

Abstract
Idiopathic pulmonary fibrosis (IPF) is a complex, heterogeneous genetic disorder that is associated with rare and common sequence variants in many genes (MUC5B, SFTPC, SFTPA2, RTEL1, TERT, and hTR), 11 novel loci, and multiple emerging epigenetic and transcriptional profiles. In the past 5 years, we have found that: 1) genetic risk variants play major and similar roles in the development of both familial and sporadic fibrotic idiopathic interstitial pneumonia, accounting for up to 35% of the risk of idiopathic interstitial pneumonia (a disease that was previously thought to be idiopathic); 2) a promoter variant in MUC5B rs35705950 is the strongest risk factor for the development of IIP and IPF; however, rs35705950 has a low penetrance; and 3) IPF is a complex genetic disease with 11 independent loci contributing to the development of this disease, pronounced changes in DNA methylation, and transcriptional subtypes. In aggregate, these findings suggest that IPF is a heterogeneous disease and that genetic and molecular subtypes of IPF will provide essential clues to disease pathogenesis, prognosis, treatment, and survival, all of which remain major problems in understanding and treating patients with IPF. Although the basic biological mechanisms involved in IPF are emerging, the disease is heterogeneous pathologically and the final common pathways of fibrogenesis are not well understood. These observations lead us to postulate that the etiology and severity/extent of this complex condition will best be understood through an integrated approach that accounts for inherited factors, epigenetic marks, and dynamic changes in the transcriptome.
INTRODUCTION
The fibrosing idiopathic interstitial pneumonias (IIPs) are lung diseases characterized by progressive scarring of the alveolar interstitium that lead to significant morbidity and mortality. Idiopathic pulmonary fibrosis (IPF) is the most common and most severe form of IIP with a median survival of 3 years, affects 50,000 individuals annually in the United States, and is increasing in prevalence as our population ages (1,2). Rare mutations have been associated with IPF (3−8); however, these variants account for a small proportion of the attributable risk.
In the past 5 years, we have found that: 1) genetic risk variants play major and similar roles in the development of both familial and sporadic IIP (9), accounting for 35% of the risk of IIP (previously thought to be idiopathic); 2) a promoter variant in MUC5B, rs35705950, is the strongest risk factor for the development of IIP and IPF; however, rs35705950 has a low penetrance (9,10); 3) rs35705950 can potentially be used to identify individuals earlier in the course of disease (11); 4) rs35705950 is associated with unique biological (10,12) and clinical (13) IPF phenotypes; and 5) IPF is a complex genetic disease with 11 independent loci contributing to the development of this disease (based on preliminary data and Fingerlin et al. [9]), pronounced changes in DNA methylation (14), and transcriptional subtypes (15).
RESULTS AND DISCUSSION
We identified a promoter single nucleotide polymorphism (SNP) in the MUC5B gene (rs35705950) that is strongly associated with familial and sporadic IPF (10). This finding has been validated in 8 subsequent independent cohorts (16−22), including our recent genome-wide association study (OR for T [minor] allele = 4.51; 95% confidence interval = 3.91-5.21; P = 7.21 × 1095) (9). Since this promoter variant is conserved and is in a predicted regulatory region (23−25), we explored and found that the MUC5B promoter variant is associated with a 34.1-fold increase in MUC5B expression in lung tissue among unaffected subjects (10,26) and a 5.3-fold increase among IPF patients (26) (Figure 1), with IPF patients expressing 14.1-fold more MUC5B than unaffected controls (10). Immunohistochemical staining showed dense accumulation of MUC5B in terminal bronchioles and areas of microscopic honeycombing (10,12). Based on these observations, we speculate that the MUC5B promoter SNP places individuals at risk of developing IPF via chronic mucus hypersecretion and accumulation in the peripheral airspace that impairs mucociliary transport, results in mucus adhesion in the bronchoalveolar region, and consequently induces and potentiates chronic inflammation and injury (10,27).
Fig. 1.
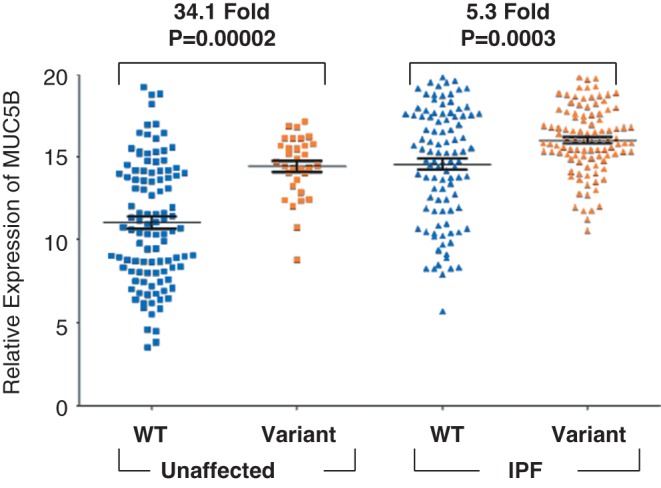
MUC5B gene expression in idiopathic pulmonary fibrosis (IPF) (N = 203) and unaffected subjects (N = 139) stratified by MUC5B promoter single nucleotide polymorphism (SNP) (rs35705950) genotype.
With the goal of identifying additional genetic risk factors that collectively further our understanding of pulmonary fibrosis, we conducted a case-control genome-wide association study (1,616 non-Hispanic white cases [IPF = 1,251 (77%)] and 4,683 controls) and replication study (876 non-Hispanic white cases [IPF = 774 (88%)] and 1890 controls) of IIP (9). Under an additive model for the minor allele at each SNP, we identified 19 SNPs, representing seven chromosomal locations (5p15, 6p24, 7q22, 11p15, 15q14-15, 17q21, and 19p13), with genome-wide significant associations (P < 5 × 108; Figure 2). We genotyped the 19 genome-wide significant SNPs in addition to 178 SNPs with 5 × 108 < P < .0001 (SNPs between red and blue lines in Figure 2) and found that four additional loci (3q26, 4q22, 10q24, and13q34) had genome-wide significant SNPs in the meta-analysis.
Fig. 2.

Genome-wide association study results at 439,828 single nucleotide polymorphisms (SNPs) with 1,616 cases and 4,683 controls under additive model. SNPs above red line were genome-wide significant at P < 5 × 10–8. These SNPs and SNPs between red and blue lines, corresponding to 5 × 10–8 < P < .0001 were followed-up in 876 cases and 1,890 controls. Adapted from Fingerlin et al. (9).
Epigenetic mechanisms are likely to be involved in the control of gene expression in IPF, especially given the association of IPF with cigarette smoking and the relationship between cigarette smoke and changes in the epigenome. Moreover, these epigenetic changes are likely to be important factors in determining transcriptional profiles that directly contribute to pathogenic features of this disease. Several targeted studies have shown that epigenetic modulation regulates expression of genes involved in IPF, namely, cyclooxygenase-2 (28), chemokine IP-10 (29), Thy-1 (CD90) (30,31), p14(ARF) (32), and a-smooth muscle actin (33,34).
Epigenomic studies of DNA methylation profiles in IPF are just emerging. Two genomic studies of DNA methylation in IPF lung have revealed extensive DNA methylation changes both within (35) and outside (36) cytosine guanine deoxynucleotide (CpG) islands. To identify methylation marks that modify gene expression in IPF lung, we assessed genomic methylation and gene expression in 94 IPF and 67 control subjects (14). We identified 2,130 differentially methylated regions (<5% false discovery rate) of which 738 are associated with significant changes in gene expression and enriched for expected inverse relationship between methylation and expression (P < 2.2 × 1016). We validated 13 of 15 differentially methylated regions (DMRs) by targeted analysis of methylation (Epityper; Sequenom, San Diego, CA). Methyl-expression quantitative trait loci (eQTL) analysis identified methylation marks that control cis and trans gene expression, with an enrichment for cis relationships (P < 2.2 × 1016). We identified five trans methyl-eQTLs where a methylation change at a single DMR is associated with transcriptional changes in a substantial number of genes; four of these DMRs are near transcription factors (CASZ1, FOXC1, MXD4, and ZDHHC4). We studied the in vitro effects of change in CASZ1 expression and validated its role in regulation of target genes in the methyl-eQTL. These results suggest that DNA methylation may be involved in the pathogenesis of IPF.
Gene expression profiling studies have demonstrated that transcriptional changes are present in lung parenchyma from individuals with IPF (37−43). IPF associated gene expression changes are dramatic and involve a few thousand differentially expressed genes. In aggregate, these studies have consistently identified the same genes and pathways that are differentially expressed in fibrotic lungs, namely, genes associated with extracellular matrix formation, degradation, and signaling, smooth muscle markers, growth factors, developmental pathways, and genes encoding immunoglobulins, complement, chemokines, and host defense/innate immune genes. We recently identified two molecular subtypes of IPF (Figure 3) based on a strong gene expression signature (transcript clusters A-C in Figure 3) in 119 Lung Tissue Research Consortium IPF subjects (15). Transcript cluster B contains 80 unique genes upregulated in group II compared to group I IPF that include a number of genes that have been previously shown to be upregulated in IPF, namely, osteopontin, MMP1, MMP7, PLUNC, MUC5B, collagen COL17A1, and keratins 5, 6C, 15, and 17. Our results demonstrate that these IPF-associated genes differentiate two subpopulations of subjects with IPF. Cluster C contains 71 unique genes that are downregulated in group II compared to group I IPF with a few previously implicated in IPF (advanced glycosylation end product−specific receptor [43]) or other chronic lung diseases (hedgehog interacting protein [44]) and many novel IPF candidate genes. Functional enrichment analysis, using Fisher’s exact test, of the 121 unique transcripts in cluster A showed it to be strongly enriched in transcripts associated with the cilium genes (Gene Ontology category 0005929; Benjamini corrected P = 3.7 × 1011) and their structural components (axoneme, P = 3.9 × 1011; dynein, P = 9.4 × 107). The cilium gene signature was validated in multiple lung specimens from the same subjects (expression profiles of middle and upper lobes from group I and group II IPF are distinguishable but differences are attenuated compared to lower lobes, consistent with disease localization to lower lobes) and in an independent cohort of subjects with IPF (same findings in 111 National Jewish Health IPF subjects). In addition to being associated with higher expression of MUC5B and MMP7, higher cilium gene expression is also associated with the presence of microscopic honeycombing (P < 0.0001) but not fibroblastic foci (P = 1.0) (15). The most significant network of genes upregulated in group II compared to group I IPF contains cilium genes as well as transcription factors that regulate expression of cilium genes (FOXJ1, RFX2, and RFX3).
Fig. 3.
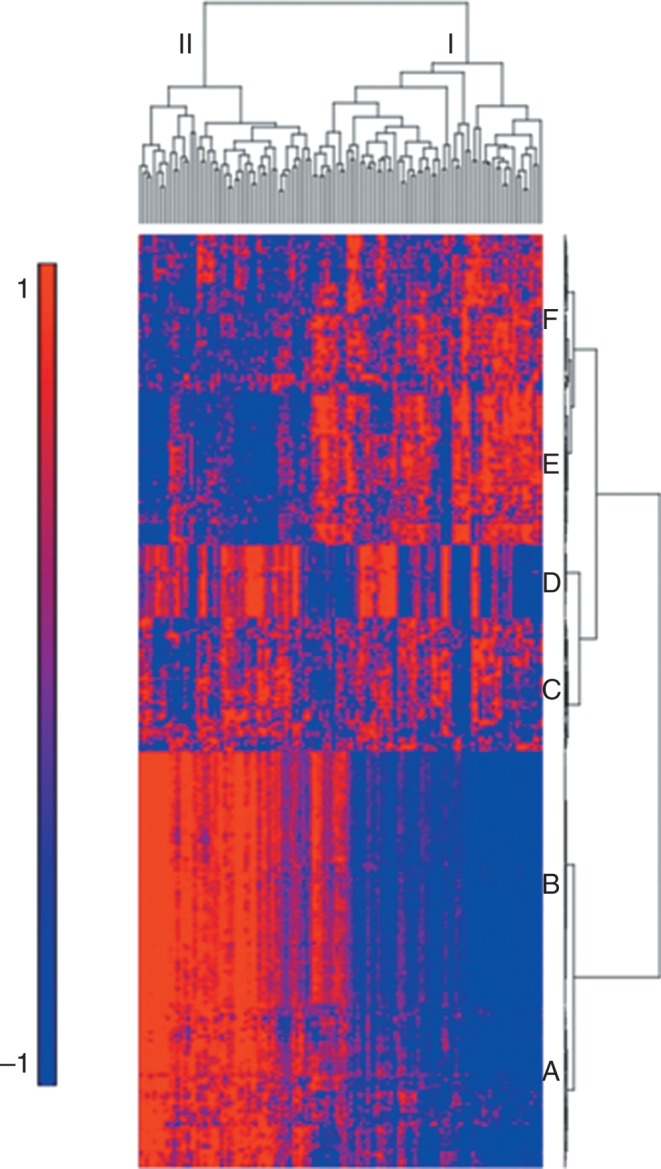
Gene expression profiling identifies two subtypes of idiopathic pulmonary fibrosis (IPF). mRNA profiles from 119 IPF lungs were subject to hierarchical clustering based on the expression of 472 transcripts that are differentially expressed at 5% false discover rate (FDR) and with greater than 2-fold change in IPF compared to control lung. Decreased expression is represented by blue color, whereas red indicates increased expression. Adapted from Figure 1 in Yang et al. (15).
Isolated genetics, epigenetics, transcriptomics, and clinical attributes have so far provided only a partial picture regarding risk and severity/extent of IPF. We believe that the proposed comprehensive, multidimensional model of IPF will: 1) establish the basic molecular profiles to develop novel molecular insights into disease etiology and clinical severity/extent; 2) facilitate a strategic approach to the development of primary and secondary forms of disease prevention; and 3) provide the rationale and targets/biomarkers for intervention in this fatal disease.
ACKNOWLEDGMENTS
Supported by the National Heart, Lung and, Blood Institute (R01-HL097163, R01-HL130140, R21-HL120770, and UH2-HL123442) and the Veteran’s Administration (1I01BX001534). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Potential Conflicts of Interest: None disclosed.
DISCUSSION
Anderson, Baltimore: David that was a great story. So I am just thinking as a non-lung person about the phenotypic divergent of various lung disorders that share this feature of impaired mucociliary clearance, you know, too much mucus. Is there a connectedness? Or how do you think about those differences? Why doesn’t asthma become pulmonary fibrosis and why doesn’t CF become pulmonary fibrosis?
Schwartz, Denver: When I consider CF and COPD, both diseases associated with increased mucus production, I think the type and location of disease is, in part, dependent on where the mucus is being produced, what type of mucus is overproduced, and other contextual features of the disease. For example, if CF mucus is overproduced in the proximal airway and the submucosal glands, mucociliary transport is severely impaired due to excessive salt in the airway, and bacteria persistently infect the airway. In COPD, MUC5AC, rather than MUC5B, is produced in the proximal airway and is associated with persistent mucosal inflammation. For IPF, overproduction of mucus is in the distal airway and is primarily MUC5B, potentially altering the balance of injury and repair at the bronchoalveolar junction.
Johnson, Denver: Wonderful talk…..two questions for you, one is: in people with sarcoidosis or silica exposure who are at risk for pulmonary fibrosis, does this polymorphism increase their risk to develop their fibrosis? And then the second question is if this is due to retention of bacteria in certain areas leading to low grade inflammation, has anyone ever tried prophylactic antibiotics to prevent or to reduce the risk for progression to fibrosis?
Schwartz, Denver: Regarding the relationship of the MUC5B promoter polymorphism to other fibro-proliferative diseases of the lung, we have looked at many of them: sarcoidosis, scleroderma, rheumatoid arthritis associated in lung disease, and asbestosis; and have found that the MUC5B polymorphism is specific to idiopathic pulmonary fibrosis. We have also looked at COPD and asthma and neither of those diseases appear to be associated with this particular gene variant; the MUC5B promoter variant appears to be specific to idiopathic pulmonary fibrosis. Prophylactic antibiotics have not been tried in idiopathic pulmonary fibrosis; however, pirfenidone, an agent recently found to slow the progression of idiopathic pulmonary fibrosis, has antimicrobial properties.
Sorscher, Atlanta: That was a wonderful talk. I just wondered if you have any direct connection pathophysiological or otherwise, between MUC5B and the TGF beta pathway that signals to myofibroblast and fibrosis and so forth. Have you looked at that?
Schwartz, Denver: We haven’t studied this and we don’t understand the relationship between MUC5B overproduction in the distal airway and the fibroproliferative activity in the alveoli and interstitium. However, this is an active area of investigation.
Annex, Charlottesville: Spectacular talk….really terrific….two quick genetic questions; one is: in the human expressions study, there is enormous amount of overlap between the disease in the control group in regard to expression. So, thoughts about confounders or other factors involved? And the second is, is there any evidence in mice, of strain differences and susceptibility?
Schwartz, Denver: Cigarette smoking is an important risk factor for the development idiopathic pulmonary fibrosis and we’re in the process of studying the relationship between cigarette smoking, excess MUCB production, and the development of pulmonary fibrosis. In terms of mouse strains, while we know that different strains of mice demonstrate differential susceptibility to bleomycin, asbestos, and radiation, we don’t yet know whether mucins are involved in this determining these different patterns of susceptibility. We have created genetically engineered mice that have either no expression of MUC5b or have cell specific overexpression of MUC5b, and we’re in the process of determining how the concentration of Muc5b in mice affects susceptibility to fibroproliferative agents.
Duffy, New Haven: In your lovely paper, you describe the association of MUC5B with exaggerated mucus secretion. And then the question was asked with regard to: how does that create fibrosis in the TGF-beta pathway. My question is, there is a cell within the pulmonary tree which lines the entire epithelium of tree….which is the mast cell…and the mast cell is probably one of the most potent cells in inducing fibrosis…..do you know anything with regard to the mast cell in this particular pathway?
Schwartz, Denver: While there are reasons to believe that the mast cell may be important in the persistent inflammation and fibrosis that is observed in idiopathic pulmonary fibrosis, the mast cell has not been a focus of study in pulmonary fibrosis. We haven’t looked at the mast cell, however, we’re beginning to study stem cells and, in particular, the effect of excess MUC5B production on stem cell development in the lung.
Duffy, New Haven: Lovely work, thank you.
Crystal, New York City: Dave, lovely body of work….one of the interesting aspects of the MUC5B promoter variant is that it’s also frequent in your control group….do you have any thoughts in terms why they are resistant to development of the disease.
Schwartz, Denver: The high frequency of the MUC5B promoter variant in our controls tells us a few things. First, it’s likely that in addition to the MUC5B promoter variant, another factor (genetic, environmental, other) needs to be present to actually cause disease. Second, the group of susceptible individuals is large and it’s likely that this disease is underdiagnosed and much more common than we currently appreciate. Third, it’s likely that there is a positive benefit to having the MUC5B promoter variant that selected that variant into the gene pool and maintains it in the gene pool. It’s quite possible that this variant and increased MUC5B production early in life improves host defense, preventing or reducing the severity of a potentially lethal childhood respiratory disease.
Bray, Philadelphia: My question deals with the last one: does the allele frequency vary by geographical ancestry? Who would you screen for this? And do you think this is the causative variant? You showed it was associated with RNA levels, but do you think it’s the causative variant?
Schwartz, Denver: We don’t know whether it’s the causative variant yet. What I can tell you is it’s the strongest risk factor for idiopathic pulmonary fibrosis, genetic or otherwise. However, since 19% of the non-Hispanic White population has one or two copies of the MUC5B promoter polymorphism and far fewer develop pulmonary fibrosis, it’s much more likely that the variant represents a susceptibility allele that is not causative. In terms of ancestry, I find that to be an absolutely fascinating question…it turns out while the variant is present in 19% of non-Hispanic whites, the variant is present in 2% to 3% of Asians and less than 1% of Africans. And the prevalence of this allele in different ancestries is consistent with the prevalence of idiopathic pulmonary fibrosis in different ethnic groups. We have been able to actually ancestrally map this variant back to the British Isles and we think that this was a variant that was introduced into the genome in that population, and is under positive selection with potential benefit early in life. It probably has an effect in terms of viral illness early in life. I think the ancestral origin of the MUC5B promoter variant is the key to understanding why this variant has been maintained in non-Hispanic whites and also a key to understanding how this variant alters homeostatic properties in the distal lung that lead to the development of idiopathic pulmonary fibrosis.
Bray, Philadelphia: So should all non-Hispanic whites be screened for it?
Schwartz, Denver: Not yet. While screening would identify individuals with either early interstitial lung disease or individuals at risk of developing idiopathic pulmonary fibrosis, currently we don’t have anything to offer these individuals. So while genetic screening companies are offering this screen, I’m not convinced that we’re doing our patients a service by screening them for this variant. We and others are involved in developing therapeutic approaches for individuals with early interstitial lung disease, treatment that would prevent the development of pulmonary fibrosis.
Boxer, Ann Arbor: As you are well aware, dyskeratosis congenita is associated with pulmonary fibrosis, so I like the thought about it being a stem cell defect….what proportion of your patients in large clinic have dyskeratosis congenita and you screen for that?
Schwartz, Denver: Dyskeratosis congenita is an uncommon finding in pulmonary fibrosis and is associated with rare variants in the telomerase genes. Rare variants in TERT and TERC are unusual causes of idiopathic pulmonary fibrosis, and the association of pulmonary fibrosis with dyskeratosis congenita is even more uncommon.
REFERENCES
- 1.Raghu G, Weycker D, Edelsberg J, Bradford WZ, et al. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174(7):810–6. doi: 10.1164/rccm.200602-163OC. [DOI] [PubMed] [Google Scholar]
- 2.Olson AL, Swigris JJ, Lezotte DC, Norris JM, et al. Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am J Respir Crit Care Med. 2007;176(3):277–84. doi: 10.1164/rccm.200701-044OC. Epub 2007/05/05. [DOI] [PubMed] [Google Scholar]
- 3.Armanios MY, Chen JJ, Cogan JD, Alder JK, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356(13):1317–26. doi: 10.1056/NEJMoa066157. Epub 2007/03/30. [DOI] [PubMed] [Google Scholar]
- 4.Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci U S A. 2007;104(18):7552–7. doi: 10.1073/pnas.0701009104. Epub 2007/04/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas AQ, Lane K, Phillips J, 3rd, Prince M, et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med. 2002;165(9):1322–8. doi: 10.1164/rccm.200112-123OC. Epub 2002/05/07. [DOI] [PubMed] [Google Scholar]
- 6.Lawson WE, Grant SW, Ambrosini V, Womble KE, et al. Genetic mutations in surfactant protein C are a rare cause of sporadic cases of IPF. Thorax. 2004;59(11):977–80. doi: 10.1136/thx.2004.026336. Epub 2004/11/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y, Kuan PJ, Xing C, Cronkhite JT, et al. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet. 2009;84(1):52–9. doi: 10.1016/j.ajhg.2008.11.010. Epub 2008/12/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Moorsel CH, van Oosterhout MF, Barlo NP, de Jong PA, et al. Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a dutch cohort. Am J Respir Crit Care Med. 2010;182(11):1419–25. doi: 10.1164/rccm.200906-0953OC. Epub 2010/07/27. [DOI] [PubMed] [Google Scholar]
- 9.Fingerlin TE, Murphy E, Zhang W, Peljto AL, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. 2013;45(6):613–20. doi: 10.1038/ng.2609. Epub 2013/04/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seibold MA, Wise AL, Speer MC, Steele MP, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364(16):1503–12. doi: 10.1056/NEJMoa1013660. Epub 2011/04/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hunninghake GM, Hatabu H, Okajima Y, Gao W, et al. MUC5B promoter polymorphism and interstitial lung abnormalities. N Engl J Med. 2013;368(23):2192–200. doi: 10.1056/NEJMoa1216076. Epub 2013/05/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seibold MA, Smith RW, Urbanek C, Groshong SD, et al. The idiopathic pulmonary fibrosis honeycomb cyst contains a mucocilary pseudostratified epithelium. PLoS One. 2013;8(3):e58658. doi: 10.1371/journal.pone.0058658. Epub 2013/03/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peljto AL, Zhang Y, Fingerlin TE, Ma SF, et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA. 2013;309(21):2232–9. doi: 10.1001/jama.2013.5827. Epub May 21, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang IV, Pedersen BS, Rabinovich E, Hennessy CE, et al. Relationship of DNA methylation and gene expression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2014;190(11):1263–72. doi: 10.1164/rccm.201408-1452OC. Epub 2014/10/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang IV, Coldren CD, Leach SM, Seibold MA, et al. Expression of cilium-associated genes defines novel molecular subtypes of idiopathic pulmonary fibrosis. Thorax. 2013;68(12):1114–21. doi: 10.1136/thoraxjnl-2012-202943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Noth I, Garcia GN, Kaminski N. A variant in the promoter of MUC5B and idiopathic pulmonary fibrosis. N Engl J Med. 2011;364(16):1576–7. doi: 10.1056/NEJMc1013504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stock CJ, Sato H, Fonseca C, Banya WA, et al. Mucin 5B promoter polymorphism is associated with idiopathic pulmonary fibrosis but not with development of lung fibrosis in systemic sclerosis or sarcoidosis. Thorax. 2013 doi: 10.1136/thoraxjnl-2012-201786. Epub 2013/01/17. [DOI] [PubMed] [Google Scholar]
- 18.Noth I, Zhang Y, Ma SF, Flores C, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med. 2013;1(4):309–17. doi: 10.1016/S2213-2600(13)70045-6. Epub April 17, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borie R, Crestani B, Dieude P, Nunes H, et al. The MUC5B Variant is associated with idiopathic pulmonary fibrosis but not with systemic sclerosis interstitial lung disease in the European Caucasian population. PLoS One. 2013;8(8):e70621. doi: 10.1371/journal.pone.0070621. Epub 2013/08/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wei R, Li C, Zhang M, Jones-Hall YL, et al. Association between MUC5B and TERT polymorphisms and different interstitial lung disease phenotypes. Transl Res. 2014;163(5):494–502. doi: 10.1016/j.trsl.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horimasu Y, Ohshimo S, Bonella F, Tanaka S, et al. MUC5B promoter polymorphism in Japanese patients with idiopathic pulmonary fibrosis. Respirology. 2015;20(3):439–44. doi: 10.1111/resp.12466. [DOI] [PubMed] [Google Scholar]
- 22.Peljto AL, Selman M, Kim DS, Murphy E, et al. The MUC5B promoter polymorphism is associated with idiopathic pulmonary fibrosis in a Mexican cohort but is rare among Asian ancestries. Chest. 2015;147(2):460–4. doi: 10.1378/chest.14-0867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fujisawa T, Chang MM, Velichko S, Thai P, et al. NF-κB Mediates IL-1β and IL-17A-induced MUC5B expression in the airway epithelial cells. Am J Respir Cell Mol Biol. 2010 doi: 10.1165/rcmb.2009-0313OC. Epub 2010/10/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15(8):1034–50. doi: 10.1101/gr.3715005. Epub 2005/07/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.King DC, Taylor J, Elnitski L, Chiaromonte F, et al. Evaluation of regulatory potential and conservation scores for detecting cis-regulatory modules in aligned mammalian genome sequences. Genome Res. 2005;15(8):1051–60. doi: 10.1101/gr.3642605. Epub 2005/07/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Helling BA, Yang IV, Steele MP, Brown KA, et al. MUC5B is a common link between idiopathic pulmonary fibrosis and non-specific interstitial pneumonia. paper in preparation, 2016 [Google Scholar]
- 27.Boucher RC. Idiopathic pulmonary fibrosis—a sticky business. N Engl J Med. 2011;364(16):1560–1. doi: 10.1056/NEJMe1014191. Epub 2011/04/22. [DOI] [PubMed] [Google Scholar]
- 28.Coward WR, Watts K, Feghali-Bostwick CA, Knox A, et al. Defective histone acetylation is responsible for the diminished expression of cyclooxygenase 2 in idiopathic pulmonary fibrosis. Mol Cell Biol. 2009;29(15):4325–39. doi: 10.1128/MCB.01776-08. Epub 2009/06/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coward WR, Watts K, Feghali-Bostwick CA, Jenkins G, et al. Repression of IP-10 by interactions between histone deacetylation and hypermethylation in idiopathic pulmonary fibrosis. Mol Cell Biol. 2010;30(12):2874–86. doi: 10.1128/MCB.01527-09. Epub 2010/04/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanders YY, Pardo A, Selman M, Nuovo GJ, et al. Thy-1 promoter hypermethylation: a novel epigenetic pathogenic mechanism in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2008;39(5):610–8. doi: 10.1165/rcmb.2007-0322OC. Epub 2008/06/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanders YY, Tollefsbol TO, Varisco BM, Hagood JS. Epigenetic regulation of thy-1 by histone deacetylase inhibitor in rat lung fibroblasts. Am J Respir Cell Mol Biol. 2011;45(1):16–23. doi: 10.1165/rcmb.2010-0154OC. Epub 2010/08/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cisneros J, Hagood J, Checa M, Ortiz-Quintero B, et al. Hypermethylation-mediated silencing of p14(ARF) in fibroblasts from idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012;303(4):L295–303. doi: 10.1152/ajplung.00332.2011. Epub 2012/06/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu B, Gharaee-Kermani M, Wu Z, Phan SH. Epigenetic regulation of myofibroblast differentiation by DNA methylation. Am J Pathol. 2010;177(1):21–8. doi: 10.2353/ajpath.2010.090999. Epub 2010/05/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu B, Gharaee-Kermani M, Wu Z, Phan SH. Essential role of MeCP2 in the regulation of myofibroblast differentiation during pulmonary fibrosis. Am J Pathol. 2011;178(4):1500–8. doi: 10.1016/j.ajpath.2011.01.002. Epub 2011/03/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rabinovich EI, Kapetaneli M, Steinfeld I, Gibson KF, et al. Global methylation patterns in idiopathic pulmonary fibrosis. PLoS One. 2012;7(4):e33770. doi: 10.1371/journal.pone.0033770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanders YY, Ambalavanan N, Halloran B, Zhang X, et al. Altered DNA methylation profile in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;186(6):525–35. doi: 10.1164/rccm.201201-0077OC. Epub 2012/06/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zuo F, Kaminski N, Eugui E, Allard J, et al. Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc Natl Acad Sci U S A. 2002;99(9):6292–7. doi: 10.1073/pnas.092134099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaminski N. Microarray analysis of idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2003;29(3 Suppl :S):32–6. [PubMed] [Google Scholar]
- 39.Selman M, Pardo A, Barrera L, Estrada A, et al. Gene expression profiles distinguish idiopathic pulmonary fibrosis from hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2006;173(2):188–98. doi: 10.1164/rccm.200504-644OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Selman M, Carrillo G, Estrada A, Mejia M, et al. Accelerated variant of idiopathic pulmonary fibrosis: clinical behavior and gene expression pattern. PLoS ONE. 2007;2(5):e482. doi: 10.1371/journal.pone.0000482. Epub 2007/05/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang IV, Burch LH, Steele MP, Savov JD, et al. Gene expression profiling of familial and sporadic interstitial pneumonia. Am J Respir Crit Care Med. 2007;175(1):45–54. Epub 2006/09/26. [Google Scholar]
- 42.Boon K, Bailey NW, Yang J, Steel MP, et al. Molecular phenotypes distinguish patients with relatively stable from progressive idiopathic pulmonary fibrosis (IPF) PLoS One. 2009;4(4):fe5134. doi: 10.1371/journal.pone.0005134. Epub 2009/04/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Konishi K, Gibson KF, Lindell KO, Richards TJ, et al. Gene expression profiles of acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;180(2):167–75. doi: 10.1164/rccm.200810-1596OC. Epub 2009/04/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pillai SG, Kong X, Edwards LD, Cho MH, et al. Loci identified by genome-wide association studies influence different disease-related phenotypes in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;182(12):1498–505. doi: 10.1164/rccm.201002-0151OC. Epub 2010/07/27. [DOI] [PMC free article] [PubMed] [Google Scholar]