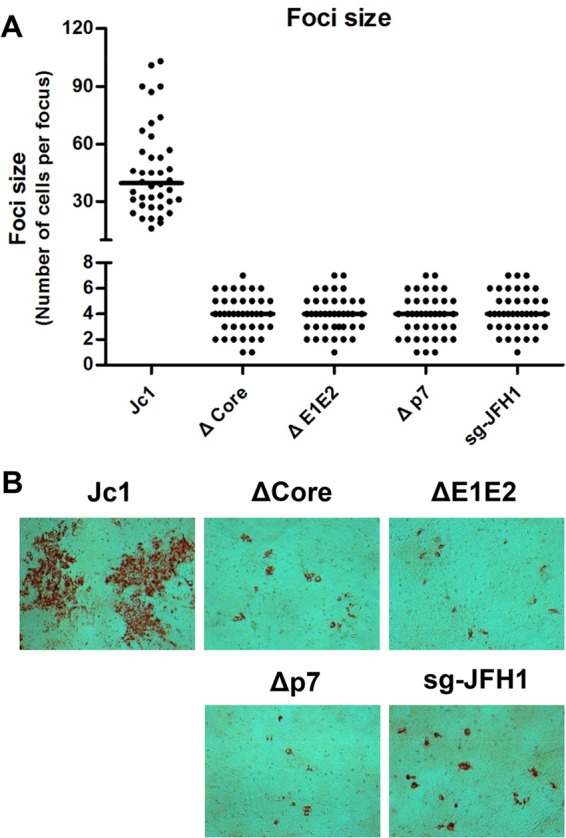
Confirmation of essential role of HCV structural proteins and p7 in HCV cell-to-cell transmission. Huh7.5.1 cells were cocultured with Huh7.5.1 cells electroporated with wild-type or mutant HCV RNA in the presence of neutralizing antibody. After coculture for 96 h, cells were fixed with methanol and probed with HCV NS5A specific antibody to visualize HCV-positive foci. (A) Numbers of HCV-positive cells per focus were counted by using bright-field microscopy. In the scatter plot, each black dot represents the number of HCV-positive cells per focus and horizontal lines represent the median for 40 randomly selected foci. (B) Bright-field microscope images of representative HCV-positive foci formed in the presence of AR3A neutralizing antibody.
ApoE depletion in donor cells but not recipient cells is important for efficient cell-to-cell transmission.
The HCV life cycle is intimately associated with the lipoprotein assembly pathway, and ApoE was consistently found to be important for infectious cell-free virion production. However, whether ApoE is involved in cell-to-cell transmission is under debate. Therefore, we applied Huh7.5.1-VISI cells to reexamine the role of ApoE in HCV cell-to-cell transmission. ApoE expression was stably knocked down in both Huh7.5.1-VISI-GFP donor cells and Huh7.5.1-VISI-mCherry recipient cells, and knockdown efficiency was checked by Western analysis (Fig. 8A). Huh7.5.1-VISI-GFP cells with or without ApoE knockdown were electroporated with Jc1 in vitro transcripts. Individual electroporated donor cells were cocultured with Huh7.5.1-VISI-mCherry recipient cells. After coculture for 72 h, cell-to-cell transmission was examined by autofluorescence. Our data showed that the ApoE expression level was not important for proliferation of HCV-infected donor cells. Depletion of ApoE from donor cells led to a significantly decreased cell-to-cell transmission efficiency, whereas ApoE expression knockdown in recipient cells has no significant effect (Fig. 8B and C). This result suggested that ApoE expression in donor cells was important for efficient HCV cell-to-cell viral spread.
FIG 8.

Effect of ApoE on HCV cell-to-cell transmission. (A) ApoE knockdown efficiency in Huh7.5.1-VISI-GFP cells and Huh7.5.1-VISI-mCherry cells. Tubulin was used as a loading control. Proteins are specified on the right of each panel. Positions of molecular mass standards are indicated on the left. (B and C) sh-NT Huh7.5.1-VISI-mCherry cells (sh-NT-m) or sh-ApoE Huh7.5.1-VISI-mCherry cells (sh-ApoE-m) were cocultured with electroporated sh-NT Huh7.5.1-VISI-GFP cells (sh-NT-G) or sh-ApoE Huh7.5.1-VISI-GFP cells (sh-ApoE-G) in the presence of neutralizing antibody. (B) After coculture for 96 h, numbers of HCV-positive donor cells or HCV-positive recipient cells per focus were counted. In the scatter plot, green triangles represent number of HCV-positive donor cells per focus; red dots represent number of HCV-positive recipient cells per focus; horizontal lines represent the median for 40 randomly selected foci. Statistical significance was determined by Student's t test (***, P < 0.001; ns, not significant). (C) HCV cell-to-cell transmission phenomena were observed by fluorescence microscopy after coculture for 96 h. The green fluorescence represents HCV-positive donor cells, and the red fluorescence represents HCV-positive recipient cells.
DISCUSSION
The aim of this study was to establish an optimized live-cell visualization system to monitor HCV infection and to use this system to analyze the functional role of the complete virion assembly machinery in HCV cell-to-cell transmission. We devised a simple viral infection-activated split-intein-mediated reporter system in Huh7.5.1 cells and showed that Huh7.5.1 cells harboring the VISI-GFP or VISI-mCherry system do not affect cell permissiveness to authentic HCVcc infection. Through both immunofluorescence and Western analysis, we observed excellent correlations between fluorescent signal intensity and HCV protein expression kinetics. Combining Huh7.5.1-VISI-GFP and Huh7.5.1-VISI-mCherry cells, we achieved live-cell monitoring of HCV cell-to-cell transmission with clear-cut recognition of donor and recipient cells. Deletion of core, E1E2, and p7 separately or deletion of core to NS2 resulted in a smaller but significant amount of genome secretion; however, none of these deletions supported functional HCV cell-to-cell transmission. Rab27a overexpression conferred more efficient exosome secretion but still did not support cell-to-cell transmission of the HCV subgenome (data not shown). Additionally, we confirmed that ApoE is important for efficient HCV cell-to-cell transmission, as ApoE knockdown in donor but not recipient cells resulted in significantly reduced HCV cell-to-cell transmission efficiency. In general, our data do not support the concept of virion assembly-independent HCV transmission. In agreement with our findings, adaptive assembly-enhancing mutations conferred significantly increased cell-to-cell transmission (41).
To achieve live-cell visualization of HCV infection at the cell culture level, strategies modifying either the virus genome or permissive cells have been applied successfully (37–39, 42). Amendment of the HCV genome by inserting a fluorescent reporter gene into NS5A, often with adaptive mutations, enabled easy detection of infected cells but was accompanied by attenuated virus fitness. Moreover, the reporter intensity of the genetically modified HCV genome could not be guaranteed during virus passaging. The HCV-dependent fluorescence relocalization (HDFR) system offered a novel avenue to live-cell monitoring of HCV infection, avoiding direct viral genome modification. However, application of the HDFR system requires careful recognition of fluorescence relocalization for its high fluorescence signal in the cytoplasm. The VISI system optimized the HDFR system by minimizing the cytoplasmic fluorescence, which helped identification of HCV-infected cells. This new live-cell detection system could enable successful sorting of HCV-infected living cells. SEC14L2 was recently identified to enable serum-derived HCV replication in cell culture (43). The VISI system might facilitate direct selection of cells replicating HCV isolates from clinical samples and assist efficient development of a more versatile HCVcc toolbox.
HCV infects hepatocytes through two distinct entry mechanisms: cell-free particle spread and cell-to-cell direct transmission. It was well established that cell-free particle assembly is intimately associated with the host lipoprotein pathway (29). Various apolipoproteins, including ApoB, ApoE, ApoAI, and ApoC, are identified on the surface of affinity-purified HCV lipoviral particles (LVP) (10, 44). After prior attachment to the cell surface mediated through virion surface ApoE binding to heparan sulfate proteoglycan (HSPG), cell-free HCV LVP enter hepatocytes, which is dependent on sequential engagement of several coreceptors, including CD81, SRBI, NPC1L1, and tight junction-associated proteins claudin-1 and occludin. In contrast, HCV cell-to-cell transmission is less well defined due to controversial observations. Besides the disagreement on the role of CD81 in HCV cell-to-cell transmission (45, 46), the requirements for complete HCV assembly machinery (from both virus and host sides) are under debate.
HCV structural proteins and the p7 ion channel protein are essential for infectious virion morphogenesis and secretion (47); however, HCV genome replication and release are not dependent on these proteins. Consistently, Dreux and colleagues reported that HCV-infected Huh7.5.1 cells or HCV subgenomic replicon (SGR) cells selectively package the viral genome into CD63- and CD81-coated exosomes, which triggers plasmacytoid dendritic cells (pDC) to produce IFN-α through cell-to-cell contact (48). Our data showed that deletion of structural proteins individually or collectively does not block the genome secretion pathway. This observation confirmed that a virus assembly-independent pathway for genome RNA secretion, possibly through exosomes, exists.
Exosomes are extracellular vesicles originating as intraluminal vesicles during the process of multivesicular body (MVB) formation and are released upon MVB fusion with the cellular plasma membrane. A link between exosomes and HCV was first suggested by electron microscopy analysis of HCV virion samples for the similarity in morphology of exosomes and observed virion structure (7). Exosomes prepared by ultracentrifugation were further reported to transmit HCV infection productively (49). However, these observations are not conclusive as morphological and biophysical characteristics of exosomes are very comparable to those of cell-free infectious HCV LVP. To avoid infectious HCV virion assembly, Ramakrishnaiah and colleagues prepared exosomes from SGR cells and found that these exosomes had mediated viral genome transmission and replication in HCV-permissive naive Huh7.5.1 cells (27). However, this finding was challenged by another report in which Longatti and colleagues found that SGR cell-derived exosomes were not able to transmit HCV RNA to target cells directly to initiate RNA replication (28). Our data based on individual structural protein deletion confirmed that a complete set of structure proteins and p7 is essential for cell-free infectivity, although genome release is less dependent on them. Moreover, Longatti and colleagues succeeded in demonstrating that subgenomic HCV RNA can be transferred to naive permissive cells through cell-to-cell contact to establish viral RNA replication, suggesting that a virion-independent transfer of replication-competent RNA between permissive cells did occur with extremely low efficiency. In our assay, not a single cell with a red nucleus designating successful cell-to-cell transmission was detected, suggesting that structural genes and p7 are essential viral factors for successful cell-to-cell transmission of the HCV genome. The divergence between our findings and those of Longatti et al. could be due to the following reasons: (i) the double-resistant selection strategy applied in cell-to-cell transmission is much more sensitive than our live-cell monitoring assay and (ii) this selection strategy recovers a significant amount of cell colonies led by DNA transfer due to cell-cell fusion.
ApoE is a well-characterized important determinant of HCV cell-free infectious virion production. However, the role of ApoE in HCV cell-to-cell transmission is unclear because of contradictory observations. One report showed that lack of ApoE in 293T cells completely prevents HCV cell-to-cell transmission, suggesting an essential role (31), whereas another report using Huh7.5.1 cells described that knockdown of ApoE is not involved in this type of HCV transmission because of the observation of successful cell-to-cell transmission, albeit with smaller focus size (32). Recently, another report confirmed that knockdown of ApoE in Huh7.5 cells significantly decreased focus size, and this effect is caused only by ApoE expression depletion in infection donor cells (50), which is in agreement with our finding. For HCV cell-to-cell transmission, ApoE was found to be an important but not an essential factor in Huh7.5 or Huh7.5.1 cells, which is in contrast to the finding derived from 293T cells. Besides ApoE, Huh7.5 and Huh7.5.1 cells express other exchangeable apolipoproteins which could also support infectious HCV virion assembly (51); this might explain why the complete block of cell-to-cell transmission from 293T cells was not reproduced in Huh7.5 and Huh7.5.1 cells. An ApoE expression level in uninfected cells is required for HCV cell-free transmission to maintain the ApoE level on infectious HCV virions (52). However, in the cell-to-cell transmission system, the ApoE level in recipient cells is not important. This difference suggested that ApoE exchange equilibrium is not required for short-range dissemination of HCV infectivity through cell-to-cell contact.
In conclusion, we developed a clear-cut viral infection-activated split-intein-mediated reporter system (VISI) in Huh7.5.1 cells. Huh7.5.1-VISI cells show limited to no cytoplasm signal, while HCV infection illuminates the nuclei of infected Huh7.5.1-VISI cells with a different fluorescent signal. Combining VISI-GFP and VISI-mCherry systems, we monitored HCV cell-to-cell transmission. We demonstrated that HCV structural genes and the p7 gene are essential for not only cell-free virion production but also cell-to-cell transmission. Additionally, in agreement with the most recent report by Gondar et al. (50), depletion of ApoE from donor cells but not recipient cells significantly reduced HCV cell-to-cell transmission efficiency. Our data indicate that the complete HCV virion assembly machinery determined by viral and host factors is critical for both HCV cell-free and cell-to-cell transmission, although the pH dependencies of these two transmission pathways might be different.
MATERIALS AND METHODS
Cell lines and antibodies.
Human hepatocyte-derived cell line Huh7.5.1 and its derivatives were cultured in Dulbecco's modified minimal essential medium (DMEM; Invitrogen) supplemented with 2 mM l-glutamine, nonessential amino acids, 100 U penicillin per ml, 100 μg streptomycin per ml, and 10% fetal calf serum (complete DMEM). Cells were routinely passaged twice a week at a split ratio of 1:7. Rabbit anticore was generated in-house. Mouse anti-NS5A and anti-NS3 were kindly provided by J. Zhong's lab at the Institut Pasteur of Shanghai. AR3A antibody was kindly provided by M. Law. The following commercial antibodies have been used: goat anti-human ApoE (ab947; Abcam), mouse antitubulin (Santa Cruz), and mouse anti-GFP (AE012; ABclone).
Plasmid construction.
Plasmids encoding full-length HCV chimera Jc1, JcR2a (a Jc1-derived construct carrying a Renilla luciferase gene), and Jc1 with deletion of core gene, E1E2, and p7 were kindly provided by R. Bartenschlager's lab at Heidelberg University. The coding sequences of GFPn (aa 1 to 157)/GFPc (aa 158 to 238) from pEGFP-N1 (GenBank accession no. U55762.1) were amplified with the primer pairs GFPn-F/GFPn-R and GFPc-F/GFPc-R (Table 1), respectively; the primer pairs intein-N-F/intein-N-R and intein-C-F/intein-C-R were used to amplify the coding sequences of intein-N (aa 1 to 113)/intein-C (aa 1 to 38) from Sel-intein (synthetic DNA fragment; GenBank accession no. CP000100.1). The coding sequence of IPS1 (aa 462 to 540; GenBank accession no. AB232371.1) was amplified by primer pair IPS-F/IPS-R.
TABLE 1.
Primers used in this studya
| Primer | Sequence (5′–3′) | Introduced restriction site |
|---|---|---|
| NLS-GFPn-F | GTACCTGCAGGATGGATCCAAAGAAAAAGAGAAAAGTTGATCCAAAAAAGAAAAGAAAGGTTGATCCAAAGAAAAAGAGAAAAGTTGTGAGCAAGGGCGAG | SbfI |
| GFPn-R | CTCTCTAGACTGCTTGTCGGC | XbaI |
| INTEINn-F | GAGTCTAGATGTTTGGCGGCAG | XbaI |
| INTEINn-R | CTCACTAGTTTACGCCGTCGC | SpeI |
| INTEINc-F | ATACCTGCAGGATGGTCAAAATTG | SbfI |
| INTEINc-R | CGCTCTAGAGTTGAAACAGTTG | XbaI |
| GFPc-F | GAGTCTAGAAAGAACGGCATC | XbaI |
| GFPc-R | TCCGCCACCCTTGTACAGCTC | |
| IPS(GFP)-F | CTGTACAAGGGTGGCGGAGGTTCTGGAGATCCAAAGAAAAAGAGAAAAGTTTCCGAGGGCACCTTTG | |
| IPS-R | CGCACTAGTTTAGTGCAGAC | SpeI |
| NLS-Cherry(n)-F | GTACCTGCAGGATGGACCCAAAGAAAAAGAGAAAAGTTGATCCAAAAAAGAAAAGAAAGGTTGATCCAAAGAAAAAGAGAAAAGTTGTGAGCAAGGGCGAG | SbfI |
| Cherry(n)-R | AGATCTAGAGTCCTCGGGGTAC | XbaI |
| Cherry(c)-F | ATATCTAGAGGCGCCCTGAAG | XbaI |
| Cherry(c)-R | CTCCAGAACCTCCGCCACCCTTGTACAGCTCGTCCATG | |
| IPS(cherry)-F | GAGCTGTACAAGGGTGGCGGAGGTTCTGGAGATCCAAAGAAAAAGAGAAAAGTTTCCGAGGGCACCTTTG |
F and R, sense and antisense primers, respectively; n and c, N- and C-terminal parts, respectively. Underlining indicates restriction sites.
The 3 nucleus localization signals (NLS; DPKKKRKV) were added to the coding sequence of GFPn by using long primer NLS-GFPn-F and GFPn-R. The PCR product of NLS-GFPn was digested with SbfI and XbaI. The INTEINn fragment was PCR amplified by using the INTEINn-F and INTEINn-R primer pair and digested with XbaI and SpeI. Digested NLS-GFPn and INTEINn were inserted into the SbfI- and SpeI-doubly digested pWPI-Blr vector, resulting in pWPI-blr-NLS-GFPn-INTEINn.
INTEINc was PCR amplified by using the INTEINc-F and INTEINc-R primer pair. The PCR product was double digested with SbfI and XbaI. The GFPc fragment was PCR amplified by using the GFPc-F and GFPc-R primer pair; the IPS1 sequence was PCR amplified by using the IPS-F and IPS-R primer pair. GFPc-IPS1 was obtained by overlap PCR by using the GFPc-F and IPS-R primer pair and was double digested with XbaI and SpeI. Digested INTEINc and GFPc-IPS1 were inserted into the SbfI- and SpeI-doubly digested pWPI-puro vector, resulting in pWPI-puro-INTEINc-GFPc-NLS-IPS.
The same strategy was applied to generate mCherry plasmids (GenBank accession no. KP238582.1). The coding sequences of mCherryn (aa 1 to 159)/mCherryc (aa 160 to 236) were amplified with the primer pairs Cherryn-F/Cherryn-R and Cherryc-F/Cherryc-R (Table 1), respectively.
Generation of Huh7.5.1-VISI by lentiviral transduction.
Briefly, lentivirus encoding VISI N-terminal and C-terminal halves was used to establish Huh7.5.1-VISI-GFP and Huh7.5.1-VISI-mCherry cell lines. 293T cells were cotransfected with the following vectors: first, transfer vector, carrying the puromycin or blasticidin resistance gene and the N-terminal half or C-terminal half of VISI; second, the HIV-1 packaging plasmid (psPAX2); third, a vesicular stomatitis virus glycoprotein (VSVg) expression vector (pMD2.G). Huh7.5.1-VISI-GFP and Huh7.5.1-VISI-mCherry cell lines were selected by both puromycin and blasticidin pressure.
For stable knockdown of ApoE expression in Huh7.5.1-VISI-GFP and Huh7.5.1-VISI-mCherry cell lines, lentiviruses carrying short hairpin RNA (shRNA) that were used to establish cell lines sh-NT and sh-ApoE and 293T cells were cotransfected with the following vectors: first, transfer vector pAPM, carrying the puromycin resistance gene and an shRNA sequence targeting the 3′ untranslated region of ApoE or control shRNA (sh-NT); second, the HIV-1 packaging plasmid (psPAX2); third, a VSVg expression vector (pMD2.G). The following shRNA targeting sequences were used: shmirAPOE, 5′-TGCTGTTGACAGTGAGCGCGGACCCTAGTTTAATAAAGATTAGTGAAGCCACAGATGTAATCTTTATTAAACTAGGGTCCATGCCTACTGCCTCGGA-3′; sh-NT, 5′-TGCTGTTGACAGTGAGCGCTCTCGCTTGGGCGAGAGTAAGTAGTGAAGCCACAGATGTACTTACTCTCGCCCAAGCGAGATAGTGAAGCCACAGATGTA-3′.
In vitro transcription and RNA transfection.
In vitro transcripts generated with pFK-based plasmids were transfected into cells by electroporation as described previously (9). The concentration of purified RNA was determined by photometry, and the integrity of the transcripts was verified by agarose gel electrophoresis. Transcripts were stored as 10-μg aliquots at −80°C for further use. For RNA transfection, subconfluent Huh7.5 cell monolayers were detached from the culture dish by trypsinization, washed once with phosphate-buffered saline (PBS), and resuspended at a concentration of 1.5 × 107 cells per ml in Cytomix containing 2 mM ATP and 5 mM glutathione. Ten micrograms of in vitro transcript was mixed with 400 μl of the cell suspension and transfected by electroporation at 960 mF and 270 V using a GenePulser system (Bio-Rad) and a cuvette with a gap width of 0.4 cm (Bio-Rad). Immediately after electroporation, cells were resuspended in complete DMEM and seeded as required.
Cytosol and nuclear fractionation.
Huh7.5.1 cells were grown in a 6-well plate and washed one time with cold PBS at 72 h postinfection. Cells were treated with 300 μl hypotonic buffer (10 mM HEPES, pH 7.6, 1.5 mM MgCl2, 10 mM KCl, 1 mM EDTA, proteinase inhibitors) and incubated on ice for 10 min. After that, the cell suspension was centrifuged at 3,000 rpm for 5 min at 4°C; the resulting supernatants were recovered as cytoplasmic fraction. The nuclear pellets were washed with hypotonic buffer twice and lysed with 100 μl high-salt buffer (20 mM HEPES, pH 7.6, 0.5 M NaCl, 1.5 mM MgCl2, 1 mM EDTA, proteinase inhibitors) by vigorous vortexing for 10 s. After incubation on ice for 20 min and centrifugation at 13,000 rpm for 15 min at 4°C, supernatants were collected as nuclear fraction.
Immunofluorescence staining.
For NS3 immunostaining, cells grown on glass coverslips were fixed in formaldehyde (3% [wt/vol] in PBS) after prior washing with PBS and incubated in blocking reagents (3% bovine serum albumin [BSA] in PBS). After overnight incubation at 4°C with monoclonal antibody against NS3 and 1 h of incubation at room temperature with Alexa Fluor 594-conjugated secondary antibody to mouse antibody (Invitrogen), cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Images were taken and analyzed with a Leica TCS-SP5 (Leica Microsystems) confocal microscope.
Quantification of HCV infectivity.
Infectivity titers were determined by using a limiting dilution assay. In brief, Huh7.5.1 cells were seeded into 96-well plates. Cells were infected and fixed 3 days after infection. For immunohistochemistry, we used a 1:100 dilution of an antibody reacting specifically with the JFH1 NS5A. Bound antibody was detected with a 1:300 dilution of a peroxidase-conjugated secondary antibody against mouse IgG (Sigma-Aldrich).
FACS assay.
Huh7.5.1-VISI-GFP cells were infected with HCVcc or HCV NS3-4A-expressed lentivirus. Cells were washed in PBS and treated with 0.005% trypsin at 48 h postinfection. Harvested cells were washed in PBS twice and then resuspended with FACS buffer (PBS-2% fetal bovine serum [FBS]). GFP signal was analyzed with a BD Fortessa flow cytometer.
Western blot analysis.
Samples for Western blotting were denatured in Laemmli buffer (125 mM Tris-HCl, 2% [wt/vol] SDS, 5% [vol/vol] 2-mercaptoethanol, 10% [vol/vol] glycerol, 0.001% [wt/vol] bromophenol blue, pH 6.8) and separated by SDS-12% polyacrylamide gel electrophoresis. Proteins were transferred onto a polyvinylidene difluoride (PVDF) membrane. The membrane was blocked overnight in PBS supplemented with 0.5% Tween (PBS-T) and 5% dried milk (PBS-M) at 4°C prior to 1 h of incubation with primary antibody diluted in PBS-M. The membrane was washed 3 times with PBS-T and incubated for 1 h with horseradish peroxidase-conjugated secondary antibody diluted 1:10,000 in PBS-M. Bound antibodies were detected after being washed 3 times with the enhanced chemiluminescence (ECL) reagent (PerkinElmer).
Cell-to-cell transmission assays.
Huh7.5.1-VISI-GFP cells were electroporated with in vitro transcripts of Jc1 wild type, deletion mutant, or subgenomic JFH1 (sgJFH1). These cells were individualized and mixed with Huh7.5.1-VISI-mCherry cells at a ratio of 1:30. One thousand Huh7.5.1-VISI-GFP cells plus 30,000 Huh7.5.1-VISI-mCherry cells were plated in single wells of 96-well plates. Cells were cultured in the presence of HCV-neutralizing antibody AR3A at 50 μg/ml to block completely cell-free infectivity. Antibody-containing medium was replaced every 24 h. After coculture for 96 h, HCV cell-to-cell transmission efficiency was evaluated by examining the number of GFP or mCherry glowing cells.
Statistical analysis.
Differences between samples were analyzed using the two-tailed, unpaired Student t test available in the GraphPad Prism 5 software package. P values of less than 0.05 (indicated by asterisks) were considered statistically significant. The following categories were used: ***, P ≤ 0.001; **, P ≤ 0.01; *, P ≤ 0.05.
Accession number(s).
Newly determined sequences are available in GenBank under the following accession numbers: KY067203 (for NLS-GFPn-INTEINn), KY067204 (INTEINc-GFPc-NLS-IPS), KY067205 (NLS-mCherry-INTEINn), and KY067206 (INTEINc-mCherry-NLS-IPS).
ACKNOWLEDGMENTS
We thank F. Chisari for the gift of Huh7.5.1 cells; M. Law for the gift of AR3A antibody; and T. Wakita, C. M. Rice, J. Bukh, and R. Bartenschlager for providing HCV strains. We apologize to many respected colleagues whom we could not cite because of space limitations.
F.Z., T.Z., J.X., and G.L. designed experiments. F.Z., T.Z., L.D., D.L., and X.Z. performed experiments. F.Z., X.P., and G.L. analyzed data. F.Z., T.Z., X.P., and G.L. wrote the manuscript.
This work was supported by the National Key R&D program of China (2016YFC1200400), the “100 talents program” from the Chinese Academy of Sciences, and the National Science and Technology Major Project of the Ministry of Science and Technology of China (2014ZX10002002-001-004 and 2015CB554300).
We disclose no conflicts.
REFERENCES
- 1.Gravitz L. 2011. Introduction: a smouldering public-health crisis. Nature 474:S2–S4. doi: 10.1038/474S2a. [DOI] [PubMed] [Google Scholar]
- 2.Chung RT, Baumert TF. 2014. Curing chronic hepatitis C—the arc of a medical triumph. N Engl J Med 370:1576–1578. doi: 10.1056/NEJMp1400986. [DOI] [PubMed] [Google Scholar]
- 3.Pawlotsky JM. 2014. New hepatitis C therapies: the toolbox, strategies, and challenges. Gastroenterology 146:1176–1192. doi: 10.1053/j.gastro.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Baumert TF, Fauvelle C, Chen DY, Lauer GM. 2014. A prophylactic hepatitis C virus vaccine: a distant peak still worth climbing. J Hepatol 61:S34–S44. doi: 10.1016/j.jhep.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 5.Dubuisson J, Cosset FL. 2014. Virology and cell biology of the hepatitis C virus life cycle: an update. J Hepatol 61:S3–S13. doi: 10.1016/j.jhep.2014.06.031. [DOI] [PubMed] [Google Scholar]
- 6.Chang KS, Jiang J, Cai Z, Luo G. 2007. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J Virol 81:13783–13793. doi: 10.1128/JVI.01091-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gastaminza P, Cheng G, Wieland S, Zhong J, Liao W, Chisari FV. 2008. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J Virol 82:2120–2129. doi: 10.1128/JVI.02053-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang H, Sun F, Owen DM, Li W, Chen Y, Gale M Jr, Ye J. 2007. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc Natl Acad Sci U S A 104:5848–5853. doi: 10.1073/pnas.0700760104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Long G, Hiet MS, Windisch MP, Lee JY, Lohmann V, Bartenschlager R. 2011. Mouse hepatic cells support assembly of infectious hepatitis C virus particles. Gastroenterology 141:1057–1066. doi: 10.1053/j.gastro.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 10.Merz A, Long G, Hiet MS, Brugger B, Chlanda P, Andre P, Wieland F, Krijnse-Locker J, Bartenschlager R. 2011. Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J Biol Chem 286:3018–3032. doi: 10.1074/jbc.M110.175018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meunier JC, Russell RS, Engle RE, Faulk KN, Purcell RH, Emerson SU. 2008. Apolipoprotein c1 association with hepatitis C virus. J Virol 82:9647–9656. doi: 10.1128/JVI.00914-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nielsen SU, Bassendine MF, Burt AD, Martin C, Pumeechockchai W, Toms GL. 2006. Association between hepatitis C virus and very-low-density lipoprotein (VLDL)/LDL analyzed in iodixanol density gradients. J Virol 80:2418–2428. doi: 10.1128/JVI.80.5.2418-2428.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ding Q, von Schaewen M, Ploss A. 2014. The impact of hepatitis C virus entry on viral tropism. Cell Host Microbe 16:562–568. doi: 10.1016/j.chom.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ogden SC, Tang H. 2015. The missing pieces of the HCV entry puzzle. Future Virol 10:415–428. doi: 10.2217/fvl.15.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, Traboni C, Nicosia A, Cortese R, Vitelli A. 2002. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J 21:5017–5025. doi: 10.1093/emboj/cdf529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, Abrignani S. 1998. Binding of hepatitis C virus to CD81. Science 282:938–941. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- 17.Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wolk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801–805. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- 18.Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, Rice CM. 2009. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457:882–886. doi: 10.1038/nature07684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L, Davis C, Mee CJ, Turek M, Gorke S, Royer C, Fischer B, Zahid MN, Lavillette D, Fresquet J, Cosset FL, Rothenberg SM, Pietschmann T, Patel AH, Pessaux P, Doffoel M, Raffelsberger W, Poch O, McKeating JA, Brino L, Baumert TF. 2011. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med 17:589–595. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sainz B Jr, Barretto N, Martin DN, Hiraga N, Imamura M, Hussain S, Marsh KA, Yu X, Chayama K, Alrefai WA, Uprichard SL. 2012. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat Med 18:281–285. doi: 10.1038/nm.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi Q, Jiang J, Luo G. 2013. Syndecan-1 serves as the major receptor for attachment of hepatitis C virus to the surfaces of hepatocytes. J Virol 87:6866–6875. doi: 10.1128/JVI.03475-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamamoto S, Fukuhara T, Ono C, Uemura K, Kawachi Y, Shiokawa M, Mori H, Wada M, Shima R, Okamoto T, Hiraga N, Suzuki R, Chayama K, Wakita T, Matsuura Y. 2016. Lipoprotein receptors redundantly participate in entry of hepatitis C virus. PLoS Pathog 12:e1005610. doi: 10.1371/journal.ppat.1005610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Timpe JM, Stamataki Z, Jennings A, Hu K, Farquhar MJ, Harris HJ, Schwarz A, Desombere I, Roels GL, Balfe P, McKeating JA. 2008. Hepatitis C virus cell-cell transmission in hepatoma cells in the presence of neutralizing antibodies. Hepatology 47:17–24. [DOI] [PubMed] [Google Scholar]
- 24.Xiao F, Fofana I, Heydmann L, Barth H, Soulier E, Habersetzer F, Doffoel M, Bukh J, Patel AH, Zeisel MB, Baumert TF. 2014. Hepatitis C virus cell-cell transmission and resistance to direct-acting antiviral agents. PLoS Pathog 10:e1004128. doi: 10.1371/journal.ppat.1004128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feneant L, Potel J, Francois C, Sane F, Douam F, Belouzard S, Calland N, Vausselin T, Rouille Y, Descamps V, Baumert TF, Duverlie G, Lavillette D, Hober D, Dubuisson J, Wychowski C, Cocquerel L. 2015. New insights into the understanding of hepatitis C virus entry and cell-to-cell transmission by using the ionophore monensin A. J Virol 89:8346–8364. doi: 10.1128/JVI.00192-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bukong TN, Momen-Heravi F, Kodys K, Bala S, Szabo G. 2014. Exosomes from hepatitis C infected patients transmit HCV infection and contain replication competent viral RNA in complex with Ago2-miR122-HSP90. PLoS Pathog 10:e1004424. doi: 10.1371/journal.ppat.1004424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramakrishnaiah V, Thumann C, Fofana I, Habersetzer F, Pan Q, de Ruiter PE, Willemsen R, Demmers JA, Stalin Raj V, Jenster G, Kwekkeboom J, Tilanus HW, Haagmans BL, Baumert TF, van der Laan LJ. 2013. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc Natl Acad Sci U S A 110:13109–13113. doi: 10.1073/pnas.1221899110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Longatti A, Boyd B, Chisari FV. 2015. Virion-independent transfer of replication-competent hepatitis C virus RNA between permissive cells. J Virol 89:2956–2961. doi: 10.1128/JVI.02721-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bartenschlager R, Penin F, Lohmann V, Andre P. 2011. Assembly of infectious hepatitis C virus particles. Trends Microbiol 19:95–103. doi: 10.1016/j.tim.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 30.Fukuhara T, Ono C, Puig-Basagoiti F, Matsuura Y. 2015. Roles of lipoproteins and apolipoproteins in particle formation of hepatitis C virus. Trends Microbiol 23:618–629. doi: 10.1016/j.tim.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 31.Hueging K, Doepke M, Vieyres G, Bankwitz D, Frentzen A, Doerrbecker J, Gumz F, Haid S, Wolk B, Kaderali L, Pietschmann T. 2014. Apolipoprotein E codetermines tissue tropism of hepatitis C virus and is crucial for viral cell-to-cell transmission by contributing to a postenvelopment step of assembly. J Virol 88:1433–1446. doi: 10.1128/JVI.01815-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barretto N, Sainz B Jr, Hussain S, Uprichard SL. 2014. Determining the involvement and therapeutic implications of host cellular factors in hepatitis C virus cell-to-cell spread. J Virol 88:5050–5061. doi: 10.1128/JVI.03241-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 34.Pietschmann T, Kaul A, Koutsoudakis G, Shavinskaya A, Kallis S, Steinmann E, Abid K, Negro F, Dreux M, Cosset FL, Bartenschlager R. 2006. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc Natl Acad Sci U S A 103:7408–7413. doi: 10.1073/pnas.0504877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A 102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koutsoudakis G, Kaul A, Steinmann E, Kallis S, Lohmann V, Pietschmann T, Bartenschlager R. 2006. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J Virol 80:5308–5320. doi: 10.1128/JVI.02460-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schaller T, Appel N, Koutsoudakis G, Kallis S, Lohmann V, Pietschmann T, Bartenschlager R. 2007. Analysis of hepatitis C virus superinfection exclusion by using novel fluorochrome gene-tagged viral genomes. J Virol 81:4591–4603. doi: 10.1128/JVI.02144-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones CT, Catanese MT, Law LM, Khetani SR, Syder AJ, Ploss A, Oh TS, Schoggins JW, MacDonald MR, Bhatia SN, Rice CM. 2010. Real-time imaging of hepatitis C virus infection using a fluorescent cell-based reporter system. Nat Biotechnol 28:167–171. doi: 10.1038/nbt.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen L, Zhang Y, Li G, Huang H, Zhou N. 2010. Functional characterization of a naturally occurring trans-splicing intein from Synechococcus elongatus in a mammalian cell system. Anal Biochem 407:180–187. doi: 10.1016/j.ab.2010.08.018. [DOI] [PubMed] [Google Scholar]
- 41.Mathiesen CK, Prentoe J, Meredith LW, Jensen TB, Krarup H, McKeating JA, Gottwein JM, Bukh J. 2015. Adaptive mutations enhance assembly and cell-to-cell transmission of a high-titer hepatitis C virus genotype 5a core-NS2 JFH1-based recombinant. J Virol 89:7758–7775. doi: 10.1128/JVI.00039-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moradpour D, Evans MJ, Gosert R, Yuan Z, Blum HE, Goff SP, Lindenbach BD, Rice CM. 2004. Insertion of green fluorescent protein into nonstructural protein 5A allows direct visualization of functional hepatitis C virus replication complexes. J Virol 78:7400–7409. doi: 10.1128/JVI.78.14.7400-7409.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saeed M, Andreo U, Chung HY, Espiritu C, Branch AD, Silva JM, Rice CM. 2015. SEC14L2 enables pan-genotype HCV replication in cell culture. Nature 524:471–475. doi: 10.1038/nature14899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Catanese MT, Uryu K, Kopp M, Edwards TJ, Andrus L, Rice WJ, Silvestry M, Kuhn RJ, Rice CM. 2013. Ultrastructural analysis of hepatitis C virus particles. Proc Natl Acad Sci U S A 110:9505–9510. doi: 10.1073/pnas.1307527110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Catanese MT, Loureiro J, Jones CT, Dorner M, von Hahn T, Rice CM. 2013. Different requirements for scavenger receptor class B type I in hepatitis C virus cell-free versus cell-to-cell transmission. J Virol 87:8282–8293. doi: 10.1128/JVI.01102-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Witteveldt J, Evans MJ, Bitzegeio J, Koutsoudakis G, Owsianka AM, Angus AG, Keck ZY, Foung SK, Pietschmann T, Rice CM, Patel AH. 2009. CD81 is dispensable for hepatitis C virus cell-to-cell transmission in hepatoma cells. J Gen Virol 90:48–58. doi: 10.1099/vir.0.006700-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Steinmann E, Penin F, Kallis S, Patel AH, Bartenschlager R, Pietschmann T. 2007. Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PLoS Pathog 3:e103. doi: 10.1371/journal.ppat.0030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dreux M, Garaigorta U, Boyd B, Decembre E, Chung J, Whitten-Bauer C, Wieland S, Chisari FV. 2012. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe 12:558–570. doi: 10.1016/j.chom.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu Z, Zhang X, Yu Q, He JJ. 2014. Exosome-associated hepatitis C virus in cell cultures and patient plasma. Biochem Biophys Res Commun 455:218–222. doi: 10.1016/j.bbrc.2014.10.146. [DOI] [PubMed] [Google Scholar]
- 50.Gondar V, Molina-Jimenez F, Hishiki T, Garcia-Buey L, Koutsoudakis G, Shimotohno K, Benedicto I, Majano PL. 2015. Apolipoprotein E, but not apolipoprotein B, is essential for efficient cell-to-cell transmission of hepatitis C virus. J Virol 89:9962–9973. doi: 10.1128/JVI.00577-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fukuhara T, Wada M, Nakamura S, Ono C, Shiokawa M, Yamamoto S, Motomura T, Okamoto T, Okuzaki D, Yamamoto M, Saito I, Wakita T, Koike K, Matsuura Y. 2014. Amphipathic alpha-helices in apolipoproteins are crucial to the formation of infectious hepatitis C virus particles. PLoS Pathog 10:e1004534. doi: 10.1371/journal.ppat.1004534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang Z, Wang X, Chi X, Zhao F, Guo J, Ma P, Zhong J, Niu J, Pan X, Long G. 2016. Neglected but important role of apolipoprotein E exchange in hepatitis C virus infection. J Virol 90:9632–9643. doi: 10.1128/JVI.01353-16. [DOI] [PMC free article] [PubMed] [Google Scholar]