

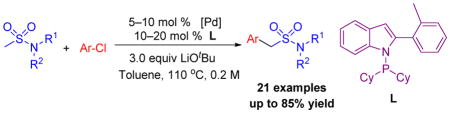
Abstract
A Palladium-catalyzed α-arylation of sulfonamides with aryl chlorides is presented. A Buchwald type precatalyst formed with Kwong’s indole-based ligand enabled this transformation to be compatible with a large variety of methyl sulfonamides and aryl chlorides in good to excellent yields. Importantly, under the optimized reaction conditions, only mono-arylated products were observed. This method has been applied to the efficient synthesis of sumatriptan, which is used to treat migraines.
Keywords: Palladium-catalyzed, arylation, sulfonamide, aryl chloride, sumatriptan
Graphical Abstract
Introduction
Sulfonamides are widely occurring in synthetic intermediates and bioactive compounds.[1] They are also important structural motifs in drugs such as Almotriptan,[2] Avitriptan[3] and Sumatriptan[4] (Figure 1). Previous methods to access sulfonamides range from simple N–S bond formation,[5] to tandem reactions that also include S=O,[6] C–S,[7] and C–N bond-forming reactions.[8] Although α-arylation of unfunctionalized methyl sulfonamides with aryl halides to form C–C bonds is an attractive route to these important compounds, such transformations have met with limited success.
Figure 1.

Sulfonamides motifs in drugs.
A viable approach to the α-arylation of sulfonamides is that used in the arylation of ketones and esters.[9] Due to the synthetic utility of α-arylated carbonyl compounds, the arylation of these substrates has been investigated by numerous research groups.
Despite similarities between α-arylation of ketones and α-arylation of sulfonamides, the pKa values of the α-hydrogens of sulfonamides, such as CH3SO2NR2, are estimated to be in the range of 32–35.[10], [11] This high pKa value is approaching those of the α-hydrogens of acetamides, CH3CONR2, which have proven very challenging substrates in arylation reactions.[10]
Early studies on the α-arylation of methyl sulfonamides by the Parkinson group[11] (Scheme 1, A) employed Pd(OAc)2/PtBu3, NaOtBu, and aryl bromides and gave moderate yields (average yield with Ph–Br of 44%). The dominant byproducts in these reactions were derived from deprotonation of the more acidic monoarylation product and subsequent arylation to provide diarylated products. Recently, René and coworkers explored the arylation of cyclic sulfonamides (sultams).[12] They noted that the arylation did not provide product in the presence of LiN(SiMe3)2, KN(SiMe3)2, NaOtBu, or TMP–MgCl•LiCl bases. Arylation of sultams was observed with TMP–ZnCl•LiCl, aryl iodides, and 10 mol % of a Pd(RuPhos)-based catalyst (Scheme 1, B). One example with chlorobenzene was presented in this study that gave 39% yield. The results from these two groups suggest that the direct C–H arylation of sulfonamides is a challenging reaction.
Scheme 1.

Palladium-catalyzed α-arylation of sulfonamides.
The Zhou group also reported difficulties in the direct C–H functionalization of sulfonamides.[13] They found that the reaction of 1-(methylsulfonyl)piperidine, bromobenzene, and LiN(SiMe3)2 in the presence of a palladium catalyst failed to give coupling product. Switching to a Negishi coupling with intermediate transmetallation to zinc, they discovered that combination of 1.25 equiv LiN(SiMe3)2 and 1.5 equiv of ZnCl2 with methyl sulfonamides and a Pd(X-Phos)-based catalyst resulted in cross-coupling to furnish the arylation product in good to excellent yields (Scheme 1, C).[14] Although the transmetallation to zinc facilitated the coupling process, introduction of the zinc salts increases the cost and lowers the overall atom economy.
Despite these efforts, general direct intermolecular C–H α-arylation of methyl sulfonamides with more economical and abundant aryl chlorides is scarcely reported. Herein, we report the first chemoselective mono-arylation of methyl sulfonamides with aryl chlorides (Scheme 1, D). A Buchwald type precatalyst[15] formed with Kwong’s indole-based ligand[16] and alkoxide base led to formation of mono-arylation products, with no diarylation observed. This method has been applied to the efficient synthesis of the marketed medication sumatriptan.
Results and Discussion
Two important variables in the development of arylation of weakly acidic C(sp3)–H’s are 1) identification of a suitable ligand and 2) choice of a palladium precursor that will efficiently generate the catalyst. Based on our previous experience in the palladium-catalyzed α-arylation of sulfoxides,[17] sulfones,[18] and amides,[19] we initiated studies of the coupling between 1-(methylsulfonyl)piperidine 1a and aryl chlorides employing Buchwald’s palladium precatalysts (see Table 2 for structures).
Table 2.
Substrate scope of aryl chlorides in the α-arylation of sulfonamide 1a with Kwong’s indole-based phosphine ligand.a

|
Reactions conducted on a 0.2 mmol scale using 1 equiv of 1a, 3 equiv of LiOtBu, and 2 equiv of ArCl. Isolated yields after chromatographic purification.
10 mol % Pd loading in CPME.
10 mol % Pd loading after 48 h.
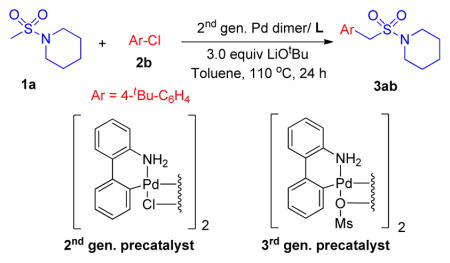
Using microscale (10 umol) High Throughput Experimentation (HTE) techniques [20] we examined the coupling between 1-(methylsulfonyl)piperidine 1a and 1-(tert-butyl)-4-chlorobenzene 2b by screening ligands known to perform well in many coupling reactions. A total of 37 electronically diverse mono– and bidentate phosphines were examined using Buchwald’s 2nd generation palladium dimer, LiOtBu as base, toluene as solvent at 110° C for 24 h (see the Supporting Information for details). In the ligand screen, the reaction using Kwong’s indole-based ligand exhibited the highest assay yield based on the product/internal standard ratio (2.05) (determined by integration of the product against the biphenyl internal standard by HPLC (UV/vis, see Supporting information for details). Other ligands that showed good assay yields were cataCXium ABn (1.83), JosiPhos (1.75), PtBu3 (1.42), DavePhos (1.36), MePhos (1.33) in HPLC.
With the best ligand hit, we next conducted a second microscale screen (0.01 mmol) focusing on six bases [LiOtBu, NaOtBu, KOtBu, LiN(SiMe3)2, NaN(SiMe3)2, and KN(SiMe3)2] and four solvents [THF, CPME, DME, toluene] with Buchwald’s 2nd and 3rd generation palladium dimers (see Supporting Information for details). The most promising hit from this screen was LiOtBu, Buchwald-type 2nd generation palladium dimer in toluene at 110 °C. On laboratory scale (0.1 mmol), such conditions led to the desired arylation product in 96% assay yield (determined by 1H NMR analysis, Table 1, entry 1). Changing the concentration from 0.2 to 0.1 and 0.05 M resulted in decreased assay yields to 88% and 80%, respectively (entries 2–3). A significant decrease in assay yield to 66% was observed when lowering the temperature to 80 °C (entry 4). Further optimization of the catalyst loading indicated that the assay yield fell to 87% at 5 mol % Pd (entry 5) and dropped to 75% at 2.5 mol % (entry 6). Using 5.0 mol % palladium, the isolated yield was 85% (entry 5).
Table 1.
| ||||
|---|---|---|---|---|
| Entry | dimer/L (mol %) | Conc. (M) | Temp (°C) | Assay yield (%) |
| 1 | 5/20 | 0.2 | 110 | 96 |
| 2 | 5/20 | 0.1 | 110 | 88 |
| 3 | 5/20 | 0.05 | 110 | 80 |
| 4 | 5/20 | 0.2 | 80 | 66 |
| 5 | 2.5/10 | 0.2 | 110 | 87 (85)c |
| 6 | 1.25/5 | 0.2 | 110 | 75 |
Reactions conducted on a 0.1 mmol scale using 1 equiv of 1a, 3 equiv of LiOtBu, and 2 equiv of ArCl.
Yields determined by 1H NMR spectroscopy of the crude reaction mixtures on a 0.1 mmol scale.
Isolated yield after chromatographic purification.
Starting from the optimized conditions (Table 1, entry 5), we explored the scope of the reaction (Table 2). In general, mono-arylated products were formed with very good yields. Chlorobenzene rendered product 3aa in 80% yield. Aryl chlorides bearing electron donating 4-tBu (2b) and 4-methoxy (2c) groups provided products in excellent yields at 5 and 10 mol % Pd loading (85% and 79% yield, respectively, for 3ab and 3ac). 4-Fluoro-1-chlorobenzene was successfully coupled with 1a to afford 3ad in 69% yield. Electron poor 4-chlorobenzophenone afforded 3ae in 57% yield at 10 mol % Pd loading in CPME. Sterically hindered 2-chlorotoluene (2f) delivered desired product 3af in 70% yield. Aryl chlorides bearing various substituents at the 3-position were good substrates. For example, 3-chlorotoluene furnished the product (3ag) in 75% yield, 3-chloro-N,N-dimethylaniline provided 3ah in 78% yield (10 mol % Pd in CPME), 1-chloro-3,5-dimethoxybenzene led to 3ai in 79% yield (10 mol % Pd in CPME), and 1-chloro-3,5-difluorobenzene resulted in formation of 3aj in 60% yield. Hetero aryl chlorides 3-chloropyridyl and 5-chloroquinoline were coupled with 1a to afford 3ak and 3al in 72% and 63% yield, respectively, after extending reaction times to 48 h.
We next examined the substrate scope of sulfonamides with different substituents on the nitrogen (Table 3). Arylation of pyrrolidine-substituted sulfonamide (1b) was sluggish under the optimized conditions, and required increasing Pd loading from 5 to 10 mol % to afford product 3ba in 75% yield. Morpholine substituted sulfonamide 1c underwent coupling with chlorobenzene to give product 3ca in 69% yield under the standard conditions. Acyclic substrates N,N-dimethyl (1d) and N,N-diethyl (1e) methyl sulfonamides coupled with chlorobenzene to form the mono-arylated products 3da and 3ea in 78% and 63% yield, respectively. Extending the alkyl group to N,N-diisopropyl rendered product 3fa in 80% yield. Furthermore, N-benzyl-N-methyl, N-benzyl-N-isopropyl, and N-benzyl-N-phenyl-substituted sulfonamides led to satisfactory yields of arylation products 3ga, 3ha, 3ia in 83, 70 and 70% yield, respectively, without any arylation at the benzylic positions observed.[21]
Table 3.
Substrate scope of sulfonamides in the α-arylation of aryl chloride 2a.a

|
Reactions conducted on a 0.2 mmol scale using 1 equiv of sulfonamide, 3 equiv of LiOtBu, and 2 equiv of PhCl. Isolated yield after chromatographic purification.
10 mol % Pd loading.
We desired to evaluate the scalability of our method (Scheme 3). We conducted the arylation of N,N-diisopropylmethane sulfonamide 1f with chlorobenzene 2a on an 8 mmol scale with 5.0 mol % Pd loading. The product 3fa was isolated in 82% yield (1.67 g).
Scheme 3.

Gram scale synthesis of 3fa.
We next applied our method in the synthesis of a marketed pharmaceutical. Sumatriptan is a 5-HT receptor agonist[22] used in the treatment of migraines. The synthesis of sumatriptan (3ja) was envisioned through the coupling of N-benzyl-N-methylmethanesulfonamide (1g) with tert-butyl 5-chloro-3-(2-(dimethylamino)ethyl)-1H-indole-1-carboxylate (2m). The coupling between 1g and 2m under standard conditions followed by removal of the protecting group gave rise to sumatriptan in 75% yield (Scheme 4).
Scheme 4.

Synthesis of sumatriptan (3ja).
Conclusion
In summary, benzylic sulfonamides are an important class of bioactive compounds and marketed pharmaceuticals. One can envision rapid assembly of an array of such compounds from methyl sulfonamides and commercially available aryl halides, however, a α-arylation of methylsulfonamides that avoides an intermediate transmetallation step remained elusive. Herein, we advance the first general palladium-catalyzed α-arylation of sulfonamides with aryl chlorides. Under the optimized reaction conditions, good to excellent yields of mono-arylated methyl sulfonamides have been achieved.
A significant finding of our research efforts into the α-arylation of sulfonamides, sulfones, and sulfoxides is that Kwong’s indole-based phosphine exhibits excellent selectivity with these challenging substrates. Despite the increased acidity of the products over the starting materials by at lease 5 pKa units, we observe mono-arylation with high selectivity. We hypothesize that the large size of Kwong’s ligand is responsible for the remarkable selectivity for the monoarylation product in these reactions. We attribute the observed selectivity to a slow rate of transmetallation of the deprotonated benzyl sulfonamide with Pd(II) ligated to Kwong’s bulky phosphine. It will be interesting to see if our hypothesis above can be supported by successful application of Kwong’s ligand to other systems where diarylation is problematic.
Experimental Section
General Information
All reactions were carried out under an atmosphere of dry nitrogen. Anhydrous dioxane, CPME, and toluene were purchased from Sigma-Aldrich and used without further purification. THF was dried through activated alumina columns. Unless otherwise stated, all reagents were commercially available and used without further purification. Flash chromatography was performed with silica gel (300–400 mesh). The NMR spectra were obtained using a Brüker 500 MHz Fourier-transform NMR spectrometer. High-resolution mass spectrometry (HRMS) data were obtained on a Waters LC-TOF mass spectrometer (model LCT-XE Premier) using chemical ionization (CI) or electrospray ionization (ESI) in positive or negative mode, depending on the analyte.
General Experimental Procedure: High-throughput Experimentation Screening for Palladium-Catalyzed α-Arylation of Methyl Sulfonamides with Aryl Chlorides
(1) Screening of ligand
Set up
Two 24-Well Plate
Experiments were set up inside a glovebox under a nitrogen atmosphere. A 24-well aluminum block containing 1 mL glass vials was predosed with Buchwald-type 2nd generation palladium dimer (0.5 μmol) and the phosphine ligands (2 μmol for monodentate ligands and 1 μmol for bidentate ligands) in THF. The solvent was removed to dryness using a GeneVac and LiOtBu (30 μmol) in THF was added to the ligand/catalyst mixture. The solvent was removed on the GeneVac and a parylene stir bar was then added to each reaction vial. 1-(methylsulfonyl)piperidine 1a (10 μmol/reaction) and 1-(tert-butyl)-4-chlorobenzene 2b (20 μmol) were then dosed together into each reaction vial as a solution in toluene (50 μL, 0.2 M). The 24-well plate was then sealed and stirred for 24 h at 110 °C.
Work up
Upon opening the plate to air, 500 μL of a solution of biphenyl (used as internal standard to measure HPLC yields) in acetonitrile (0.002 mol/L) was added into each vial. The plate was covered again and the vials stirred for 10 min to ensure good homogenization. Into a separate 96-well LC block was added 700 μL of acetonitrile, followed by 25 μL of the diluted reaction mixtures. The LC block was then sealed with a silicon-rubber storage mat and mounted on an automated HPLC instrument for analysis.
(2) Screening of Pd Source, base and solvent
Set up
Experiments were set up inside a glovebox under a nitrogen atmosphere. A 24-well aluminum block containing 1 mL glass vials was predosed with 2nd gen. Pd dimer (0.5 μmol) and the N-(dicyclohexylphosphino)-2-2′-tolylindole (2 μmol) in THF. The solvent was removed to dryness using a GeneVac and 6 different bases (LiOtBu, NaOtBu, KOtBu, LiN(SiMe3)2, NaN(SiMe3)2, KN(SiMe3)2 30 μmol) in THF were added to the ligand/catalyst mixture. The solvent was removed on the GeneVac and a parylene stir bar was then added to each reaction vial. 1-(Methylsulfonyl)piperidine 1a (10 μmol/reaction) and 4-tert-butyl chlorobenzene 2b (20 μmol/reaction) were then dosed together into each reaction vial as a solution in 4 different solvents (CPME, THF, DME, toluene, 50 μL, 0.2 M). The 24-well plate was then sealed and stirred for 24 h at 110 °C then cooled to room temperature.
Work up
Upon opening the plate to air, 500 μL of a solution of biphenyl (used as internal standard to measure HPLC yields) in acetonitrile (0.002 mol/L) was added into each vial. The plate was covered again and the vials stirred for 10 min to ensure good homogenization. Into a separate 96-well LC block was added 700 μL of acetonitrile, followed by 25 μL of the diluted reaction mixtures. The LC block was then sealed with a silicon-rubber storage mat and mounted on an automated HPLC instrument for analysis.
Procedures for the Pd-Catalyzed Arylation of sulfonamides
An oven-dried microwave vial equipped with a stir bar was charged with 2nd generation Pd dimer (7.2 mg, 0.010 mmol) and ligand L (16.2 mg, 0.040 mmol) under a nitrogen atmosphere, followed by 1 mL dry toluene via syringe. After the catalyst solution stirred for 120 min at 25 °C, LiOtBu (48.3 mg, 0.60 mmol, 3.0 equiv) was added to the reaction vial and 1-(methylsulfonyl) piperidine (32.6 mg, 0.20 mmol, 1.0 equiv) was added dropwise. The microwave vial was sealed and chlorobenzene (40.6 μL, 0.40 mmol, 2.0 equiv) was added by syringe while under a nitrogen atmosphere. The reaction was stirred at 110 °C for the specified time then allowed to cool to room temperature. The reaction mixture was quenched with H2O (0.2 mL) and passed through a short pad of silica gel and eluted with ethyl acetate. The combined organics were dried over Na2SO4 and concentrated in vacuo. The crude residue was purified by flash column chromatography to yield the monoarylated sulfonamides derivatives 3.
Synthesis of sumatriptan 3ja
The reaction was performed following General Procedure with 2m (128.9 mg, 0.40 mmol), LiOtBu (48.3 mg, 0.60 mmol) and 1g (39.8 mg, 0.20 mmol). Then the crude material was reacted in CF3COOH for 12 h., After workup following the General Procedure, the product was purified by flash chromatography on silica gel (eluted with MeOH:CH2Cl2 = 2:1) to give the product 3ja (44.2 mg, 75% yield) as a white solid. The 1H and 13C{1H} NMR data for this compound match the literature data.
Supplementary Material
Scheme 2.

Ligand screening.
Acknowledgments
We thank the National Science Foundation (CHE-1464744) and National Institutes of Health (NIGMS 104349) for financial support. BZ thanks the Chinese Universities Scientific Fund (2015QC001) and Peking College Students Innovation Training Program (2015bj127 and 2015bj145)
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/adsc.201######.
References
- 1.a) Bosch J, Roca T, Armengol M, Fernández-Forner D. Tetrahedron. 2001;57:1041–1048. [Google Scholar]; (b) Uno H, Kurokawa M, Masuda Y, Nishimura H. J Med Chem. 1979;22:180–183. doi: 10.1021/jm00188a011. [DOI] [PubMed] [Google Scholar]; (c) Mujumdar P, Poulsen SA. J Nat Prod. 2015;78:1470–1477. doi: 10.1021/np501015m. [DOI] [PubMed] [Google Scholar]; (d) Law V, Knox C, Djoumbou Y, Jewison T, Guo AC, Liu Y, Maciejewski A, Arndt D, Wilson M, Neveu V, Tang A, Gabriel G, Ly C, Adamjee S, Dame ZT, Han B, Zhou Y, Wishart DS. Nucleic Acids Res. 2014;42:D1091–1097. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Diao X. Handb Metab Pathways Xenobiot. 2014;3:851–854. [Google Scholar]
- 3.Saxena PR, De Vries P, Wang W, Heiligers JP, Maassen vandenBrink A, Bax WA, Yocca FD. Naunyn Schmiedebergs Arch Pharmacol. 1997;355:295–302. doi: 10.1007/pl00004946. [DOI] [PubMed] [Google Scholar]
- 4.Derry CJ, Derry S, Moore RA. Cochrane Database Syst Rev. 2014;5:CD009108. doi: 10.1002/14651858.CD009108.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Veisi H, Ghorbani-Vaghei R, Hemmati S, Mahmoodi J. Synlett. 2011:2315–2320. [Google Scholar]; b) Bahrami K, Khodaei MM, Soheilizad M. J Org Chem. 2009;74:9287–9291. doi: 10.1021/jo901924m. [DOI] [PubMed] [Google Scholar]; c) DeBergh JR, Niljianskul N, Buchwald SL. J Am Chem Soc. 2013;135:10638–10641. doi: 10.1021/ja405949a. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Nguyen B, Emmett EJ, Willis MC. J Am Chem Soc. 2010;132:16372–16373. doi: 10.1021/ja1081124. [DOI] [PubMed] [Google Scholar]; e) Ye S, Wu J. Chem Comm. 2012;48:7753–7755. doi: 10.1039/c2cc33725h. [DOI] [PubMed] [Google Scholar]
- 6.a) Ruano JLG, Parra A, Yuste F, Mastranzo VM. Synthesis. 2008:311–319. [Google Scholar]; b) Johnson MG, Gribble MW, Jr, Houze JB, Paras NA. Org Lett. 2016;16:6248–6251. doi: 10.1021/ol503208z. [DOI] [PubMed] [Google Scholar]
- 7.a) Woolven H, Gonzalez-Rodriguez C, Marco I, Thompson AL, Willis MC. Org Lett. 2011;13:4876–4878. doi: 10.1021/ol201957n. [DOI] [PubMed] [Google Scholar]; b) Colombe JR, DeBergh JR, Buchwald SL. Org Lett. 2015;17:3170–3173. doi: 10.1021/acs.orglett.5b01540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Rosen BR, Ruble JC, Beauchamp TJ, Navarro A. Org Lett. 2011;13:2564–2567. doi: 10.1021/ol200660s. [DOI] [PubMed] [Google Scholar]; (b) Moon SY, Nam J, Rathwell K, Kim WS. Org Lett. 2014;16:338–341. doi: 10.1021/ol403717f. [DOI] [PubMed] [Google Scholar]
- 9.a) Palucki M, Buchwald SL. J Am Chem Soc. 1997;119:11108–11109. [Google Scholar]; b) Hamann BC, Hartwig JF. J Am Chem Soc. 1997;119:12382–12383. [Google Scholar]; c) Novak P, Martin R. Curr Org Chem. 2011;18:3233–3262. [Google Scholar]
- 10.a) Bordwell FG, Harrelson JA, Zhang X. J Org Chem. 1991;56:4448–4450. [Google Scholar]; b) Bordwell FG. Acc Chem Res. 1988;21:456–463. [Google Scholar]
- 11.a) Zeevaart JG, Parkinson CJ, de Koning CB. Tetrahedron Lett. 2005;46:1597–1599. [Google Scholar]; b) Niwa T, Yorimitsu H, Oshima K. Tetrahedron. 2009;65:1971–1976. [Google Scholar]; c) Grimm JB, Katcher MH, Witter DJ, Northrup AB. J Org Chem. 2007;72:8135–8138. doi: 10.1021/jo701431j. [DOI] [PubMed] [Google Scholar]; d) Nambo M, Crudden CM. Angew Chem, Int Ed. 2014;53:742–746. doi: 10.1002/anie.201307019. [DOI] [PubMed] [Google Scholar]
- 12.Rene O, Fauber BP, Malhotra S, Yajima H. Org Lett. 2014;16:3468–3471. doi: 10.1021/ol501389k. [DOI] [PubMed] [Google Scholar]
- 13.Zhou G, Ting P, Aslanian R, Piwinski JJ. Org Lett. 2008;10:2517–2520. doi: 10.1021/ol800785g. [DOI] [PubMed] [Google Scholar]
- 14.In a reversed polarity approach, beginning with α-bromo sulfonamides, Fu and coworkers performed a beautiful stereoconvergent arylation with aryl zinc reagents to furnish enantioenriched sulfonamides. ( Choi J, Martin-Gago P, Fu GC. J Am Chem Soc. 2014;136:12161–12165. doi: 10.1021/ja506885s.
- 15.Bruno NC, Tudge MT, Buchwald SL. Chem Sci. 2013;4:916–920. doi: 10.1039/C2SC20903A.Kinzel T, Zhang Y, Buchwald SL. J Am Chem Soc. 2010;132:14073–14705. doi: 10.1021/ja1073799.For other example of applying Buchwald type in arylation reaction of sulfoxides, 2-azaallyl anions and diphenylmethanes with aryl halides, see: Jia T, Zhang M, Sagamanova IK, Wang CY, PJ Org Lett. 2015;17:1168–1171. doi: 10.1021/acs.orglett.5b00092.Li M, Berritt S, Walsh PJ. Org Lett. 2014;16:4312–4315. doi: 10.1021/ol502043j.Li M, Yuecel B, Adrio J, Bellomo A, Walsh PJ. Chem Sci. 2014;5:2383–2391. doi: 10.1039/C3SC53526F.Zhang J, Bellomo A, Trongsiriwat N, Jia T, Carroll PJ, Dreher SD, Tudge MT, Yin H, Robinson JR, Schelter EJ, Walsh PJ. J Am Chem Soc. 2014;136:6276–6287. doi: 10.1021/ja411855d.Li M, González-Esguevillas M, Berritt S, Yang X, Bellomo A, Walsh PJ. Angew Chem Int Ed. 2016;55:2825. doi: 10.1002/anie.201509757.Li M, Yucel B, Jimenez-Hernandez J, Rotella M, Fu Y, Walsh PJ. Adv Synth Catal. doi: 10.1002/adsc.201600075. in press.
- 16.a) Lee HW, Lam FL, So CM, Lau CP, Chan ASC, Kwong FY. Angew Chem Int Ed. 2009;48:7436–7439. doi: 10.1002/anie.200904033. [DOI] [PubMed] [Google Scholar]; b) So CM, Lau CP, Kwong FY. Org Lett. 2007;9:2795–2798. doi: 10.1021/ol070898y. [DOI] [PubMed] [Google Scholar]; c) So CM, Lau CP, Kwong FY. Chem Eur J. 2011;17:761–765. doi: 10.1002/chem.201002354. [DOI] [PubMed] [Google Scholar]; d) Wong SM, So CM, Chung KH, Lau CP, Kwong FY. Eur J Org Chem. 2012;2012:4172–4177. [Google Scholar]; e) Yeung PY, Chung KH, Kwong FY. Org Lett. 2011;13:2912–2915. doi: 10.1021/ol2009522. [DOI] [PubMed] [Google Scholar]; f) So CM, Kwong FY. Chem Soc Rev. 2011;40:4963–4972. doi: 10.1039/c1cs15114b. [DOI] [PubMed] [Google Scholar]
- 17.Jia T, Bellomo A, Baina KEL, Dreher SD, Walsh PJ. J Am Chem Soc. 2013;135:3740–3743. doi: 10.1021/ja4009776. [DOI] [PubMed] [Google Scholar]
- 18.Zheng B, Jia T, Walsh PJ. Org Lett. 2013;15:1690–1693. doi: 10.1021/ol400472v. [DOI] [PubMed] [Google Scholar]
- 19.a) Zheng B, Jia T, Walsh PJ. Org Lett. 2013;15:4190–4193. doi: 10.1021/ol4019002. [DOI] [PubMed] [Google Scholar]; b) Zheng B, Jia T, Walsh PJ. Adv Synth Catal. 2014;356:165–178. doi: 10.1002/adsc.201300851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Montel S, Raffier L, He Y, Walsh PJ. Org Lett. 2014;16:1446–1449. doi: 10.1021/ol5002413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hussain N, Kim BS, Walsh PJ. Chem Eur J. 2015;21:11010–11013. doi: 10.1002/chem.201502017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.a) Pete B, Bitter I, Harsanyi K, Toke L. Heterocycles. 2000;53:665–673. [Google Scholar]; b) Bosch J, Roca T, Armengol M, Fernandez-Forner D. Tetrahetron. 2001;57:1041–1048. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.